

Abstract
Transforming growth factor β (TGF-β) plays a critical role in wound healing and the pathogenesis of fibrosis (scarring). Three isoforms of TGF-β have been identified in mammals. Previous studies have shown that the addition of TGF-β1 (T1) or -β2 (T2) to human corneal fibroblasts (HCF) cultured in a 3-dimensional construct resulted in a fibrotic matrix, while the addition of TGF-β3 (T3) resulted in the production of enhanced non-fibrotic matrix as compared to control (Vitamin C [VitC] only). In the current investigation, we undertook the molecular comparison of fibrosis-related gene expression in T1 or T3-treated HCF to gain further insights into the regulation and roles of these two isoforms on the fibrotic response. HCF were cultured in 100mm dishes in basic medium (Eagles minimum essential medium [EMEM] with 10% fetal bovine serum [FBS]). At 70-80% confluency, cells were exposed to basic medium with 0.5mM 2-O-α-D-glucopyranosyl-L-ascorbic acid (VitC) ± 2ng/ml of T1 or T3. After 4 hours or 3 days, cells were harvested, and mRNA or protein was isolated. Fibrosis related mRNA levels were assayed using a commercial qRT-PCR Array. Selected proteins were examined using Western blotting (WB). Experiments were performed 6 times for the qRT-PCR and 4 times for WB for each condition. qRT-PCR results showed that most of the fibrosis-related genes were up or downregulated in HCF exposed to T1 or T3 as compared with VitC control. At 4 hours, only Smad7 expression was significantly altered in T3-treated HCF, compared to T1, and at 3 days, five genes were altered. WB confirmed that T1 significantly decreased Smad7 expression compared to T3 and control, and that the expression of thrombospondin-1 in T3-stimulated HCF was enhanced compared to T1-treated cells. Finally, both T1 and T3 decreased Smad3 expression dramatically at both time points. At early time points, T1 and T3 have similar effects on expression of fibrosis related genes; however, with a longer exposure, an increasing number of genes were differentially expressed. Interestingly, most of the differentially expressed gene products are secreted by the cells and may be related to the modulation of extracellular matrix.
Keywords: TGF-beta1, TGF-beta3, human corneal fibroblasts, gene array, Smad7, Smad3, Thrombospondin-1, fibrosis
1. Introduction
The cornea is a transparent tissue covering the front of the eye. It not only serves as a barrier against dirt and germs from the environment, but also plays a key role in vision. There are three main layers in the cornea: 1) epithelium, the most superficial layer; 2) stroma, the middle and thickest layer populated with quiescent keratocytes; and 3) endothelium, a single layer of cells located between the stroma and the aqueous humor (Wilson et al., 2012). In the cornea, quiescent stromal keratocytes play a critical role in keeping the cornea clear and refractive (Nishida, 1995). Injury or wounds to the cornea, especially to the stroma, frequently lead to corneal keratocyte activation, migration, and differentiation into fibroblasts and myofibroblasts, which then produce corneal haze or fibrosis (scarring). These complications of wound healing are the major causes of decreased visual quality and vision loss worldwide (Jester, 2008; Jester et al., 1999; Wilson et al., 2012).
Corneal wound healing is regulated by various growth factors, including, but not exclusive to, epidermal growth factor (EGF), fibroblast growth factor (FGF), transforming growth factor β (TGF-β), keratinocyte growth factor (KGF), hepatocyte growth factor (HGF), platelet-derived growth factor (PDGF), insulin-like growth factor (IGF), the interleukins (IL-1, IL-6, IL-8), tumor necrosis factor alpha (TNF-α), and secreted protein, acidic and rich in cystine (SPARC) (Imanishi et al., 2000; Schultz et al., 1992; Wilson et al., 2012; Yu et al., 2010). Evidence has accumulated indicating that TGF-β is one of the key regulators of fibrosis in the cornea, as well as in other tissues (Branton and Kopp, 1999; Cutroneo, 2007; Karamichos et al., 2010; Leask and Abraham, 2004; Park et al., 2001; Ruiz-Ortega et al., 2007; Tatler and Jenkins, 2012; Wilson et al., 2012). Three closely related isoforms of TGF-β—TGF-β1, -β2, and -β3 (T1, T2, and T3, respectively)—have been identified in mammals and are encoded by three distinct genes with 64-85% amino acid sequence homology (Schiller et al., 2004; Wilson et al., 2012). All three isoforms are synthesized as precursor molecules containing a propeptide region and the TGF-β homodimer. During processing, they are cleaved to form an active 25kDa growth factor and a latency-associated peptide (LAP). Although cleaved, these two pieces remain a small complex attached by noncovalent bonds. This small complex then binds another protein, latent TGF-β-binding protein (LTBP), to form a large complex before it is secreted. After its secretion, it remains in the extracellular matrix (ECM) as an inactive complex containing the LAP and LTBP, which prevents the complex from binding to its receptors. The inactive complex of TGF-β is further processed through proteases, acid, reactive oxygen species (ROS), thrombospndin-1 (THBS1), and αV containing integrins in order to release active TGF-β (Annes et al., 2004; Carrington et al., 2006; Daniel et al., 2007; Shi et al., 2011; Taylor, 2009). Since different cellular mechanisms require distinct levels of TGF-β signaling, the inactive complex of this growth factor gives opportunity for a proper mediation of TGF-β signaling (Annes et al., 2003).
All three TGF-β isoforms signal through the same cell surface receptors and have similar cellular targets (Letterio and Roberts, 1998). Signaling occurs once the activated TGF-β is directly bound to its receptor (TGF-βRII or TRII), which then forms a complex with the type I receptor (TRI). The canonical signaling pathway for TGF-β is the Smad-cascade. Three functional classes of Smads are involved in this pathway: the receptor-activated Smad (R-Smads: Smad1, 2, 3, 5, and 8), the common mediator Smad (Co-Smad: Smad4), and the inhibitory Smads (I-Smads: Smad6 and 7) (Euler-Taimor and Heger, 2006; Massague et al., 2005). T1 and T3 exhibit a substantial overlap in biological functions, such as inhibition of proliferation of different cell types, regulation of immune response, and stimulation of ECM formation (Olsson et al., 2000). Among them, T1 is the most commonly documented isoform. It has been found to promote the transition of quiescent corneal keratocytes to activated fibroblasts to myofibroblasts in a rabbit corneal wound (Jester et al., 2002), and induce fibrosis and scar formation after wounding (Ask et al., 2008; Shah et al., 1995); while T3 has been shown in skin, lung, and kidney models to be anti-fibrotic, and in some cases, to stimulate scar-free healing (Ask et al., 2008; Carrington et al., 2006; Eslami et al., 2009; Hosokawa et al., 2003; Occleston et al., 2008; Ohno et al., 2012; Saika, 2006; So et al., 2011; Waddington et al., 2010). We have developed a cell-based 3-dimensional (3D) corneal stromal construct with a self-assembled matrix that mimics the in vivo stromal development (Karamichos et al., 2011). Interestingly, addition of T1 or T2 to the culture resulted in a fibrotic matrix; however, addition of T3 resulted in enhanced matrix production compared to control, with no evidence of fibrotic markers, such as collagen type III and α-smooth muscle actin (SMA, also known as ACTA2). It remains unclear why T1 and T3 stimulate different responses. Insights of molecular mechanisms of T1 and T3 and their relationship with ECM deposition by HCF would provide effective options for understanding the potential anti-scarring effects of T3. In the current study, we compared the fibrosis-related mRNA and protein level changes in HCFs treated with T1 or T3.
2. Material and Methods
This study adheres to the Declaration of Helsinki and was approved by the institutional review board for human subjects at the Schepens Eye Research Institute/Massachusetts Eye and Ear.
2.1. Cell Culture
The isolation and culture of human corneal fibroblasts (HCF) was described previously (Guo et al., 2007b). Briefly, human corneal stromal explants from eyes received from National Disease Research Interchange (NDRI; Philadelphia, PA) were put in 6-well plates in basic medium (Eagles minimum essential medium with 10% FBS) and incubated at 37°C with 5% CO2 until sufficient HCF migrated from the explants. The HCF then were seeded in 100mm dishes in basic medium until they reached 70-80% confluency, at which time the cells were exposed to Vitamin C (VitC) medium (basic medium plus 0.5mM 2-O-α-D-glucopyranosyl-L-ascorbic acid) ± 2ng/ml of T1 or T3, and harvested after 4 hours or 3 days. This concentration of TGF-β has been previously used by us and shown to activate TGF-β receptor (Zieske et al., 2001). Experiments were performed 6 times for quantitative real-time polymerase chain reaction (qRT-PCR) and 4 times for western blot for each condition.
2.2. Indirect Immunofluorescence
HCF (1×105) were plated on 4-well chamber slides, and grown for 3 days in VitC medium ± T1 or T3. The medium was removed and cells were fixed with 100% Methanol at -20°C for 10 minutes. Indirect-immunofluorescence (IF) was performed as previously described (Zieske et al., 1994). Cells were incubated with primary antibody against SMA (Dako North America, Inc.; Carpinteria, CA) and then the corresponding secondary antibody (donkey anti-mouse IgG-FITC: Jackson ImmunoResearch; West Grove, PA). After which, the cells were coverslipped with Vectashield mounting media containing DAPI (Vector Laboratory; Burlingame, CA), a nuclear counterstain. Cells were then examined with a Nikon Eclipse E800 equipped with an Andor Clara E camera and Nikon NIS Elements for Basic Research (Micro Video Instruments, Inc.; Avon, MA) and photographed. ImageJ (v. 1.45s; National Institutes of Health; Bethesda, MD: http://imagej.nih.gov/ij) was used to count the number of SMA-positive cells and nuclei stained with DAPI. The experiment was performed 4 times and for each time, at least 5 areas in each well were counted and averaged.
2.3. RNA Isolation and qRT-PCR
qRT-PCR experiments were performed to screen for fibrosis-related genes that may be differentially regulated in HCF stimulated with either T1 or T3. Total RNA was isolated from HCF using Trizol (Invitrogen; Carlsbad, CA) according to manufacturer's protocol, and further total RNA purification was performed using RNeasy Mini kit (Qiagen; Valencia, CA). Total RNA (1μg) then was reverse transcribed to cDNA using the RT2 first-strand kit (Qiagen), and qRT-PCR was performed according to manufacturer's instructions for the Human Fibrosis RT2 PCR Array (Qiagen). The Array was run on an Eppendorf Multiplex 2 Real Time PCR machine (Eppendorf; Hauppauge, NY), and the data was analyzed with the SABiosciences RT2 Profiler PCR Array Data Analysis Software, v.3.4.
2.4. SDS-PAGE and Western Blot Analysis
Protein isolation and western blot analysis were performed as previously described (Zieske et al., 2001). In brief, protein from HCF treated ± T1 or T3 in VitC medium was extracted with RIPA buffer (10mM Tris, 150nM NaCl, 1% deoxycholic acid, 1% Triton X, 0.1% SDS, 1mM EDTA) plus protease inhibitors (aprotinin, PMSF, and sodium orthovanadate). Protein concentration was determined using a protein assay kit (Bio-Rad Protein Assay; Hercules, CA), and equal amount of protein (15-30μg/lane) from each sample was loaded onto 4-20% gradient Tris-Glycine Gels (Invitrogen). Proteins were transferred onto PVDF membranes (Invitrogen), and the transfer was confirmed by staining the membrane with 0.1% Ponceau S solution (Sigma-Aldrich; St. Louis, MO). Membranes then were incubated with primary antibodies against either type I collagen (abcam; Cambridge, MA), Smad3 (Zymed; Grand Island, NY), Smad7 (LifeSpan BioSciences, Inc.; Seattle, WA), or thrombospondin-1 (THBS1: Thermo Scientific; Waltham, MA). Protein bands were detected by Chemiluminescence (Milipore; Billerica, MA) after exposure to film. Band intensities were quantified with imaging software (ImageJ, v.1.45s: National Institutes of Health, USA; http://imagej.nih.gov/ij).
2.5. Statistical Analysis
All experiments were repeated 4-6 times and data was analyzed for significance (p<0.05 to p<0.001) using the Student's t-test and Dunnett's Multiple Comparison test (GraphPad Prism v.5.0b; La Jolla, CA).
3. Results
3.1. Indirect-Immunofluorescence
We have previously observed that the addition of T1 to 4-week HCF cultures stimulated an increase of SMA-positive cells that was far greater than seen upon T3 stimulation (Karamichos et al., 2011). To confirm that this effect was also seen in short term cultures, T1 or T3 were added to HCF and the number of SMA-positive cells were observed by indirect-immunofluorescence and quantified (Figure 1). Figure 1 shows that the number of SMA-positive cells was significantly increased (p<0.05) by both T1 (Fig. 1B and D) and T3 (Fig. 1C and D) as compared with control (Fig. 1A and D), with T1 having a higher number of SMA-positive cells than T3.
Figure 1. Indirect-immunofluorescent analysis of α-smooth muscle actin (SMA) in human corneal fibroblasts (HCF) ± TGF-β1 (T1) or TGF-β3 (T3).
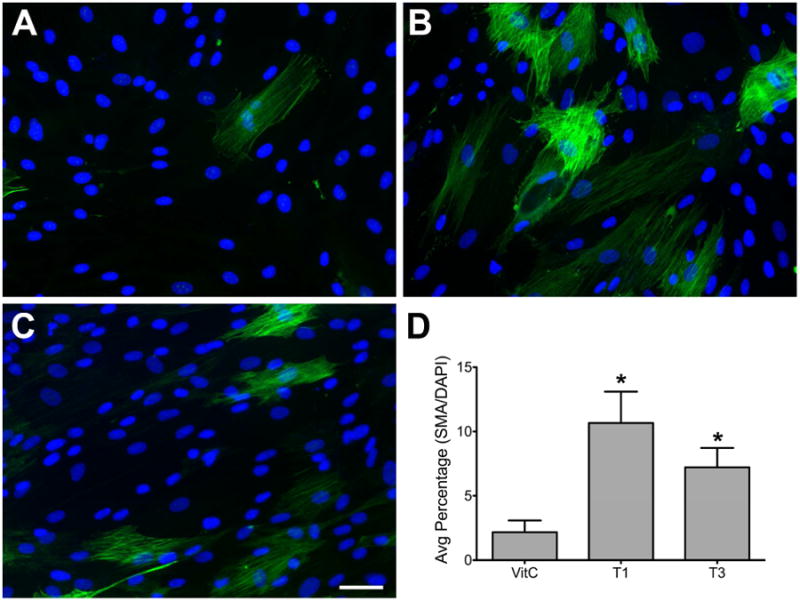
Representative indirect-imuunofluorescent images show that in Control (VitC) samples (A), there are only a few SMA-positive cells present; however, with the addition of T1 (B), the number of SMA-positive cells increased significantly (D: *p<0.05) as compared with control (A and D). T3-stimulated cells (C) also showed a significant increase of SMA-positive cells (D: *p<0.05) as compared with control (A and D), however, to a lesser extent than T1 (B and D).
3.2. qRT-PCR
Since T1 and T3 appeared to have differing effects on the increase in the number of SMA-positive cells, we used a commercial PCR array to examine genes associated with fibrosis (Table 1). The Human Fibrosis PCR Array contained 84 key genes involved in the dysregulated tissue remodeling during repair and healing of wounds. Numerous statistically significant changes were seen in all conditions examined. As seen in Table 2, in response to T1 compared with Control, 25 genes were upregulated and 4 downregulated by 4 hours, while 10 were upregulated and 12 downregulated by 3 days. In response to T3, 16 genes were upregulated and 7 were downregulated by 4 hours, and 19 were upregulated and 8 were downregulated by 3 days. Several patterns of gene regulation were observed. One of the most dramatic was INHBE (inhibin, beta E), whose mRNA in T1-treated samples changed dramatically from a decrease of about 3 fold at 4 hours to an increase of 127 fold at 3 days. Other genes, such as Smad 7, ITGB6 (Integrin, beta 6), CTGF (connective tissue growth factor), PDGFA (platelet-derived growth factor alpha), and SERPINE 1 (serpin peptidase inhibitor, clade E [nexin, plasminogen activator inhibitor type 1], member 1), maintained a significant upregulation in both 4-hour and 3-day T1-stimulated samples. Still others went from a significant increase at 4 hours to a significant decrease by 3 days with T1 stimulation, these genes include, ITGB3 (Integrin, beta 3), MMP1 (matrix metalloproteinase 1), PLAU (Plasminogen Activator, Urokinase), CCL2 (Chemokine [C-C motif] ligand 2) and CCL11. In T3-treated samples, COL3A1 (collagen type III, alpha 1) mRNA changed from -2 fold at 4 hours to +4 fold at 3 days, whereas, ITGB6, ITGAV (integrin, alpha V), EDN1 (endothelin 1), CTGF, VEGFA (vascular endothelial growth factor A), PDGFA, SERPINE1, THBS1 all remained upregulated in 4-hour and 3-day samples. Only CCL11 mRNA decreased with time, changing from 6.3 fold at 4 hours to -8.7 fold by 3 days.
Table 1. Genes on the Human Fibrosis RT2 PCR Array (Qiagen).
| Symbol | Description | Gname |
|---|---|---|
| ACTA2 | Actin, alpha 2, smooth muscle, aorta | AAT6/ACTSA |
| AGT | Angiotensinogen (serpin peptidase inhibitor, clade A, member 8) | ANHU/FLJ92595/FLJ97926/SERPINA8 |
| AKT1 | V-akt murine thymoma viral oncogene homolog 1 | AKT/MGC99656/PKB/PKB-ALPHA/PRKBA/RAC/RAC-ALPHA |
| BCL2 | B-cell CLL/lymphoma 2 | Bcl-2 |
| BMP7 | Bone morphogenetic protein 7 | OP-1 |
| CAV1 | Caveolin 1, caveolae protein, 22kDa | BSCL3/CGL3/MSTP085/VIP21 |
| CCL11 | Chemokine (C-C motif) ligand 11 | MGC22554/SCYA11 |
| CCL2 | Chemokine (C-C motif) ligand 2 | GDCF-2/HC11/HSMCR30/MCAF/MCP-1/MCP1/MGC9434/SCYA2/SMC-CF |
| CCL3 | Chemokine (C-C motif) ligand 3 | G0S19-1/LD78ALPHA/MIP-1-alpha/MIP1A/SCYA3 |
| CCR2 | Chemokine (C-C motif) receptor 2 | CC-CKR-2/CCR2A/CCR2B/CD192/CKR2/CKR2A/CKR2B/CMKB R2/FLJ78302/MCP-1-R/MGC103828/MGC111760/MGC168006 |
| CEBPB | CCAAT/enhancer binding protein (C/EBP), beta | C/EBP-beta/CRP2/IL6DBP/LAP/MGC32080/NF-IL6/TCF5 |
| COL1A2 | Collagen, type I, alpha 2 | OI4 |
| COL3A1 | Collagen, type III, alpha 1 | EDS4A/FLJ34534 |
| CTGF | Connective tissue growth factor | CCN2/HCS24/IGFBP8/MGC102839/NOV2 |
| CXCR4 | Chemokine (C-X-C motif) receptor 4 | CD184/D2S201E/FB22/HM89/HSY3RR/LAP3/LCR1/LE STR/NPY3R/NPYR/NPYRL/NPYY3R/WHIM |
| DCN | Decorin | CSCD/DSPG2/PG40/PGII/PGS2/SLRR1B |
| EDN1 | Endothelin 1 | ET1/HDLCQ7/PPET1 |
| EGF | Epidermal growth factor | HOMG4/URG |
| ENG | Endoglin | CD105/END/FLJ41744/HHT1/ORW/ORW1 |
| FASLG | Fas ligand (TNF superfamily, member 6) | APT1LG1/CD178/CD95-L/CD95L/FASL/TNFSF6 |
| GREM1 | Gremlin 1 | CKTSF1B1/DAND2/DRM/GREMLIN/IHG-2/MGC126660 |
| HGF | Hepatocyte growth factor (hepapoietin A; scatter factor) | DFNB39/F-TCF/HGFB/HPTA/SF |
| IFNG | Interferon, gamma | IFG/IFI |
| IL10 | Interleukin 10 | CSIF/IL-10/IL10A/MGC126450/MGC126451/TGIF |
| IL13 | Interleukin 13 | ALRH/BHR1/IL-13/MGC116786/MGC116788/MGC116789/P600 |
| IL13RA2 | Interleukin 13 receptor, alpha 2 | CD213A2/CT19/IL-13R/IL13BP |
| IL1A | Interleukin 1, alpha | IL-1A/IL1/IL1-ALPHA/IL1F1 |
| IL1B | Interleukin 1, beta | IL-1/IL1-BETA/IL1F2 |
| IL4 | Interleukin 4 | BCGF-1/BCGF1/BSF-1/BSF1/IL-4/MGC79402 |
| IL5 | Interleukin 5 (colony-stimulating factor, eosinophil) | EDF/IL-5/TRF |
| ILK | Integrin-linked kinase | DKFZp686F1765/ILK-2/P59 |
| INHBE | inhibin, beta E | MGC4638 |
| ITGA1 | Integrin, alpha 1 | CD49a/VLA1 |
| ITGA2 | Integrin, alpha 2 (CD49B, alpha 2 subunit of VLA-2 receptor) | BR/CD49B/GPIa/VLA-2/VLAA2 |
| ITGA3 | Integrin, alpha 3 (antigen CD49C, alpha 3 subunit of VLA-3 receptor) | CD49C/FLJ34631/FLJ34704/GAP-B3/GAPB3/MSK18/VCA-2/VL3A/VLA3a |
| ITGAV | Integrin, alpha V (vitronectin receptor, alpha polypeptide, antigen CD51) | CD51/DKFZp686A08142/MSK8/VNRA |
| ITGB1 | Integrin, beta 1 (fibronectin receptor, beta polypeptide, antigen CD29 includes MDF2, MSK12) | CD29/FNRB/GPIIA/MDF2/MSK12/VLA-BETA/VLAB |
| ITGB3 | Integrin, beta 3 (platelet glycoprotein IIIa, antigen CD61) | CD61/GP3A/GPIIIa |
| ITGB5 | Integrin, beta 5 | FLJ26658 |
| ITGB6 | Integrin, beta 6 | - |
| ITGB8 | Integrin, beta 8 | - |
| JUN | Jun proto-oncogene | AP-1/AP1/c-Jun |
| LOX | Lysyl oxidase | MGC105112 |
| LTBP1 | Latent transforming growth factor beta binding protein 1 | MGC163161 |
| MMP1 | Matrix metallopeptidase 1 (interstitial collagenase) | CLG/CLGN |
| MMP13 | Matrix metallopeptidase 13 (collagenase 3) | CLG3/MANDP1 |
| MMP14 | Matrix metallopeptidase 14 (membrane-inserted) | 1/MMP-14/MMP-X1/MT-MMP/MT-MMP 1/MT1-MMP/MT1MMP/MTMMP1 |
| MMP2 | Matrix metallopeptidase 2 (gelatinase A, 72kDa gelatinase, 72kDa type IV collagenase) | CLG4/CLG4A/MMP-II/MONA/TBE-1 |
| MMP3 | Matrix metallopeptidase 3 (stromelysin 1, progelatinase) | CHDS6/MGC126102/MGC126103/MGC126104/MMP-3/SL-1/STMY/STMY1/STR1 |
| MMP8 | Matrix metallopeptidase 8 (neutrophil collagenase) | CLG1/HNC/MMP-8/PMNL-CL |
| MMP9 | Matrix metallopeptidase 9 (gelatinase B, 92kDa gelatinase, 92kDa type IV collagenase) | CLG4B/GELB/MANDP2/MMP-9 |
| MYC | V-myc myelocytomatosis viral oncogene homolog (avian) | MRTL/bHLHe39/c-Myc |
| NFKB1 | Nuclear factor of kappa light polypeptide gene enhancer in B-cells 1 | DKFZp686C01211/EBP-1/KBF1/MGC54151/NF-kappa-B/NF-kappaB/NFKB-p105/NFKB-p50/NFkappaB/p105/p50 |
| PDGFA | Platelet-derived growth factor alpha polypeptide | PDGF-A/PDGF1 |
| PDGFB | Platelet-derived growth factor beta polypeptide | FLJ12858/PDGF2/SIS/SSV/c-sis |
| PLAT | Plasminogen activator, tissue | DKFZp686I03148/T-PA/TPA |
| PLAU | Plasminogen activator, urokinase | ATF/UPA/URK/u-PA |
| PLG | Plasminogen | DKFZp779M0222 |
| SERPINA1 | Serpin peptidase inhibitor, clade A (alpha-1 antiproteinase, antitrypsin), member 1 | A1A/A1AT/AAT/MGC23330/MGC9222/PI/PI1/PRO2275/ alpha1AT |
| SERPINE1 | Serpin peptidase inhibitor, clade E (nexin, plasminogen activator inhibitor type 1), member 1 | PAI/PAI-1/PAI1/PLANH1 |
| SERPINH1 | Serpin peptidase inhibitor, clade H (heat shock protein 47), member 1, (collagen binding protein 1) | AsTP3/CBP1/CBP2/HSP47/PPROM/RA-A47/SERPINH2/gp46 |
| SMAD2 | SMAD family member 2 | JV18/JV18-1/MADH2/MADR2/MGC22139/MGC34440/hMAD-2/hSMAD2 |
| SMAD3 | SMAD family member 3 | DKFZp586N0721/DKFZp686J10186/HSPC193/HsT174 36/JV15-2/MADH3/MGC60396 |
| SMAD4 | SMAD family member 4 | DPC4/JIP/MADH4 |
| SMAD6 | SMAD family member 6 | HsT17432/MADH6/MADH7 |
| SMAD7 | SMAD family member 7 | CRCS3/FLJ16482/MADH7/MADH8 |
| SNAI1 | Snail homolog 1 (Drosophila) | SLUGH2/SNA/SNAH/SNAIL/SNAIL1/dJ710H13.1 |
| SP1 | Sp1 transcription factor | - |
| STAT1 | Signal transducer and activator of transcription 1, 91kDa | DKFZp686B04100/ISGF-3/STAT91 |
| STAT6 | Signal transducer and activator of transcription 6, interleukin-4 induced | D12S1644/IL-4-STAT/STAT6B/STAT6C |
| TGFB1 | Transforming growth factor, beta 1 | CED/DPD1/LAP/TGFB/TGFbeta |
| TGFB2 | Transforming growth factor, beta 2 | MGC116892/TGF-beta2 |
| TGFB3 | Transforming growth factor, beta 3 | ARVD/FLJ16571/TGF-beta3 |
| TGFBR1 | Transforming growth factor, beta receptor 1 | AAT5/ACVRLK4/ALK-5/ALK5/LDS1A/LDS2A/SKR4/TGFR-1 |
| TGFBR2 | Transforming growth factor, beta receptor II (70/80kDa) | AAT3/FAA3/LDS1B/LDS2B/MFS2/RIIC/TAAD2/TGFR-2/TGFbeta-RII |
| TGIF1 | TGFB-induced factor homeobox 1 | HPE4/MGC39747/MGC5066/TGIF |
| THBS1 | Thrombospondin 1 | THBS/THBS-1/TSP/TSP-1/TSP1 |
| THBS2 | Thrombospondin 2 | TSP2 |
| TIMP1 | TIMP metallopeptidase inhibitor 1 | CLGI/EPA/EPO/FLJ90373/HCI/TIMP |
| TIMP2 | TIMP metallopeptidase inhibitor 2 | CSC-21K |
| TIMP3 | TIMP metallopeptidase inhibitor 3 | HSMRK222/K222/K222TA2/SFD |
| TIMP4 | TIMP metallopeptidase inhibitor 4 | - |
| TNF | Tumor necrosis factor | DIF/TNF-alpha/TNFA/TNFSF2 |
| VEGFA | Vascular endothelial growth factor A | MGC70609/MVCD1/VEGF/VPF |
| B2M | Beta-2-microglobulin | |
| HPRT1 | Hypoxanthine phosphoribosyltransferase 1 | HGPRT/HPRT |
| RPL13A | Ribosomal protein L13a | L13A/TSTA1 |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase | G3PD/GAPD/MGC88685 |
| ACTB | Actin, beta | PS1TP5BP1 |
Shaded = Housekeeping genes
Table 2. qRT-PCR array data comparing human corneal fibroblasts (HCF) stimulated with either TGF-β1 (T1) or -β3 (T3) to Control.
| Gene Symbol | T1 vs Control | T3 vs Control | ||||||
|---|---|---|---|---|---|---|---|---|
| 4 hours | 3 days | 4 hours | 3 days | |||||
| Fold Regulation | P | Fold Regulation | P | Fold Regulation | P | Fold Regulation | P | |
| AGT | 2.4051 | ** | 2.7865 | ** | ||||
| BCL2 | 2.3968 | *** | 2.1363 | *** | ||||
| CAV1 | (-4.6375) | *** | (-2.5319) | *** | (-3.1986) | *** | ||
| CCL11 | 5.1615 | ** | (-8.187) | *** | 6.3252 | ** | (-8.7147) | *** |
| CCL2 | 2.0108 | *** | (-8.604) | *** | (-4.0731) | ** | ||
| COL1A2 | 2.2983 | *** | ||||||
| COL3A1 | 2.1019 | ** | (-2.0082) | ** | 3.8563 | *** | ||
| CTGF | 10.2398 | *** | 18.4856 | * | 9.0303 | *** | 24.3563 | * |
| DCN | (-4.7623) | *** | (-2.6204) | *** | ||||
| EDN1 | 25.7728 | *** | 7.2518 | *** | 26.0602 | *** | 10.4196 | ** |
| FASLG | 6.1807 | *** | 4.9983 | ** | ||||
| GREM1 | 2.0721 | *** | 2.3891 | *** | ||||
| HGF | (-73.5167) | *** | (-2.2872) | *** | (-55.9388) | *** | ||
| IL13RA2 | (-7.586) | *** | (-4.7047) | *** | ||||
| IL1A | 2.6502 | *** | 2.055 | *** | ||||
| IL1B | (-3.6092) | *** | ||||||
| INHBE | (-2.8558) | *** | 127.2628 | *** | 158.9272 | *** | ||
| ITGA1 | 2.1852 | *** | 3.7883 | *** | ||||
| ITGA2 | 2.372 | *** | ||||||
| ITGAV | 4.9512 | *** | 3.8445 | *** | 3.7405 | ** | ||
| ITGB3 | 2.3775 | *** | (-3.1167) | * | ||||
| ITGB5 | 2.6182 | *** | ||||||
| ITGB6 | 61.8676 | *** | 16.1486 | ** | 53.8837 | *** | 32.9884 | ** |
| JUN | (-2.5501) | *** | (-2.9729) | *** | ||||
| LOX | 3.0455 | *** | 4.266 | *** | ||||
| MMP1 | 4.4314 | *** | (-3.1203) | ** | 4.3483 | *** | ||
| MMP13 | 3.8984 | ** | ||||||
| MMP2 | 2.1658 | ** | ||||||
| MMP3 | (-3.9816) | ** | ||||||
| PDGFA | 5.3127 | *** | 8.6638 | *** | 3.8712 | *** | 9.5924 | *** |
| PLAT | 2.4415 | *** | ||||||
| PLAU | 2.3475 | *** | (-15.8345) | *** | (-4.9478) | ** | ||
| SERPINA1 | 3.4929 | *** | 2.3907 | * | ||||
| SERPINE1 | 4.3352 | *** | 5.6503 | *** | 3.8649 | *** | 7.1464 | *** |
| SERPINH1 | (-2.0499) | *** | (-2.8243) | *** | ||||
| SMAD3 | (-10.0329) | *** | (-2.6961) | *** | (-7.3723) | *** | ||
| SMAD4 | 2.4107 | *** | ||||||
| SMAD6 | (-3.3571) | ** | (-3.6273) | *** | ||||
| SMAD7 | 3.4809 | *** | 3.2603 | * | 5.7846 | * | ||
| SNAI1 | 2.1212 | ** | ||||||
| TGFB2 | 2.1523 | ** | ||||||
| THBS1 | 4.4314 | *** | 2.5393 | *** | 4.265 | ** | ||
| TIMP3 | 2.9542 | ** | ||||||
| VEGFA | 7.5655 | *** | 2.5997 | * | 7.9234 | *** | 3.6056 | *** |
Upregulated (black) or downregulated (red) genes that were statistically significant in HCF when stimulated with either T1 or T3 as compared with Control samples at 4 hours and 3 day. By 4 hours with T1 stimulation, there were 25 genes that were significantly upregulated and 4 downregulated as compared with Control. By 3 days, the upregulated quantity decreased to 10 genes and the downregulated increased to 12 genes. With T3 by 4 hours, there were 16 genes significantly upregulated and 7 genes downregulated, and by 3 days there were 19 genes upregulated and 8 genes downregulated.
P<0.05,
P<0.01 and
P<0.001
Only genes that were altered by 2-fold or greater are included.
One of the goals of our study was to determine if differential alterations in gene expression could be determined between T1- and T3-stimulated samples as compared with control. As seen in Table 2, many of the genes showed the same regulation by both T1 and T3. The 3 genes that were most highly stimulated by both T1 and T3 as compared with control at 4 hours were ITGB6, END1, and CTGF, while at 3 days they were INHBE, ITGB6, and CTGF. ITGB6 and CTGF mRNA levels were maintained at both time points. In Table 3, the effect of T3 was compared with T1. Although both growth factors stimulated numerous changes, at 4 hours only Smad7 mRNA levels were significantly different (-2.0 fold) between the two groups. At the longer time point (3 days) an increased number of changes were observed, MMP1, PLAU, IL1B (Interleukin-1 beta), ITGA1, and THBS1 were all significantly higher in T3 samples as compared with T1 (Table 3).
Table 3. qRT-PCR array data comparing human corneal fibroblasts (HCF) stimulated with TGF-β3 (T3) to those stimulated with TGF-β1 (T1).
| T3 vs T1 | ||||
|---|---|---|---|---|
|
| ||||
| Gene Symbol | 4 hours | 3 days | ||
| Fold Regulation | P | Fold Regulation | P | |
| IL1B | 2.7155 | ** | ||
| ITGA1 | 2.3948 | ** | ||
| MMP1 | 3.2736 | * | ||
| PLAU | 3.2003 | * | ||
| SMAD7 | (-2.0284) | ** | ||
| THBS1 | 2.339 | ** | ||
Statistically significant regulation of genes that were either up- or downregulated in HCF when stimulated with T3 as compared to those stimulated with T1 at 4 hours and 3 days. By 4 hours, only Smad7 appeared in T3 samples to have a statistically significant decrease in mRNA as compared with T1; however, by 3 days, 5 genes were statistically significant in T3 samples compared with T1, and there were no decreases of note.
P<0.05,
P<0.01 and
P<0.001
Only genes that were altered by 2-fold or greater are included.
3.3. Protein expression
From the qRT-PCR data, we found that compared to control, mRNA levels of many fibrosis-related genes were significantly altered by 4-hour or 3-day treatment with T1 or T3. To determine if protein levels reflected the changes seen in mRNA levels, we analyzed by western blot the protein product of two genes that were differentially regulated, THBS1 and Smad7, and two genes that were similarly regulated, Collagen I and Smad3. Only 3-day protein samples were analyzed due to insufficient time for the 4-hour samples to be substantially altered. As shown in Figure 2, by 3-days exposure to T1 or T3, Smad7 was significantly decreased by T1 (p<0.001); however, T3 had no significant effect. THBS1 was significantly increased by T3, 2.5-fold higher than control (p<0.05); however, T1 had no significant effect. Finally, Collagen I was significantly increased (p<0.05) and Smad3 was dramatically inhibited (p<0.001). These data agree with the qRT-PCR data.
Figure 2. Western blot analysis of selected genes.
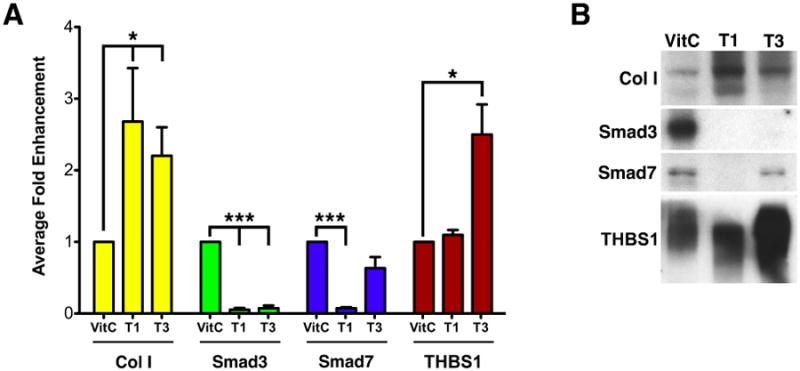
Selected genes of interest (type I collagen [Col I], Smad3, Smad7, and thrombospondin-1 [THBS1]) were examined by western blot to determine if the protein reflected the changes seen in the qRT-PCR data at 3 days. (A) Graph showing Average Fold Enhancement of samples treated either with Vitamin C only (Control, VitC), TGF-β1 (T1) or TGF-β3 (T3). (*P<0.05 and ***P<0.001) (B) Representative western blots for the data presented in (A).
4. Discussion
TGF-β plays a critical role in wound healing and the pathogenesis of fibrosis (or scarring). Therefore, determining how TGF-β regulates the expression of fibrosis-related genes is essential to understanding the mechanisms of these processes. Three isoforms of TGF-β have been identified in mammals—T1, T2, and T3. Previous studies (Karamichos et al., 2011) have shown that the addition of T1 or T2 to our 3D HCF culture resulted in a fibrotic matrix, including increased expression of type III collagen and SMA. When T3 was added to these cultures, an enhanced matrix was produced compared to control, with relative thickness similar to that of the T1-treated construct; however, there was no evidence of fibrotic markers. Identifying the genes regulated by T1 and T3 in HCF may help us to understand the mechanism of corneal fibrosis associated with TGF-β. Here we undertook the molecular comparison of fibrosis-related gene expression in HCF exposed to T1 or T3 in the presence of VitC to gain further insights into the regulation and roles of these two isoforms on the fibrotic response. We choose to examine 2D cultures rather than 3D in order to reduce variability and increase sample amounts. In our initial studies, mRNA levels of 84 key genes involved in human fibrosis were tested using qRT-PCR array technology. After which, Western blot was performed to compare the protein expression of several genes that showed dramatic changes of mRNA. Two time points, 4 hours and 3 days of TGF-β treatment, were chosen in the present study to examine both rapid and slow responses.
Surprisingly, although many genes were regulated by both T1 and T3 at the early 4-hour time point, only one gene (Smad7, an I-Smad) appeared to be differentially regulated. At 4 hours as compared to control, Smad7 mRNA expression was upregulated by T1 (∼3 fold) and maintained at that level through at least 3 days. With T3 stimulation, Smad7 mRNA did not show any significant increase by 4 hours; however, by 3 days, Smad7 mRNA levels were dramatically upregulated (∼6 fold). The western blot data (Fig. 2) agrees with the qRT-PCR data in that there is a difference in Smad7 expression in HCF stimulated with T1 and T3. At 3 days, Smad7 protein expression significantly decreased with T1, but significantly increased with T3; however, the changes in mRNA and protein levels due to T1 stimulation of the HCF were not consistent. This may be due to the fact that Smad7 is known to be an intracellular antagonist of TGF-beta signaling and has been associated with decreased fibrosis (Chung et al., 2013; Dooley et al., 2003; Guo et al., 2007a; He et al., 2009; Liu et al., 2013; Ljubimov and Saghizadeh, 2015; Nakao et al., 1999; Saika et al., 2005). Our data suggests that T3, not T1, stimulates accumulation of a non-fibrotic matrix in HCF, at least partially, through upregulating Smad7 expression.
Smad3, an R-Smad, has been observed to play a critical role as a mediator of fibrotic response in models for various tissues and organs both in vitro and in vivo (Flanders, 2004). Our qRT-PCR data shows that compared to control, HCF Smad3 mRNA started to decrease by 4 hours of T1 treatment and was significantly downregulated (10 fold) by 3 days. The decrease in Smad3 mRNA caused by T3, however, was more significant earlier on. By 4 hours of T3 exposure, Smad3 mRNA was significantly decreased by 2.7 fold and continued to decrease to 7.4 by 3 days. The inhibitory effect of T1 and T3 on Smad3 expression was confirmed by examining protein levels (Fig. 2). Our western blot results showed that Smad3 protein expression was at an almost non-detectable level after HCF were exposed to T1 or T3 for 3 days, which agrees with previous studies (Poncelet et al., 2007; Wang et al., 2005; Yanagisawa et al., 1998). Yanagisawa et al. showed that the downregulation of Smad3 mRNA in human lung epithelial cells was observed as early as 4 hours after exposure to TGF-β, with a 50% reduction at 9.3 hours (Yanagisawa et al., 1998). Other studies showed that Smad3 protein levels in tubular epithelial cells (Poncelet et al., 2007) and rat bone marrow-derived mesenchymal stem cells (Wang et al., 2005) were unaffected by T1 at early time points (less than 24 hours), but decreased markedly after 24 hours. Constitutive expression (Yanagisawa et al., 1998) or overexpression of Smad3 (Cao et al., 2007) in the presence of TGF-β has been shown to induce apoptosis in epithelial cells, and the decline in Smad3 levels has been shown to help the cells avoid apoptotic death (Poncelet et al., 2007). Indeed, in our 3D construct model, after 4 weeks of exposure to TGF-β isoforms, HCF cell number was significantly increased compare to control (Karamichos et al., 2011). We speculate that with T1 or T3 stimulation, HCF proliferate and escape from apoptosis by lowering the Smad3 expression.
At the 3-day time point, we observed five genes that were differentially expressed between the T1 and T3 groups. Although none of these genes explain the differential affect of T1 and T3 on fibrosis, they do demonstrate that the two isoforms do have differing affects. Interestingly, all of the five genes MMP1 (matrix metalloproteinase 1), PLAU (plasminogen activator, urokinase), IL1B (Interleukin-1 beta), ITGA1 (integrin alpha 1) and THBS1 either interact with or are part of the extracellular matrix. This would suggest that the differential effect of T1 and T3 might involve interaction with matrix. Since, our previous studies (Blanco-Mezquita et al., 2013; Matsuba et al., 2011; Zieske et al., 2001) demonstrated that THBS1 was upregulated in wounded cornea and healing in THBS1-deficient corneas was impaired, we further examined this protein. THBS1 is a multifunctional matrix glycoprotein that plays an important role in the healing response. Here we give direct evidence showing that both T1 and T3 upregulate THBS1 expression in HCF. Our qRT-PCR results showed that THBS1 mRNA was elevated at 4-hour treatment with both T1 and T3, indicating an early THBS1 transcriptional response to both T1 and T3 stimulation. After 3 days, THBS1 mRNA decreased to normal in T1 but continued to increase in T3, showing significant difference between T1 and T3 treated cells, which were in agreement with the western blot results. Although peak expression of THBS1 in HCF was observed at 24 hours of treatment with T1 (unpublished observations), in current studies (Fig. 2), the amount of THBS1 protein was not significantly higher than control after 3 days; however, T3 dramatically stimulated THBS1 protein expression by 3 days. It remains to be elucidated why T1 and T3 have such a different regulation on THBS1 expression in HCF.
One of our original observations that stimulated our project was the finding that in long-term HCF cultures (4 weeks), T1 stimulated fibrosis and SMA expression, while T3 did not. We anticipated that our current study would agree with this finding. With IF, we found that in short-term cultures (Fig. 1), there was a greater number of SMA-positive cells in the T1-treated HCF than the T3, which agrees with the long-term culture data; however, both T1 and T3 had significantly more SMA-positive cells than controls. With our array data, we also observed that T1 and T3 both stimulated SMA-gene expression at short time points. These data are in agreement with Agarwal and Wang (Agarwal and Wang, 2005) who found in ligament fibroblasts that T1 and T3 had similar effects at short time points. The array data may simply reflect one of the pitfalls of gene arrays in that not all probes may be equally effective, and that gene arrays should only be considered a starting point to direct the initiation of studies. However, we have also observed in our laboratory that SMA mRNA levels do not always reflect protein level and the fact that with short-term IF cultures, the data agreed somewhat with the array data. Therefore, these findings may suggest that the differential effect of T1 and T3 is not a simple matter of relative level of SMA mRNA, but in fact T1 and T3's effect may be slow to develop and complex. This may in part help explain why T3 has shown to be beneficial in some animal models, but has failed clinical trials to improve scarring (Akhurst and Hata, 2012; Ferguson et al., 2009; Finnson et al., 2013).
In conclusion, the alteration of fibrosis-related molecular expression in HCF caused by T1 or T3 stimulation was compared using qRT-PCR. Based on the PCR results, four genes were chosen to test the protein level change. Although none of the genes totally explain the differential effect of T1 and T3 on fibrosis, the results are novel in that they do identify gene targets that are differentially regulated. We found that compared to T1, T3 may stimulate non-fibrotic matrix by upregulating Smad7 and THBS1. Further work is needed to clarify why T1 and T3 have different regulation on the other genes and how they may differentially regulate fibrosis.
Highlights.
TGF-β1 and -β3 have similar effects on the expression of fibrotic genes at 4 hours.
At 3 days, there were more differentially expressed genes between TGF-β1 and -β3.
Both TGF-β1 and -β3 decreased HCF Smad3 mRNA at both time points.
TGF-β3 may stimulate non-fibrotic matrix by upregulating Smad7 and THBS1.
Acknowledgments
This study was funded by grants from National Institute of Health (EY03790 and EY005665) and Department of Defense (W81XWH-11-1-0477). Authors have no commercial interests to disclose. Data in this manuscript has been previously presented at ARVO 2013 as a poster.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Audrey E. K. Hutcheon, Email: Audrey_Hutcheon@meei.harvard.edu.
James D. Zieske, Email: James_Zieske@meei.harvard.edu.
References
- Agarwal C, Wang J. TGF-beta1 and TGF-beta3 Induce differential contraction and alpha-smooth muscle actin expression in healing ligament fibroblasts, 51st Annual Meeting of the Orthopaedic Research Society, Paper Number 0024. Washington, D.C: 2005. [Google Scholar]
- Akhurst RJ, Hata A. Targeting the TGF[beta] signalling pathway in disease. Nat Rev Drug Discov. 2012;11:790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annes JP, Chen Y, Munger JS, Rifkin DB. Integrin alphaVbeta6-mediated activation of latent TGF-beta requires the latent TGF-beta binding protein-1. The Journal of cell biology. 2004;165:723–734. doi: 10.1083/jcb.200312172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. Journal of cell science. 2003;116:217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- Ask K, Bonniaud P, Maass K, Eickelberg O, Margetts PJ, Warburton D, Groffen J, Gauldie J, Kolb M. Progressive pulmonary fibrosis is mediated by TGF-beta isoform 1 but not TGF-beta3. The international journal of biochemistry & cell biology. 2008;40:484–495. doi: 10.1016/j.biocel.2007.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Mezquita JT, Hutcheon AE, Zieske JD. Role of thrombospondin-1 in repair of penetrating corneal wounds. Investigative ophthalmology & visual science. 2013;54:6262–6268. doi: 10.1167/iovs.13-11710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branton MH, Kopp JB. TGF-beta and fibrosis. Microbes and infection / Institut Pasteur. 1999;1:1349–1365. doi: 10.1016/s1286-4579(99)00250-6. [DOI] [PubMed] [Google Scholar]
- Cao Y, Chen L, Zhang W, Liu Y, Papaconstantinou HT, Bush CR, Townsend CM, Jr, Thompson EA, Ko TC. Identification of apoptotic genes mediating TGF-beta/Smad3-induced cell death in intestinal epithelial cells using a genomic approach. American journal of physiology Gastrointestinal and liver physiology. 2007;292:G28–38. doi: 10.1152/ajpgi.00437.2005. [DOI] [PubMed] [Google Scholar]
- Carrington LM, Albon J, Anderson I, Kamma C, Boulton M. Differential regulation of key stages in early corneal wound healing by TGF-beta isoforms and their inhibitors. Investigative ophthalmology & visual science. 2006;47:1886–1894. doi: 10.1167/iovs.05-0635. [DOI] [PubMed] [Google Scholar]
- Chung AC, Dong Y, Yang W, Zhong X, Li R, Lan HY. Smad7 suppresses renal fibrosis via altering expression of TGF-beta/Smad3-regulated microRNAs. Molecular therapy : the journal of the American Society of Gene Therapy. 2013;21:388–398. doi: 10.1038/mt.2012.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutroneo KR. TGF-beta-induced fibrosis and SMAD signaling: oligo decoys as natural therapeutics for inhibition of tissue fibrosis and scarring. Wound repair and regeneration : official publication of the Wound Healing Society [and] the European Tissue Repair Society. 2007;15(1):S54–60. doi: 10.1111/j.1524-475X.2007.00226.x. [DOI] [PubMed] [Google Scholar]
- Daniel C, Schaub K, Amann K, Lawler J, Hugo C. Thrombospondin-1 is an endogenous activator of TGF-beta in experimental diabetic nephropathy in vivo. Diabetes. 2007;56:2982–2989. doi: 10.2337/db07-0551. [DOI] [PubMed] [Google Scholar]
- Dooley S, Hamzavi J, Breitkopf K, Wiercinska E, Said HM, Lorenzen J, Ten Dijke P, Gressner AM. Smad7 prevents activation of hepatic stellate cells and liver fibrosis in rats. Gastroenterology. 2003;125:178–191. doi: 10.1016/s0016-5085(03)00666-8. [DOI] [PubMed] [Google Scholar]
- Eslami A, Gallant-Behm CL, Hart DA, Wiebe C, Honardoust D, Gardner H, Hakkinen L, Larjava HS. Expression of integrin alphavbeta6 and TGF-beta in scarless vs scar-forming wound healing. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 2009;57:543–557. doi: 10.1369/jhc.2009.952572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euler-Taimor G, Heger J. The complex pattern of SMAD signaling in the cardiovascular system. Cardiovascular research. 2006;69:15–25. doi: 10.1016/j.cardiores.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Ferguson MW, Duncan J, Bond J, Bush J, Durani P, So K, Taylor L, Chantrey J, Mason T, James G, Laverty H, Occleston NL, Sattar A, Ludlow A, O'Kane S. Prophylactic administration of avotermin for improvement of skin scarring: three double-blind, placebo-controlled, phase I/II studies. Lancet (London, England) 2009;373:1264–1274. doi: 10.1016/S0140-6736(09)60322-6. [DOI] [PubMed] [Google Scholar]
- Finnson KW, McLean S, Di Guglielmo GM, Philip A. Dynamics of Transforming Growth Factor Beta Signaling in Wound Healing and Scarring. Adv Wound Care (New Rochelle) 2013;2:195–214. doi: 10.1089/wound.2013.0429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanders KC. Smad3 as a mediator of the fibrotic response. International journal of experimental pathology. 2004;85:47–64. doi: 10.1111/j.0959-9673.2004.00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Leung JC, Lam MF, Chan LY, Tsang AW, Lan HY, Lai KN. Smad7 transgene attenuates peritoneal fibrosis in uremic rats treated with peritoneal dialysis. Journal of the American Society of Nephrology : JASN. 2007a;18:2689–2703. doi: 10.1681/ASN.2007010121. [DOI] [PubMed] [Google Scholar]
- Guo X, Hutcheon AE, Melotti SA, Zieske JD, Trinkaus-Randall V, Ruberti JW. Morphologic characterization of organized extracellular matrix deposition by ascorbic acid-stimulated human corneal fibroblasts. Investigative ophthalmology & visual science. 2007b;48:4050–4060. doi: 10.1167/iovs.06-1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Sun X, Qian KQ, Liu X, Wang Z, Chen Y. Protection of cerulein-induced pancreatic fibrosis by pancreas-specific expression of Smad7. Biochimica et biophysica acta. 2009;1792:56–60. doi: 10.1016/j.bbadis.2008.10.010. [DOI] [PubMed] [Google Scholar]
- Hosokawa R, Nonaka K, Morifuji M, Shum L, Ohishi M. TGF-beta 3 decreases type I collagen and scarring after labioplasty. Journal of dental research. 2003;82:558–564. doi: 10.1177/154405910308200714. [DOI] [PubMed] [Google Scholar]
- Imanishi J, Kamiyama K, Iguchi I, Kita M, Sotozono C, Kinoshita S. Growth factors: importance in wound healing and maintenance of transparency of the cornea. Progress in retinal and eye research. 2000;19:113–129. doi: 10.1016/s1350-9462(99)00007-5. [DOI] [PubMed] [Google Scholar]
- Jester JV. Corneal crystallins and the development of cellular transparency. Seminars in cell & developmental biology. 2008;19:82–93. doi: 10.1016/j.semcdb.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jester JV, Huang J, Petroll WM, Cavanagh HD. TGFbeta induced myofibroblast differentiation of rabbit keratocytes requires synergistic TGFbeta, PDGF and integrin signaling. Experimental eye research. 2002;75:645–657. doi: 10.1006/exer.2002.2066. [DOI] [PubMed] [Google Scholar]
- Jester JV, Petroll WM, Cavanagh HD. Corneal stromal wound healing in refractive surgery: the role of myofibroblasts. Progress in retinal and eye research. 1999;18:311–356. doi: 10.1016/s1350-9462(98)00021-4. [DOI] [PubMed] [Google Scholar]
- Karamichos D, Guo XQ, Hutcheon AE, Zieske JD. Human corneal fibrosis: an in vitro model. Investigative ophthalmology & visual science. 2010;51:1382–1388. doi: 10.1167/iovs.09-3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamichos D, Hutcheon AE, Zieske JD. Transforming growth factor-beta3 regulates assembly of a non-fibrotic matrix in a 3D corneal model. Journal of tissue engineering and regenerative medicine. 2011;5:e228–238. doi: 10.1002/term.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2004;18:816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- Letterio JJ, Roberts AB. Regulation of immune responses by TGF-beta. Annual review of immunology. 1998;16:137–161. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- Liu GX, Li YQ, Huang XR, Wei L, Chen HY, Shi YJ, Heuchel RL, Lan HY. Disruption of Smad7 promotes ANG II-mediated renal inflammation and fibrosis via Sp1-TGF-beta/Smad3-NF.kappaB-dependent mechanisms in mice. PloS one. 2013;8:e53573. doi: 10.1371/journal.pone.0053573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljubimov AV, Saghizadeh M. Progress in corneal wound healing. Progress in retinal and eye research. 2015;49:17–45. doi: 10.1016/j.preteyeres.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J, Seoane J, Wotton D. Smad transcription factors. Genes & development. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- Matsuba M, Hutcheon AE, Zieske JD. Localization of thrombospondin-1 and myofibroblasts during corneal wound repair. Experimental eye research. 2011;93:534–540. doi: 10.1016/j.exer.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao A, Fujii M, Matsumura R, Kumano K, Saito Y, Miyazono K, Iwamoto I. Transient gene transfer and expression of Smad7 prevents bleomycin-induced lung fibrosis in mice. The Journal of clinical investigation. 1999;104:5–11. doi: 10.1172/JCI6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida T. Biology of Corneal Stromal Keratocytes. In: Kinoshita S, Ohashi Y, editors. Current Opinions in the Kyoto Cornea Club: Proceedings of the first Annual Meeting of Kyoto Cornea Club. Kugler Publications; New York: 1995. [Google Scholar]
- Occleston NL, Laverty HG, O'Kane S, Ferguson MW. Prevention and reduction of scarring in the skin by Transforming Growth Factor beta 3 (TGFbeta3): from laboratory discovery to clinical pharmaceutical. Journal of biomaterials science Polymer edition. 2008;19:1047–1063. doi: 10.1163/156856208784909345. [DOI] [PubMed] [Google Scholar]
- Ohno S, Hirano S, Kanemaru S, Kitani Y, Kojima T, Ishikawa S, Mizuta M, Tateya I, Nakamura T, Ito J. Transforming growth factor beta3 for the prevention of vocal fold scarring. The Laryngoscope. 2012;122:583–589. doi: 10.1002/lary.22389. [DOI] [PubMed] [Google Scholar]
- Olsson N, Piek E, ten Dijke P, Nilsson G. Human mast cell migration in response to members of the transforming growth factor-beta family. Journal of leukocyte biology. 2000;67:350–356. doi: 10.1002/jlb.67.3.350. [DOI] [PubMed] [Google Scholar]
- Park K, Ryu SB, Park YI, Ahn K, Lee SN, Nam JH. Diabetes mellitus induces vaginal tissue fibrosis by TGF-beta 1 expression in the rat model. Journal of sex & marital therapy. 2001;27:577–587. doi: 10.1080/713846811. [DOI] [PubMed] [Google Scholar]
- Poncelet AC, Schnaper HW, Tan R, Liu Y, Runyan CE. Cell phenotype-specific down-regulation of Smad3 involves decreased gene activation as well as protein degradation. The Journal of biological chemistry. 2007;282:15534–15540. doi: 10.1074/jbc.M701991200. [DOI] [PubMed] [Google Scholar]
- Ruiz-Ortega M, Rodriguez-Vita J, Sanchez-Lopez E, Carvajal G, Egido J. TGF-beta signaling in vascular fibrosis. Cardiovascular research. 2007;74:196–206. doi: 10.1016/j.cardiores.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Saika S. TGFbeta pathobiology in the eye. Laboratory investigation; a journal of technical methods and pathology. 2006;86:106–115. doi: 10.1038/labinvest.3700375. [DOI] [PubMed] [Google Scholar]
- Saika S, Ikeda K, Yamanaka O, Miyamoto T, Ohnishi Y, Sato M, Muragaki Y, Ooshima A, Nakajima Y, Kao WW, Flanders KC, Roberts AB. Expression of Smad7 in mouse eyes accelerates healing of corneal tissue after exposure to alkali. The American journal of pathology. 2005;166:1405–1418. doi: 10.1016/S0002-9440(10)62358-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller M, Javelaud D, Mauviel A. TGF-beta-induced SMAD signaling and gene regulation: consequences for extracellular matrix remodeling and wound healing. Journal of dermatological science. 2004;35:83–92. doi: 10.1016/j.jdermsci.2003.12.006. [DOI] [PubMed] [Google Scholar]
- Schultz G, Chegini N, Grant M, Khaw P, MacKay S. Effects of growth factors on corneal wound healing. Acta ophthalmologica Supplement. 1992:60–66. doi: 10.1111/j.1755-3768.1992.tb02170.x. [DOI] [PubMed] [Google Scholar]
- Shah M, Foreman DM, Ferguson MW. Neutralisation of TGF-beta 1 and TGF-beta 2 or exogenous addition of TGF-beta 3 to cutaneous rat wounds reduces scarring. Journal of cell science. 1995;108(Pt 3):985–1002. doi: 10.1242/jcs.108.3.985. [DOI] [PubMed] [Google Scholar]
- Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, Springer TA. Latent TGF-beta structure and activation. Nature. 2011;474:343–349. doi: 10.1038/nature10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So K, McGrouther DA, Bush JA, Durani P, Taylor L, Skotny G, Mason T, Metcalfe A, O'Kane S, Ferguson MW. Avotermin for scar improvement following scar revision surgery: a randomized, double-blind, within-patient, placebo-controlled, phase II clinical trial. Plastic and reconstructive surgery. 2011;128:163–172. doi: 10.1097/PRS.0b013e318217429b. [DOI] [PubMed] [Google Scholar]
- Tatler AL, Jenkins G. TGF-beta activation and lung fibrosis. Proceedings of the American Thoracic Society. 2012;9:130–136. doi: 10.1513/pats.201201-003AW. [DOI] [PubMed] [Google Scholar]
- Taylor AW. Review of the activation of TGF-beta in immunity. Journal of leukocyte biology. 2009;85:29–33. doi: 10.1189/jlb.0708415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington SN, Crossley R, Sheard V, Howe SJ, Buckley SM, Coughlan L, Gilham DE, Hawkins RE, McKay TR. Gene delivery of a mutant TGFbeta3 reduces markers of scar tissue formation after cutaneous wounding. Molecular therapy : the journal of the American Society of Gene Therapy. 2010;18:2104–2111. doi: 10.1038/mt.2010.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zheng Q, Guo X, Wu Y, Hao J. Influence of exogenous TGFbeta1 on the expression of smad2 and smad3 in rat bone marrow-derived mesenchymal stem cells. Journal of Huazhong University of Science and Technology Medical sciences = Hua zhong ke ji da xue xue bao Yi xue Ying De wen ban = Huazhong keji daxue xuebao Yixue Yingdewen ban. 2005;25:68–71. doi: 10.1007/BF02831390. [DOI] [PubMed] [Google Scholar]
- Wilson SL, El Haj AJ, Yang Y. Control of Scar Tissue Formation in the Cornea: Strategies in Clinical and Corneal Tissue Engineering. Journal of Functional Biomaterials. 2012;3:642–687. doi: 10.3390/jfb3030642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa K, Osada H, Masuda A, Kondo M, Saito T, Yatabe Y, Takagi K, Takahashi T, Takahashi T. Induction of apoptosis by Smad3 and down-regulation of Smad3 expression in response to TGF-beta in human normal lung epithelial cells. Oncogene. 1998;17:1743–1747. doi: 10.1038/sj.onc.1202052. [DOI] [PubMed] [Google Scholar]
- Yu FS, Yin J, Xu K, Huang J. Growth factors and corneal epithelial wound healing. Brain research bulletin. 2010;81:229–235. doi: 10.1016/j.brainresbull.2009.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zieske JD, Hutcheon AE, Guo X, Chung EH, Joyce NC. TGF-beta receptor types I and II are differentially expressed during corneal epithelial wound repair. Investigative ophthalmology & visual science. 2001;42:1465–1471. [PubMed] [Google Scholar]
- Zieske JD, Mason VS, Wasson ME, Meunier SF, Nolte CJ, Fukai N, Olsen BR, Parenteau NL. Basement membrane assembly and differentiation of cultured corneal cells: importance of culture environment and endothelial cell interaction. Experimental cell research. 1994;214:621–633. doi: 10.1006/excr.1994.1300. [DOI] [PubMed] [Google Scholar]