

Abstract
TEA domain transcription factors (TEAD) are essential for normal development of eukaryotes and are the downstream effectors of the Hippo tumor suppressor pathway. Whereas our earlier work established the three-dimensional structure of the highly conserved DNA binding domain using solution NMR spectroscopy, the structural-basis for regulating the DNA binding activity remains unknown. Here, we present the X-ray crystallographic structure and activity of a TEA domain mutant containing a truncated L1 loop, ΔL1 TEAD DBD. Unexpectedly, the three-dimensional structure of the ΔL1 TEAD DBD reveals a helix-swapped homodimer wherein helix 1 is swapped between monomers. Furthermore, each three-helix bundle in the domain-swapped dimer is a structural homolog of MYB-like domains. Our investigations of the DNA binding activity reveal that although the formation of the three-helix bundle by the ΔL1 TEAD DBD is sufficient for binding to an isolated M-CAT-like DNA element, multimeric forms are deficient for cooperative binding to tandemly duplicated elements, indicating that the L1 loop contributes to the DNA binding activity of TEAD. These results suggest that switching between monomeric and domain-swapped forms may regulate DNA selectivity of TEAD proteins.
Keywords: TEAD, transcription factor, X-ray crystallography, Hippo pathway, domain swapping
Graphical Abstract
Introduction
The Hippo tumor suppressor pathway is essential for normal development in eukaryotes. Signal transduction along this pathway is made possible by the DNA binding activity of the TEA domain containing transcription factors (TEAD), namely, TEC1 in fungi, scalloped in flies, and TEF/TEAD in the vertebrates. TEAD1 is the prototypical member of the family of four mammalian proteins (TEAD1-TEAD4). The highly conserved TEAD proteins comprise a N-terminal DNA binding domain (TEAD DBD) of about 75 amino acids, a C-terminal protein-protein interaction domain of about 200 amino acids, and an intervening region (Fig. 1). TEAD transcription factors are thought to be of animal origin and have not been found in the plant kingdom.
Figure 1.

The TEAD transcription factor. (A) Amino acid sequence of the DNA-binding domain of the human TEAD1 transcription enhancer factor. Amino acids deleted in the ΔL1 TEAD DBD are shown in red. N- and C-terminal amino acids resulting from cloning are in smaller font. Numbering below the sequence, in blue, correspond to the NMR structure. Black numbering and yellow cylinders correspond to the X-ray crystallographic structure (this work, PDB id: 4Z8E). (B) The corresponding three dimensional solution NMR structure of TEAD DBD (PDB id: 2HZD). The L1 loop is shown in red.
Our NMR spectroscopic investigations established that the TEAD DBD structure is made of a three-helix bundle and that the solvent exposed face of the third helix comprises the primary DNA recognition surface. [1] The helices are connected by a long L1 loop and a short L2 loop (Fig. 1B). Proteins of the TEAD family bind to double-stranded DNA elements such as the muscle-specific, M-CAT, or GTIIC enhanson. Like the full-length protein, TEAD DBD binds to M-CAT-like DNA elements with a dissociation constant of about 4–8 nM. [1–3] Furthermore, TEAD binds cooperatively to direct repeat elements. [2, 3] We found that such cooperative binding was abrogated by truncation of the L1 loop. [1, 2, 4] However, the amide spectral signatures in the 1H, 15N-HSQC spectrum of the L1 loop truncated mutant (ΔL1 TEAD DBD) were similar to that of the wild type protein, indicating that the overall fold of the domain was likely to be retained in the mutant. How, then, does the L1 loop alter the DNA selectivity of TEAD? To address this question, we investigated the three-dimensional structure of the ΔL1 TEAD DBD.
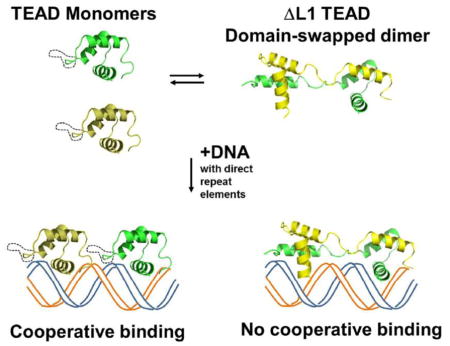
Here, we report the X-ray crystallographic structure of ΔL1 TEAD DBD. It shows that the mutant protein, like the wild type DBD, does indeed form a three-helix bundle. Unexpectedly, however, in the ΔL1 TEAD DBD, this bundle is in the context of a domain-swapped dimer. The structure and activity of the ΔL1 TEAD DBD indicate that the L1 loop controls the molecular assembly, providing an effective switch that guides DNA binding preferences of TEAD.
Results & Discussion
DNA binding activity of ΔL1 TEAD DBD
Earlier reports have established that TEADs can not only bind to isolated M-CAT-like sequences but also bind cooperatively to direct repeat, i.e., non-palindromic DNA elements arranged in tandem, such as 2xGT. [1, 2, 5] Additionally, recent ChipSeq studies have also identified genomic binding sites consisting of partially overlapping and tandem M-CAT-like elements. [6] However, to date, information that shed light on the mechanism for DNA selectivity by TEAD proteins is lacking.
To address this question, we investigated the DNA binding affinity of the ΔL1 mutant and compare it to that of the TEAD DBD. We used electrophoretic mobility shift assay (EMSA) to determine the DNA binding affinities. The formation of a 1:1 molecular complex of ΔL1 TEAD DBD with the double-stranded DNA is identified by retarded electrophoretic mobility on native gel compared to that of the free double stranded DNA (Fig. 2A). Measurement of the relative intensities of the shifted and unshifted bands at different protein concentrations and fixed DNA concentration yielded the bound and free DNA concentrations for each protein concentration. We then plotted the fraction of 1xGT DNA bound versus the total protein concentration (Fig. 2B) and carried out non-linear curve fitting to Hill equation to obtain a Kd of 7.9±1.4 nM, which is comparable to that of normal TEAD DBD. This is also comparable to the affinity of full-length human or mouse TEAD, which bind M-CAT elements with a KD of 10–24 nM. [1–4] These EMSA results establish that the mutant is as competent in DNA binding as TEAD and C27S TEAD DBD (pseudo wild type). It shows that the DNA recognition surface remains unperturbed in ΔL1 TEAD DBD.
Figure 2.

DNA binding activity of ΔL1 TEAD DBD. The mutant TEAD DBD binds to double stranded 1xGT (A, top left) and 2xGT (C, top right) DNA. Dissociation constants for binding to 1xGT (B, lower left) and 2xGT (D, lower right) were determined using the non-linear curve fitting routine. The fits for binding to one site (B) and two sites (D) are shown as red curves together with the 95% confidence limit (shaded in cyan). Panel D also shows the fraction single site occupancy on the 2xGT DNA (open circles and gray curve). Arrowhead indicates free DNA, ‘s’ refers to band consisting of one protein molecule per molecule of DNA, and ‘d’ refers to two protein molecules per DNA molecule.
Next, we carried out EMSA using the direct repeat DNA element, namely, 2xGT, which contains two tandemly duplicated M-CAT-like binding sites on a single DNA molecule. [2] Here, if the two available binding sites on a molecule of DNA were occupied by two molecules of TEAD DBD we would expect to see not only the gel shift bands corresponding to the unbound (0:1) and singly bound (1:1) bands, but also a third band that is shifted higher, as a result of the 2:1 loading of protein on to 2xGT DNA. In the case of TEAD DBD, such a 2:1 loading on 2xGT DNA appears even at low protein concentrations, and accepted as evidence of cooperative binding since all available single sites are not yet saturated under these conditions. [1, 5] In comparison, the binding of ΔL1 TEAD DBD to 2xGT predominantly yields unbound and singly bound forms at lower concentrations and the 2:1 band is observed only at higher ΔL1 TEAD DBD protein concentrations (Fig. 2C). Thus, unlike the TEAD DBD, the mutant appears to have lost the ability to bind to tandemly duplicated sites in a cooperative manner. These results are more readily appreciated in Fig. 2D, where the intensity of the band corresponding to singly occupied DNA (1:1) can be seen to peak before the intensity of the band corresponding to the doubly occupied DNA (2:1) reaches a maximum. This is in contrast to the normal TEAD DBD, where 2:1 loading of the protein to DNA proceeds well before single sites are saturated (Supplementary Fig. S1). Interestingly, the dissociation constant, Kd, for the 2:1 binding of ΔL1 on the 2xGT DNA is 8.7×10−9 ± 9.6×10−10 M (adjusted R2 = 0.978). It shows that although ΔL1 TEAD DBD can bind to single and direct repeat binding sites, the long L1 loop plays an important role in determining TEAD occupancy on DNA elements.
To learn about the structure of the L1 loop and how it might contribute to DNA selectivity of TEAD, we studied our solution NMR structure of TEAD DBD (PDB ID: 2HZD). Because the L1 loop of TEAD DBD is large, flexible, and relatively disordered, the basis for L1 loop’s influence on the DNA binding activity of TEAD could not be deduced. Therefore, in an effort to establish the structural basis for the L1 loop on TEAD activity, we determined the X-ray crystallographic structure of the ΔL1 TEAD DBD.
Structure of the ΔL1 TEAD DBD
We elucidated the three-dimensional structure of ΔL1 TEAD DBD, using X-ray crystallography, to 2.1 Å resolution. The corresponding statistics are shown in Table 1. Model building and refinement identified the presence of three molecules of ΔL1 TEAD DBD per asymmetric unit and that two adjacent unit cells contained three domain-swapped dimers (Fig. S2).
Table 1.
Crystallographic and structure refinement statistics*
| ΔL1 TEAD DBD | Se-Met ΔL1 TEAD DBD | |
|---|---|---|
| Data collection | ||
| Beamline | SSRL BL9-2 | ALS BL8.3.1 |
| Wavelenth (eV) | 12,658 | λ1=12,658 λ2=13,000 |
| Space group | C2221 | C2221 |
| Cell dimensions | ||
| a, b, c (Å) | 55.6, 96.5, 84.9 | 55.3, 95.7, 85.0 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 48.27–2.09 (2.13–2.09) | 31.77–2.99 (3.16–2.99) |
| Rmerge | 0.058 (0.752) | 0.160 (0.760) |
| Rpim | 0.023 (0.291) | 0.068 (0.309) |
| Total # of observations | 101,091 (5151) | 32,491 (4729) |
| Total # unique | 13,625 (675) | 4,782 (679) |
| I/σI | 26.3 (3.1) | 9.5 (2.5) |
| Completeness (%) | 98.2 (96.0) | 9.5 (2.5) |
| Multiplicity | 7.4 (7.6) | 99.8 (99.4) |
| CC1/2 | 1.000 (0.907) | 0.93 (0.91) |
| Refinement | ||
| Resolution (Å) | 17.83–2.09 (2.26–2.09) | |
| Number of reflections | 13,598 (2,728) | |
| Rwork/Rfree | 0.237/0.253 | |
| Number of atoms | ||
| Protein | 1322 | |
| Water | 68 | |
| B-factors | ||
| Protein | 46.6 | |
| Water | 53.4 | |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.010 | |
| Bond angles (°) | 0.98 | |
| PDB ID | 4Z8E | |
Values in parentheses denote statistics for the highest resolution shell
Each homodimer consists of two domains that are formed through domain swapping (Fig. 3). Each domain is a three-helix bundle. Overall, the domain architecture, with the three α-helices, two intervening loops, and an N-terminal arm, closely resembles the structure of the monomeric TEAD DBD that we previously elucidated using multi-dimensional solution NMR spectroscopy. In contrast to the NMR derived structure, however, each three-helix bundle consists of the H1 helix from one molecule together with H2 and H3 helices from the other molecule of the dimer (Fig. 3A). This domain swapping is made possible by the splaying out of H1 helix and repacking against the structural elements of the other chain. Thus, despite the splaying out of H1 helix, the three-helix bundle structure is formed, identifying that the three-helix bundle is the preferred domain architecture for the TEAD DBD. Retention of the native-like three-helix helix-bundle, despite domain swapping, indicates how the mutant is able to bind to 1xGT DNA with an affinity that is similar to that of TEAD. It is also consistent with the largely unchanged NMR spectrum of the ΔL1 mutant relative to that of TEAD DBD.
Figure 3. Structure of ΔL1 TEAD DBD.

A. Helix-swapped dimer (green and yellow cartoons) comprising two ΔL1 TEAD DBD monomers. Trp9 and His67 side chains are shown as stick models.
B. Closest structural homolog of ΔL1 TEAD DBD (yellow) is the Radialis MYB domain (cartoon with blue and pink helices; PDB id: 2CJJ). Superimposition of the two structures reveals that the structural homology includes the Trp-His π stacking interaction. (Superimpose was performed using ‘colorbyrmsd’ script in Pymol; blue = lowest RMSD; pink = highest RMSD, gray = not used in superimpose.)
C. Stereo image of the ΔL1 TEAD DBD three-helix bundle formed by domain swapping between two chains. Helix 1 from one chain is shown in yellow. Helices 2 and 3, shown in green, are from the second chain. Hydrophobic side chains involved in formation of the three-helix bundle are shown as sticks.
D. Inter-chain contacts between L1 loops of the two domain-swapped ΔL1 TEAD DBD monomers in the unit cell.
E. The ΔL1 TEAD DBD crystal structure (yellow), shows interaction between the N-terminal arm and DNA recognition helix through the Trp9 and His67 ring stacking interaction (shown as stick models). In contrast, in the TEAD DBD solution structure (blue) the NTA is unstructured and Trp9 is distant from the His67 side chain.
Unexpectedly, Dali analysis of a single domain of ΔL1 TEAD DBD structure identified the Radialis MYB domain to be the closest structural homolog (Fig. 3B). [7–9] MYB domain proteins are abundant in plants and and animals where they define a plethora of developmental programs [10]. MYB domains tend to contain a WWW motif comprising up to three tryptophan residues. However, the RAD MYB contains only the N-terminal Trp residue, which forms a ring stacking interaction with a histidine on the 3rd helix. Structurally, both the ΔL1 TEAD DBD and RAD MYB domain are similar, being made up of three-helix bundles and consisting the N-terminal Trp residue. Critically, as shown here, ΔL1 TEAD DBD also contains the Trp-His ring stacking interaction between Trp11 in the N-terminal arm and His67 in helix 3.
Domain swap interface
There is extensive contact between the two monomers in the domain-swapped form of ΔL1 TEAD DBD. The H1 helix of one chain is splayed out and extended towards the other chain. The core structure of each domain is stabilized through side chain interactions between helix 1 of one chain and helices 2 and 3 of the other chain (Fig. 3C). In addition, packing involves hydrogen-bonded and non-bonded contacts (Table S1). Also, the truncated L1 loop of each monomer is extended, providing H1 helices the reach needed to form the domain-swapped 3 helix bundle, while packing against one another (Fig. 3D). In all, the dimer interface consists of about 1500 Å2 of buried surface (EBI-Pisa: Interface analysis). The α-helices originating from two separate chains in the ΔL1 mutant are seamlessly integrated into one single two-domain structure, recreating the hydrophobic core and DNA recognition surfaces observed for monomeric TEAD DBD.
Recent years have seen several examples of protein dimerization via domain-swapping. [11–13] FOX3P, a forkhead box protein and a DNA binding transcription factor, also dimerizes via domain swapping that involves a hinge loop region and, interestingly, dimer formation was improved by a single amino acid mutation. [14] It is intriguing that the hinge loop region is able to regulate monomer-dimer equilibrium in both TEAD and FOXP. Furthermore, dimerization led to a decreased DNA binding affinity in the case of FOXP3, and loss of cooperativity in TEAD. It is likely that structural flexibility in a hinge/loop region could provide a general mechanism for regulating the DNA binding activities of transcription factors.
X-ray crystallographic vs. solution NMR structures
We compared the three-helix bundle of TEAD DBD (lowest energy structure from 2HZD[1]) with that of a single domain (domain-swapped) of ΔL1 TEAD DBD (Fig. 3D). Secondary structure of TEAD DBD is made of 43 amino acids in alpha helices, 3 amino acids in 3–10 helix, 3 amino acids in turns, and 8 amino acids in bends; 25 amino acids are not assignable to any regular secondary structural element. In comparison, ΔL1 TEAD DBD is made of 38 amino acids in alpha helices, 5 amino acids in turns, and 3 amino acids in bends; 12 amino acids that are not ascribable to a regular secondary structural element and it lacks 12 residues in the L1 loop. The two structures were aligned using Pymol. After five rounds of automated alignment, 351 of 431 atoms aligned with an RMSD of 2.594 Å. A scrutiny of the structural differences shows that in the solution NMR structure TEAD DBD is loosely packed compared to the X-ray crystallographic structure of ΔL1 TEAD DBD, a result of altered intramolecular interactions: First, the N-terminal arm (NTA) is relatively unstructured in the solution NMR structure. In contrast, the ΔL1 TEAD DBD structure shows that the NTA is retracted towards the three-helix bundle. Second, the His67 imidazole ring in the recognition helix is rotated such that it is nearly parallel with the Trp9 indole ring in the NTA resulting in π-π stacking interaction with closest approach between the rings at 3.08 Å between Trp9 Hε1 and His67 CD2 atoms. With Trp9 stacking against His 67, several core residues are repositioned, namely, Phe30, Leu34, and Leu72 to flank the other side of His67. Third, there is a significant change in the position and angle of H1 helix relative to the other two helices.
Solution NMR structure of TEAD DBD identified a monomeric three-helix bundle whereas the X-ray crystallographic structure of ΔL1 TEAD DBD has identified a domain-swapped dimer (vide supra). To assess whether TEAD DBD exists as monomers or multimers, we first considered the available experimental evidence. 1) EMSA results with 1xGT: At sub-micromolar concentrations, we and others observe a single shifted band relative to the double-stranded DNA band. [1, 2] It corresponds to the loading of one protein molecule to the single binding site on the DNA (Fig. 2). If significant population of TEAD DBD dimers existed in solution, another band, shifted higher, would be expected in the EMSA results. Indeed, at micromolar concentration of TEAD DBD, an additional shifted band is observed in EMSA, which has been attributed to non-specific binding. 2) NMR spectroscopy: At near millimolar concentrations, the relatively narrow line widths of TEAD DBD resonances in the 1H,15N-HSQC spectrum suggest that the monomeric species dominates. [1] 3) Multiple chemical shifts: Interestingly, in the 1H, 15N-HSQC NMR spectrum of TEAD DBD, the Leu34 amide proton is found to resonate at two distinct frequencies (Leu34 at 8.57 ppm and 126.8 ppm; Leu34’ at 6.85 ppm, 115.8 ppm). [1] The Asn44 side chain similarly shows two sets of resonances. The multiplicity of resonances suggest the likelihood of alternate local conformations. Placed in the context of the crystallographic structure of ΔL1 TEAD DBD, these additional resonances for Leu34 backbone amide and Asn44 side chain could indicate the co-existence of at least two different conformational populations, such as an extended (domain-swapped) vs. compact (monomer) structure.
Size Exclusion Chromatography
Since ΔL1 TEAD DBD crystallized as a multimer, we asked whether the L1 loop truncation shifts the equilibrium from monomeric to the multimeric form. To address the molecular organization of TEAD DBD we used size exclusion chromatography. The elution profile for TEAD DBD shows that a major peak elutes at a volume corresponding to 12±0.4 kDa. Similarly, the ΔL1 TEAD DBD, showed a prominent peak corresponding to molecular weight of 8.6±0.3 kDa (Fig. 4 and Fig. S3). Each of these proteins also showed smaller peaks that corresponded to multimers (see Supplementary Fig. S3). The data suggest that although both proteins exist largely as monomers in solution (at sub-millimolar concentrations), they are capable of existing as higher order species. The deviations in the SEC derived molecular weights for ΔL1 and TEAD DBD may be indicative of the larger hydrodynamic radius of TEAD DBD, due to the relatively unstructured L1 loop, and the more compact contracted form of the mutant due to the missing L1 residues. Interestingly, when SEC was performed at the lower salt concentration of 100mM sodium chloride, both proteins eluted as broad asymmetric peaks and much later than expected, indicating retardation effects arise from interaction with the resin (data not shown). Whereas the gel filtration data show that TEAD DBD can exist in monomeric and multimeric forms, ΔL1 TEAD DBD crystals did not contain monomers; only the multimers appear to have crystallized as trimers of domain-swapped dimers.
Figure 4. ΔL1 TEAD DBD exists largely in the monomeric form.


B. DNA binding by monomeric and multimeric fractions of TEAD DBD and ΔL1 TEAD DBD. Fractions from size exclusion chromatography that correspond to monomeric TEAD DBD (a), monomeric ΔL1 TEAD DBD, multimeric TEAD DBD (c), and multimeric ΔL1 TEAD DBD (d) were used for EMSA. Arrowhead indicates free DNA at 2 fmol/lane. Protein concentrations: lane 1: 0; lanes 2–8 & 9–15: 0.3, 1, 3, 10, 30, 100, 300 nM.
C. SDS-PAGE of the four samples used for EMSA and molecular weight markers (M) (Bio-Rad, Kaleidoscope). (kD): Molecular weights of markers in kilodaltons.
Since both the TEAD DBD and the L1 mutant can exist as monomers or multimers, how did the multimerization influence the DNA binding activity in each of these proteins? To answer this question, we carried out EMSA on each peak fraction (Fig. 4B and Fig. S3). SDS-PAGE analysis of the fractions show that there is very little protein in the multimeric fractions (c, d) compared to monomeric fractions (a, b). These samples were quantified using UV-Vis spectroscopy to ensure the use of appropriately matched concentrations in the EMSA experiments. EMSA results obtained using the monomeric fraction from SEC (a: F#61 for TEAD DBD and b: F#64 for ΔL1) show that both proteins bind equally well to 1xGT and 2xGT at similar concentrations. This is consistent with the results obtained using samples before being subjected to SEC, which contained not only the monomers but also multimers. In testing the DNA binding activity of the multimers, we find that the multimeric form of TEAD DBD (c: F#50) binds just as well to 1xGT and 2xGT as the monomeric fraction. However, multimers of ΔL1 (d: F#52) bind weaker to 1xGT; even at the two highest protein concentrations only 50% of the protein is found to load as a 2:1 complex. Thus, the multimers of ΔL1 are unable to load cooperatively to the direct repeat DNA element. The data suggest that switching between monomer and multimer has the ability to alter outcomes in DNA binding activity, in particular, when the L1 loop is manipulated. Thus, the loss of cooperative binding to direct repeat sequences by ΔL1 may reflect its ability to form domain-swapped multimeric forms. These results also indicate that the L1 loop flexibility may dictate DNA selectivity of TEAD.
Implications for regulation of TEAD activity
The DNA binding activity of TEAD transcription factors resides within the N-terminal DBD. [1, 2, 4–6, 15–19] Here, we have shown that the DNA binding competent structure of TEAD DBD consists of a three-helix bundle, even in the context of the domain-swapped dimeric form. Cases of intertwined homo-oligomers have been observed in about 24% of all available structures and a majority of these are associated with dimers that crystallize in the C2 symmetry [11, 12, 20, 21]. Indeed, the ΔL1 TEAD DBD, which presents a domain-swapped dimeric structure, also crystallized in the C222 symmetry. The ΔL1 TEAD DBD homodimer results from an extension of the truncated L1 loop and the N-terminal helix to form a new closed or primary interface to mimic the interface observed in our NMR derived structure of the monomeric TEAD DBD. Our current X-ray crystallographic structure of ΔL1 TEAD DBD and the previous NMR structure and chemical shift mapping of TEAD DBD, show that although the C-terminal helix (H3) is the DNA recognition helix, the L1 loop is necessary to mediate cooperative binding to direct repeat DNA elements and that its disruption through L1 loop truncation abrogates cooperative binding.
To better understand how the L1 loop regulates the DNA binding activity of TEAD, we further examined the DNA binding behavior of TEAD proteins. The work of Halder et al. showed that a spacing of one nucleotide between tandemly duplicated sites is optimal for cooperative binding by Scalloped, the fly TEAD protein. [2] It suggests that there is a specific distance requirement between monomers that enables juxtaposition on tandemly arranged sites. To visualize the interactions, we generated cartoons consisting of normal and ΔL1 TEAD DBD structures (Fig. 5). It can be seen that the normal TEAD DBD is able to bind to one or both of the tandem sites, independently in the former case or cooperatively in the latter case (Fig. 5A–C). However, the L1 loop truncation is likely to reduce flexibility in the shortened L1 loop. Consequently, although each DNA site on 2xGT can be bound independently of the other by the dimer, steric or conformational restraints in the domain-swapped dimers could interfere with the simultaneous or cooperative loading on to both sites (Fig. 5D). Alternatively, should ΔL1 TEAD DBD monomers predominate at the lower (nanomolar-sub micromolar) concentrations used in EMSA experiments, then the inability to bind tandem sites cooperatively may be the result, not due to steric interference, but due to the loss of L1 loop specific interactions with the second monomer or with the DNA element itself. It should also be noted that ΔL1 TEAD DBD produces the 2:1 band at higher protein concentrations and may arise from a 1:1 complex formation between the ΔL1 TEAD DBD dimer and 2xGT (Fig. 5E).
Figure 5. A model of DNA binding by TEAD DBD and its ΔL1 mutant.

Top: The mutant protein can exist in the monomeric and domain-swapped dimeric forms.
Bottom: DNA binding models for monomeric and domain-swapped forms:
Bottom Left: monomers of the wild type TEAD, with an intact L1 loop (dotted black line), or ΔL1 TEAD DBD bind to the two binding sites on a direct repeat DNA element independently (A and B), or cooperatively to both sites (C).
Bottom Right: DNA binding by the domain-swapped dimer. One molecule of the domain-swapped ΔL1 TEAD DBD may bind to either of the two available sites on the direct repeat DNA (D, E). However, cooperative binding by one domain-swapped dimer to both sites is likely to be unfavorable either because the tethered second domain is likely to be out of register with regard to the adjacent binding site or because of the missing L1 mediated contact protein-protein or protein-DNA contacts (E). Cooperative binding of two molecules of oligomeric/domain-swapped form is likely disallowed, presumably due to steric constraints, and not observed experimentally (F).
Assuming that there is sufficient residual flexibility in the truncated L1 loop, and that it is possible for the dimer to undergo further structural rearrangements, it is possible that the tethered second domain may swing away from the second binding site on the DNA to make room for another monomer or domain-swapped dimer to occupy the adjacent binding site on the DNA at higher protein concentrations (Fig 5F). In such a case, one would expect to see the equivalent of 3:1 and 4:1 complexes on the EMSA results. However, the absence of higher molecular weight bands corresponding to 3:1 and 4:1 complexes consisting of monomer+dimer:DNA and dimer+dimer:DNA, suggest that ΔL1 can occupy both sites of 2xGT only upon dissociation of the domain-swapped dimer. i.e., binding to the second DNA site may depend upon kinetics of the monomer-multimer equilibrium. Together, the structural data and structure-derived models suggest that the unusually long L1 loop of TEAD DBD aids cooperative binding to tandemly duplicated DNA elements by acting as a molecular ruler. Similar analyses in the context of the full-length protein are expected to further our understanding of how the L1 loop affects DNA binding or oligomerization. Although L1 loop truncations are not known with regard to TEAD from mesophiles, we note that the L1 loop of fungal TEAD proteins, TEC1, are shortened by two amino acids when compared with TEAD DBD (see Fig. S4). Currently, the three-dimensional structures for TEC1 or its DBD are not known. Whether this naturally occurring L1 loop truncation predisposes it oligomerization or domain swapping is yet to be determined.
Using protein binding microfluidic microarrays, we previously showed that TEAD DBD is relatively promiscuous with regard to DNA selectivity when considering DNA elements comprising single binding sites. [1] This raises the question as to how such a transcription factor without stringent DNA selectivity could bring about very specific outcomes during development. It should be noted that the FOXP3 forkhead domain protein, which forms a stable domain-swapped dimer and retains its DNA binding activity allows two DNA segments that interact with each of the two domains to be brought together. [22] Similarly, with regard to TEAD, depending upon whether the protein binds to isolated elements (monomer or dimer) or tandem elements (monomer), different transcriptional outcomes may ensue. Also, combinatorial regulation of TEAD activity, though interactions with protein co-factors such as YAP/Yki/TAZ, SRF, or MEF, has been implicated in TEAD function. [2, 23, 24] How these proteins bring about the desired DNA selectivity remains a mystery. It remains to be seen whether cofactors alter TEAD activity directly, through effecting changes to the L1 loop/TEAD DBD structure, or indirectly, by altering the molecular association status of TEAD.
Materials and Methods
Sample Preparation
The ΔL1 TEAD DBD construct was generated using the QuikChange method (Stratagene, La Jolla, CA) starting with hTEAD1 DBD in pET21d. [1] The resulting protein consists of a truncated L1 loop and is lacks amino acids Pro26 to Arg37 relative to the original TEAD DBD (calculated molecular weight of 9,371.6 with molar extinction coefficient of 9,970 M−1cm−1). [25] Overexpression and protein purification were carried out as follows: Plasmid was transformed into CodonPlus Escherichia coli cells (Novagen, San Diego, CA) or Rosetta2(DE3)pLysS (EMD Millipore, MA) and grown in 2xYT medium (unlabeled) or M9 medium. Each 2.8L baffled flask containing 650 mL of medium was inoculated with 6.5 mL of overnight culture, grown with shaking at 220 rpm until the optical density measured at 600 nm was near 1.0. Cells were induced overnight with 1 mM IPTG (RPI Corp, IL) at 28°C. Enzymes and inhibitors used in the protein purification steps were purchased from Sigma Chemical Co. (MO), laboratory reagents were from VWR or Fisher Scientific.
Cells were harvested by centrifuging at 6,000 rpm for 10 minutes at 4 °C, using a Beckman Avanti equipped with JLA 8.1000 rotor. Pellets were resuspended in sonication buffer consisting of 50 mM HEPES, pH 8.0, 0.5 M sodium chloride, 10 mM imidazole, and protease inhibitors (Leupeptin, Aprotinin, Pepstatin, PMSF), 10 mM β-mercaptoethanol, and 10–20 mg DNAse I (Sigma, Mo). The crude lysate was centrifuged at 12,000 rpm for 45 min, at 4 °C. 1–2 mL of pre-equilibrated Ni-NTA resin (Qiagen) was added to the supernatant and allowed to bind for 1 hour at 4 °C, on a tilt table. The slurry was transferred to an empty glass chromatography column, washed with 50 mL sonication buffer, and eluted using manual step gradient consisting of 10 mL each of 25 mM, 50 mM, 150 mM, and 250 mM imidazole in the sonication buffer. Protein eluted at 150–250 mM imidazole and was at least 95% pure as determined by SDS-PAGE (Supplementary Fig. S3). Size exclusion chromatography was carried out using Sephacryl S-200 XK 16/60 prepacked column (Akta Explorer, GE Healthcare). Protein, at 0.2–0.5mM, was loaded on the column using a 500 μL sample loop.
Protein from Ni-NTA chromatography was buffer exchanged into IEA (IEA: ion exchange buffer A; 50 mM HEPES, pH 8.0, 1 M sodium chloride, 50 mM L-arginine and 50 mM L-glutamine) and further purified using Source S ion exchange column (16mm/100mm) chromatography using a gradient of 1 to 2 M sodium chloride. IEB consisted of 50 mM HEPES, pH 8.0, 2 M sodium chloride. About 5 mg of ΔL1 TEAD DBD was purified per liter of rich media.
Electrophoretic Mobility-Shift Assay (EMSA)
DNA-binding activity of ΔL1 TEAD was determined using synthetic 1xGT or 2xGT. Double-stranded DNA was labeled by end-filling with 32P-ATP (Perkin Elmer, Easy Tide NEG512Z250UC) using the Klenow fragment (New England Biolabs, Cat No. M0212S). Each 8 microliter reaction contained protein at 0–50 nM concentration and 2 4 fmol of labeled DNA. All 8 uL of sample was loaded into one well of a 5% native gel. DNA sequences for 1xGT and 2xGT (one strand) are as follows:
1xGT (35): 5′-TTCGATACACTTGTGGAATGTGTTTGATTTGTTAGCCCCG-3′
2xGT (35): 5′-TTCGATACACTTGTGGAATGTGTGGAATGTGTTAGCCCCG-3′;
The 1xGT consists of a single TEAD binding site (underlined) derived from the GTIIC enhanson whereas 2xGT consists of two tandemly duplicated binding sites. [2] Binding affinity was determined as described previously using non-linear curve fitting routine within the Origin data analysis software. [1]
Crystallization & Structure Determination
Purified ΔL1 TEAD DBD was buffer exchanged into 20 mM Tris.HCl, pH 7.4, and 50 mM sodium chloride with 10 mM potassium phosphate or 10 mM magnesium chloride, and concentrated to about 7.5 mg/mL and used in crystallization trials. Protein stocks left at higher than 1 mg/mL at low salt concentration tended to precipitate out in less than 1 week. Plate form crystals of ΔL1 TEAD DBD were found after about 2 months, under crystallization conditions containing 1.6–2M ammonium sulfate, 0.1M MES (pH 6.5) or Tris.HCl (pH 8.5), in the presence of 10% Dioxane or 30% PEG MME 5k. Crystals were either quickly soaked in cryo solutions and frozen for later use or immediately mounted for X-ray data collection. Cryo solutions consisted of 25–40% PEG (1000 or 3000) or MME (2000), citrate (pH 5.5) or Tris (pH 7.0), and 5–20% glycerol.
X-ray diffraction data were collected at SSRL and ALS synchrotron sources. Images were processed with AutoPROC using XDS, POINTLESS, and AIMLESS. [26–29] The effect of radiation damage was adjusted using XSCALE. [30] Molecular replacement with MOLREP used our recently solved X-ray crystallographic structure of TEAD-DNA complex (to be published elsewhere). [31] Rebuilding using BUCCANEER gave an initial model that was further refined with BUSTER. [32, 33] Secondary structures of TEAD DBD and ΔL1 TEAD DBD were computed using the DSSP plugin within Pymol (The PyMOL Molecular Graphics System, Version 1.7.4 Schrödinger, LLC). Structure quality was evaluated using the online version of PDB Sum (www.ebi.ac.uk) and validated using the RCSB validation server (http://validate.rcsb.org/).
Supplementary Material
Highlights.
Hippo pathway transcription factor, TEAD, is found to exist in monomer-multimer equilibrium
DNA binding competent structure of the monomer is preserved in domain-swapped dimer
The L1 TEAD monomer fold is similar to that of the Radialis MYB domain of plants.
Domain-swapped dimer is unable to bind cooperatively on direct repeat DNA elements
The first structure-based mechanism for DNA selectivity of TEAD is identified.
Acknowledgments
This work was supported by NIH grant R01 GM084700 to SV and CSR. X-ray data was collected at Stanford Synchrotron Radiation Laboratory (beamlines: 9-1, 9-2) and Advanced Light Source (beamline: 8.3.1). Priyanka Rauniyar assisted SV with some of the ΔL1 TEAD DBD protein preparations.
Footnotes
Author contributions:
D-SL: crystallized protein, collected X-ray data
CV: solved and refined X-ray crystallographic structure
DA: made the mutant, purified the protein used for X-ray crystallography
CSR: Participated in X-ray data collection, processing, analyses
SV: Conceived, designed, and supervised the project; also purified, characterized, and assayed proteins, analyzed structures, and prepared the manuscript
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Anbanandam A, Albarado DC, Nguyen CT, Halder G, Gao X, Veeraraghavan S. Insights into transcription enhancer factor 1 (TEF-1) activity from the solution structure of the TEA domain. Proc Natl Acad Sci U S A. 2006;103:17225–30. doi: 10.1073/pnas.0607171103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Halder G, Carroll SB. Binding of the Vestigial co-factor switches the DNA-target selectivity of the Scalloped selector protein. Development. 2001;128:3295–305. doi: 10.1242/dev.128.17.3295. [DOI] [PubMed] [Google Scholar]
- 3.Kaneko KJ, DePamphilis ML. Regulation of gene expression at the beginning of mammalian development and the TEAD family of transcription factors. Dev Genet. 1998;22:43–55. doi: 10.1002/(SICI)1520-6408(1998)22:1<43::AID-DVG5>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 4.Davidson I, Xiao JH, Rosales R, Staub A, Chambon P. The HeLa cell protein TEF-1 binds specifically and cooperatively to two SV40 enhancer motifs of unrelated sequence. Cell. 1988;54:931–42. doi: 10.1016/0092-8674(88)90108-0. [DOI] [PubMed] [Google Scholar]
- 5.Jiang SW, Desai D, Khan S, Eberhardt NL. Cooperative binding of TEF-1 to repeated GGAATG-related consensus elements with restricted spatial separation and orientation. DNA Cell Biol. 2000;19:507–14. doi: 10.1089/10445490050128430. [DOI] [PubMed] [Google Scholar]
- 6.Benhaddou A, Keime C, Ye T, Morlon A, Michel I, Jost B, et al. Transcription factor TEAD4 regulates expression of myogenin and the unfolded protein response genes during C2C12 cell differentiation. Cell Death Differ. 2012;19:220–31. doi: 10.1038/cdd.2011.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stevenson CE, Burton N, Costa MM, Nath U, Dixon RA, Coen ES, et al. Crystal structure of the MYB domain of the RAD transcription factor from Antirrhinum majus. Proteins. 2006;65:1041–5. doi: 10.1002/prot.21136. [DOI] [PubMed] [Google Scholar]
- 8.Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Research. 2010;38:W545–W9. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holm L, Sander C. Dali: a network tool for protein structure comparison. Trends Biochem Sci. 1995;20:478–80. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- 10.Prouse MB, Campbell MM. The interaction between MYB proteins and their target DNA binding sites. Biochim Biophys Acta. 2012;1819:67–77. doi: 10.1016/j.bbagrm.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 11.Gronenborn AM. Protein acrobatics in pairs--dimerization via domain swapping. Curr Opin Struct Biol. 2009;19:39–49. doi: 10.1016/j.sbi.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mackinnon SS, Malevanets A, Wodak SJ. Intertwined associations in structures of homooligomeric proteins. Structure. 2013;21:638–49. doi: 10.1016/j.str.2013.01.019. [DOI] [PubMed] [Google Scholar]
- 13.Newcomer ME. Protein folding and three-dimensional domain swapping: a strained relationship? Curr Opin Struct Biol. 2002;12:48–53. doi: 10.1016/s0959-440x(02)00288-9. [DOI] [PubMed] [Google Scholar]
- 14.Perumal K, Dirr HW, Fanucchi S. A Single Amino Acid in the Hinge Loop Region of the FOXP Forkhead Domain is Significant for Dimerisation. Protein J. 2015 doi: 10.1007/s10930-015-9603-4. [DOI] [PubMed] [Google Scholar]
- 15.Burglin TR. The TEA domain: a novel, highly conserved DNA-binding motif. Cell. 1991;66:11–2. doi: 10.1016/0092-8674(91)90132-i. [DOI] [PubMed] [Google Scholar]
- 16.Deshpande N, Chopra A, Rangarajan A, Sashidhara LS, Rogdrigues V, Krishna S. The human transcription enhancer factor-1, TEF-1, can substitute for Drosophila scalloped during wingblade development. J Biol Chem. 1997;272:10664–8. doi: 10.1074/jbc.272.16.10664. [DOI] [PubMed] [Google Scholar]
- 17.Mar JH, Ordahl CP. A conserved CATTCCT motif is required for skeletal muscle-specific activity of the cardiac troponin T gene promoter. Proc Natl Acad Sci (USA) 1988;85:6404–8. doi: 10.1073/pnas.85.17.6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mar JH, Ordahl CP. M-CAT binding factor, a novel trans-acting factor governing muscle-specific transcription. Mol Cell Biol. 1990;10:4271–83. doi: 10.1128/mcb.10.8.4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stewart AF, Larkin SB, Farrance IK, Mar JH, Hall DE, Ordahl CP. Muscle-enriched TEF-1 isoforms bind M-CAT elements from muscle-specific promoters and differentially activate transcription. J Biol Chem. 1994;269:3147–50. [PubMed] [Google Scholar]
- 20.Heringa J, Taylor WR. Three-dimensional domain duplication, swapping and stealing. Curr Opin Struct Biol. 1997;7:416–21. doi: 10.1016/s0959-440x(97)80060-7. [DOI] [PubMed] [Google Scholar]
- 21.Szilagyi A, Zhang Y, Zavodszky P. Intra-chain 3D segment swapping spawns the evolution of new multidomain protein architectures. J Mol Biol. 2012;415:221–35. doi: 10.1016/j.jmb.2011.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bandukwala HS, Wu Y, Feuerer M, Chen Y, Barboza B, Ghosh S, et al. Structure of a domain-swapped FOXP3 dimer on DNA and its function in regulatory T cells. Immunity. 2011;34:479–91. doi: 10.1016/j.immuni.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gupta M, Kogut P, Davis FJ, Belaguli NS, Schwartz RJ, Gupta MP. Physical interaction between the MADS box of serum response factor and the TEA/ATTS DNA-binding domain of transcription enhancer factor-1. J Biol Chem. 2001;276:10413–22. doi: 10.1074/jbc.M008625200. [DOI] [PubMed] [Google Scholar]
- 24.Maeda T, Gupta MP, Stewart AF. TEF-1 and MEF2 transcription factors interact to regulate muscle-specific promoters. Biochem Biophys Res Commun. 2002;294:791–7. doi: 10.1016/S0006-291X(02)00556-9. [DOI] [PubMed] [Google Scholar]
- 25.Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31:3784–8. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vonrhein C, Flensburg C, Keller P, Sharff A, Smart O, Paciorek W, et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr D Biol Crystallogr. 2011;67:293–302. doi: 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 28.Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr. 2013;69:1204–14. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–32. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diederichs K, McSweeney S, Ravelli RB. Zero-dose extrapolation as part of macromolecular synchrotron data reduction. Acta Crystallogr D Biol Crystallogr. 2003;59:903–9. doi: 10.1107/s0907444903006516. [DOI] [PubMed] [Google Scholar]
- 31.Vagin A, Teplyakov A. MOLREP: an Automated Program for Molecular Replacement. Journal of Applied Crystallography. 1997;30:1022–5. [Google Scholar]
- 32.Bricogne G, Vonrhein C, Flensburg C, Schiltz M, Paciorek W. Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta Crystallogr D Biol Crystallogr. 2003;59:2023–30. doi: 10.1107/s0907444903017694. [DOI] [PubMed] [Google Scholar]
- 33.Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr D Biol Crystallogr. 2006;62:1002–11. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.