

Abstract
Gram-negative bacterial pneumonia is a common and dangerous infection with diminishing treatment options due to increasing antibiotic resistance among causal pathogens. The mononuclear phagocyte system is a heterogeneous group of leukocytes composed of tissue-resident macrophages, dendritic cells and monocyte-derived cells that are critical in defense against pneumonia, but mechanisms that regulate their maintenance and function during infection are poorly defined. Macrophage-colony stimulating factor (M-CSF) has myriad effects on mononuclear phagocytes but its role in pneumonia is unknown. We therefore tested the hypothesis that M-CSF is required for mononuclear phagocyte-mediated host defenses during bacterial pneumonia in a murine model of infection. Genetic deletion or immunoneutralization of M-CSF resulted in reduced survival, increased bacterial burden and greater lung injury. M-CSF was necessary for the expansion of lung mononuclear phagocytes during infection but did not affect the number of bone marrow or blood monocytes, the proliferation of precursors or the recruitment of leukocytes to the lungs. In contrast, M-CSF was essential to survival and anti-microbial functions of both lung and liver mononuclear phagocytes during pneumonia and its absence resulted in bacterial dissemination to the liver and hepatic necrosis. We conclude that M-CSF is critical to host defenses against bacterial pneumonia by mediating survival and anti-microbial functions of mononuclear phagocytes in the lungs and liver.
INTRODUCTION
Pneumonia caused by aerobic Gram-negative bacilli is among the most common and dangerous nosocomial infections and is an important cause of death, prolonged hospital stay and increased healthcare costs. Gram-negative bacilli colonize the upper aerodigestive tract as part of the normal response to acute illness and cause pneumonia when introduced into the lower respiratory tract by micro-aspiration (1, 2). The progressive rise in antibiotic resistance amongst the causative organisms in recent decades has markedly diminished the available treatment options for these infections, lending new impetus to mechanistic studies that may inform the development of better therapeutic strategies.
The mononuclear phagocyte system encompasses heterogeneous populations of cells from three distinct lineages, namely yolk sac-derived and self-renewing tissue-resident macrophages, Flt-3 ligand-dependent pre-dendritic cell (DC)-derived cells and monocytes and their progeny (3). The latter lineage, derived from circulating monocytes that express high levels of the surface glycoprotein Ly6C in mice, can differentiate into diverse populations of mononuclear phagocytes depending on environmental cues: under homeostatic conditions, mouse Ly6Chi monocytes terminally differentiate into endothelial-patrolling Ly6Clo counterparts in the blood and also give rise to some short-lived populations of tissue macrophages. In response to tissue injury, Ly6Chi monocytes extravasate into the damaged tissue and differentiate into inflammatory macrophages, re-populate some tissue-resident macrophage populations and differentiate into monocyte-derived inflammatory DC that are phenotypically similar to, but transcriptionally distinct from, CD11b+ pre-DC-derived conventional DC (4). The mouse lung, in particular, contains at least 5 populations of mononuclear phagocytes under homeostatic conditions, namely the tissue-resident and long-lived alveolar macrophages, short-lived monocyte-derived interstitial macrophages, CD11b+ conventional DC, CD103+ airway DC and plasmacytoid DC (5). In the context of injury, Ly6Chi and Ly6Clo monocytes, monocyte-derived DC and macrophages enter the lungs and resident cells display accelerated efflux and death, comprising a complex and dynamic network.
Macrophage-colony stimulating factor (M-CSF) was the first cytokine described for its induction of macrophage colonies from bone marrow progenitors in soft agar (6, 7) and is a key regulator of the development and homeostasis of the mononuclear phagocyte system (8–10). In different experimental conditions, M-CSF has been shown to mediate diverse functions in the differentiation, proliferation, survival and effector functions of multiple cell types from different mononuclear phagocyte lineages (11). In addition, the sole receptor for M-CSF, CSF1R or CD115, is shared with a structurally unrelated ligand, IL-34 (12). M-CSF and IL-34 have distinct tissue expression patterns and, at least in the development of osteoclasts, microglia and skin Langerhans cells, have non-redundant roles (13). Most recently, protein-tyrosine phosphatase-ζ was described as a second receptor for IL-34 (14, 15).
Evidence from multiple experimental systems support a critical role for mononuclear phagocytes in host defenses but the mechanisms that control the development, homing and survival of these cells during specific infections are incompletely defined. In the context of bacterial pneumonia, monocytes are recruited to the lungs via a CCR2-dependent mechanism and both recruited and resident mononuclear phagocytes mediate host defenses by direct killing of bacteria and NFκB-mediated recruitment of other leukocytes (16–20). In this context, we previously defined the dynamic changes in the various mononuclear phagocyte populations during experimental Gram-negative pneumonia and reported that CCR2 was necessary not only to the recruitment of monocyte-derived lineage cells but, unexpectedly, also controlled the number and phenotype of alveolar macrophages and all DC populations in this infection (17). Little is known about the role of the M-CSF/IL-34 biologic axis in the context of infection and many of the published studies in the field pre-date current understanding of the lineages of mononuclear phagocytes (21–25). To our knowledge, this mechanism has not been investigated in pulmonary infections to date. We therefore sought to define the role of this biological axis in pneumonia by testing the hypothesis that M-CSF is required for mononuclear phagocyte-mediated host defense during Klebsiella pneumonia.
MATERIALS AND METHODS
Animals and in vivo procedures
We used a previously characterized model of experimental bacterial pneumonia (26, 27). C57Bl/6 and op/+ mice, purchased (Jackson Laboratories, Bar Harbor, ME), were bred and maintained under pathogen-free conditions and in compliance with institutional animal care regulations. Age- and sex- matched 6–8 week old mice were used in all experiments. K. pneumoniae strain 43816 (American Type Culture Collection, Manassas, VA) was grown overnight at 37º C to mid-log phase in tryptic soy broth. Bacteria were administered intratracheally as previously described (17). Unless otherwise noted, all mice received intraperitoneal injections of 100μg/100μL of anti-M-CSF neutralizing antibody (clone 5A1), anti-CSF1R neutralizing antibody (clone AFS98), IgG1 isotype control (clone HRPN) or IgG2A isotype control (clone 2A3) beginning 2 hours before intratracheal inoculation and then every 24 hours through day 2 post-infection (all from Bio X Cell, West Lebanon, NH). In some experiments, mice received 3 intraperitoneal injections of 2 mg of BrdU (BD Biosciences, San Jose, CA) in 0.2 mL of PBS, every 3 hours until 2 hours before bacterial inoculation, as described (28). At designated time points, mice were euthanized with an injection of ketamine and xylazine.
Tissue harvest
Blood was collected in heparinized syringes from the right ventricle. Lung and liver vasculature were perfused with 3 mL of PBS with 2 mM EDTA before excision. Blood and homogenized lungs and right liver lobes were serially diluted and cultured on blood agar plates for quantification of bacterial content. Bronchoalveolar lavage was performed as previously described (26). Histology was performed on lungs and left liver lobes as previously described (26, 29). In other experiments, whole lungs, left liver lobes and plasma were processed for ELISAs as described (27).
Flow cytometry
Flow cytometry was performed as previously described (26, 30, 31). Cell suspensions of whole lungs, median liver lobes, bone marrow and peripheral blood were prepared as previously described (27, 29, 32). The following reagents were used to label cells for flow cytometry (from BD Biosciences, San Jose, CA; eBiosciences, San Diego, CA or BioLegend, San Diego, CA): 7-aminoactinomycin D (7AAD), anti-CD3-PE-Cy7 (clone 17A2), anti-CD11b-APC-Cy7 (clone M1/70), anti-CD11c-PE-Cy7 (clone HL3), anti-CD19-PE-Cy7 (clone 1D3), anti-CD24-APC (clone M1/69), anti-CD45-PerCP or AmCyan (clone 30-F11), anti-CD64-BV421 (clone X54-5/7.1), anti-CD103-APC (clone 2E7), anti-CD115 (clone AFS98), anti-CD117-FITC (clone 2B8), anti-CD135-BV421 (clone A2F10.1), anti-F4/80-Pacific Blue (clone BM8), anti-F4/80-PE (clone T45-2342), Fc block (anti-CD16/CD32), anti-I-A/I-E-FITC or AmCyan (clone M5/114.15.12), Ki-67-PE and isotype control (clones B56 and MOPC-21), anti-Ly6C-PE or Pacific Blue (clone HK1.4), anti-Ly6G-PE-Cy7 (clone 1A8), anti-NK1.1-PE-Cy7 (clone PK136), anti-PDCA-1-APC (clone 129c1). In some experiments, cells were labeled, fixed and permeabilized using a commercial kit (Cytofix/Cytoperm; BD Biosciences) before staining for intra-cellular antigens. Data were acquired on a FACS Canto II instrument using BD FACSDiva software (version 8.0; BD Biosciences) and analyzed using FlowJo software (version 8.8.6; Tree Star, Ashland, OR). Leukocyte subsets were identified as previously described (4, 17, 33) with minor modifications, as depicted in the figures. The absolute number of each cell type was determined as the product of the percentage of the cell type and the total number of cells in the sample, as determined on an automated cell counter (Countess; Invitrogen, Carlsbad, CA).
ELISA and assays for alanine transaminase, aspartate aminotransferase, urea nitrogen and lactate
ELISAs for M-CSF and IL-34 were performed according to manufacturer’s instructions (R&D Systems, Minneapolis, MN). Albumin ELISA was performed using 5 μg/mL goat anti-mouse albumin (A90-134A), 0.025 μg/mL goat-HRP detection (A90-134P), mouse reference serum (rs10–101) (Bethyl Laboratories Inc.; Montgomery, TX) and 1-Step Ultra TMB-ELISA (Thermo Fisher Scientific). Plasma alanine transaminase, aspartate aminotransferase and urea nitrogen were measured using commercial kits (Liquid ALT Reagent Set and Liquid AST Reagent Set, Pointe Scientific Inc., Canton, MI; DetectX Urea Nitrogen Detection Kit, Arbor Assays, Ann Arbor, MI). Plasma lactate was measured using a commercial device (Lactate Plus Meter, Nova Biomedical; Waltham, MA).
In vitro studies
Bone marrow cells were cultured as described to generate macrophages (34). In viability assays, macrophages were washed and resuspended in DMEM with 10% fetal calf serum without antibiotics and 1x105 cells were cultured overnight in opaque flat-bottom 96-well tissue-culture plates (BD Falcon, Franklin Lakes, NJ). Cells were then incubated with or without 5 ng of M-CSF for 2 hours before the addition of K. pneumoniae. After 150 minutes, plates were washed with sterile HBSS and viability was measured using a commercial kit (LIVE/DEAD viability/cytotoxicity kit for mammalian cells, Invitrogen Molecular Probes, Eugene, OR). For phagocytosis assays, bacteria were labeled with a pH-sensitive cyanine dye that acquires fluorescence in acidic environments (35, 36): 1x105 macrophages were seeded into round bottom tissue-culture treated plates (Costar, Corning, NY) and incubated with or without M-CSF as above. Mid-log phase K. pneumoniae cells were suspended at 1x109/mL in sterile saline, fixed in 5 mL of 70% ethanol for 5 minutes, washed and resuspended in sterile HBSS; CypHer5E dye (GE Healthcare Life Sciences, Pittsburgh, PA) was added at a final concentration of 5 mM before incubation at room temperature for 30 minutes. Bacteria were then washed and 1x108 bacteria were added to wells containing macrophages. Plates were centrifuged for 30 seconds to achieve cell contact and incubated at 37º C and 5% CO2 for 30 minutes, then placed on ice, washed in cold HBSS and analyzed immediately by flow cytometry. Positive gating was set using an unstained control.
Statistical analysis
Data were analyzed using Prism statistical software (version 5.0d, GraphPad Software, San Diego, CA). Data from survival experiments were analyzed using the Log-rank test. Values between two groups over multiple time points were compared with 2-way ANOVA. Comparisons between two groups at a single time were performed using the nonparametric Mann-Whitney test. Comparisons between multiple groups at a single time point were performed using the Kruskal-Wallis nonparametric test with Dunn’s comparison post-test. Comparison of paired samples receiving different treatments was made using Wilcoxon matched-pairs signed rank test or 2-way repeated measures ANOVA. Probability values were considered statistically significant if less than 0.05.
RESULTS
Role of M-CSF during pulmonary infection with Klebsiella pneumoniae
We began by measuring the concentration of M-CSF and IL-34 protein in whole-lung homogenates during experimental pneumonia induced by K. pneumoniae. Both ligands were detectable in uninfected lungs. As compared to animals challenged with intratracheal saline vehicle, the concentration of M-CSF increased ~2-fold as the infection progressed but the concentration of IL-34 did not change significantly (Figure 1A–B). In contrast, the concentration of plasma M-CSF protein remained undetectable in plasma throughout the time course, suggesting that M-CSF was produced locally in the lungs during infection. Given these findings, we focused our studies on the role of M-CSF during Klebsiella pneumonia.
Figure 1. M-CSF level increases in the lungs of mice with pneumonia and is required for host defense.

Time-series of mean +/− SEM of lung M-CSF (A) and IL-34 (B) protein in mice infected with Klebsiella pneumoniae; n = 5–6 per time point per group from 2 experiments. Time 0 represents uninfected animals. (**** p < 0.0001, 2-way ANOVA). (C) Outcome of pneumonia in op/op mice and littermate controls. n = 9–10 per group from 2 experiments (* p < 0.05, Log-rank test). (D) Bacterial burden in lungs and blood of infected op/op mice and littermate controls on day 1 of infection. Each data point represents one animal and horizontal lines indicate medians. Data from 2 experiments (* p < 0.05, Mann-Whitney); (E) Outcome of pneumonia in animals treated with anti-M-CSF or isotype control Ab; n = 23–24 per group from 2 experiments (** p < 0.01, Log-rank test). (F) Bacterial burden in lungs and blood of animals with pneumonia on day 3 of infection, in animals treated with anti-M-CSF or isotype control Ab. n = 17–19 per group, data shown are representative of 2 experiments (** p < 0.01, Mann-Whitney). In panels D and F, samples with no recoverable bacteria are depicted as one colony-forming unit (CFU) on the logarithmic scale.
To assess the contribution of M-CSF during bacterial pneumonia, we next examined the outcome of animals deficient in M-CSF during infection. Mice homozygous for an inactivating mutation in the M-CSF locus (op/op) had notably increased mortality during experimental pneumonia as compared to littermate controls, with no animals surviving beyond 2 days; this was associated with increased incidence and severity of bacteremia on the first day of the infection (Figure 1C–D). The role of M-CSF during development of the mononuclear phagocyte system results in a number of well-documented phenotypic abnormalities in op/op mice, including reduced number of alveolar macrophages in juvenile, but not adult, animals and spontaneous production of matrix metalloproteinases by mutant alveolar macrophages (37). To discriminate between the role of M-CSF induction during bacterial pneumonia from its role during development, we tested the effect of immunoneutralization of M-CSF starting at the onset of the infection. Wildtype mice receiving anti-M-CSF neutralizing antibody had increased mortality, increased lung bacterial burden and markedly increased incidence and severity of bacteremia as compared to animals receiving an isotype control antibody (Figure 1E–F). We found similar results with antagonism of CSF1R during the infection. The administration of a neutralizing CSF1R antibody to uninfected animals did not influence the number of blood Ly6Chi monocytes or lung mononuclear phagocytes but lead to a reduction in blood Ly6Clo monocytes (Figure 2A–B), consistent with prior reports that, in steady state, M-CSF mediates differentiation of blood Ly6Chi monocytes to their Ly6Clo counterparts (38, 39). In the context of infection, administration of anti-CSF1R antibody resulted in a similar increase in mortality and bacterial burden as noted with M-CSF neutralization (Figure 2C–D). These data indicate that the M-CSF-CSF1R interaction is necessary for host defenses during bacterial pneumonia independent of their role in development.
Figure 2. CSF1R is required for host defense in pneumonia.

Concentration of circulating monocytes (A) and lung mononuclear phagocytes (B) in uninfected mice after administration of daily CSF1R antibody or isotype control for 7 days using the gating strategy depicted in Figure 3A; n = 4–5 per group (* p < 0.05, Mann-Whitney). (C) Outcome of pneumonia in animals treated with anti-CSFR1 or isotype control Ab; n = 23–24 per group from 2 experiments (** p < 0.01, Log-rank test). (D) Bacterial burden in lungs of animals with pneumonia treated with anti-CSFR1 or isotype control Ab on day 3 of infection. Data from 2 experiments. Samples with no recoverable bacteria are depicted as 1 colony-forming unit. (* p < 0.05, Mann-Whitney).
Mechanism of M-CSF-mediated host defense during pneumonia
To begin to address how M-CSF mediates its beneficial effects during bacterial pneumonia, we examined the effect of M-CSF neutralization on lung mononuclear phagocyte subsets in the lungs in the context of pneumonia (Figure 3A). M-CSF neutralization resulted in a significant reduction in the numbers of cells of monocyte lineage, namely Ly6Chi and Ly6Clo monocytes, monocyte-derived macrophages and monocyte-derived inflammatory DC, in the lungs of mice during pneumonia (Figure 3B–D). In addition, M-CSF neutralization caused a reduction in the number of alveolar macrophages and conventional CD11b+ DC, but not other DC subsets or neutrophils (Figure 3E–J), similar to our prior report in mice with CCR2 deficiency (17).
Figure 3. M-CSF is required for the maintenance of lung mononuclear phagocytes during pneumonia.

(A) Representative flow cytometry plots showing gating strategy for lung leukocytes. Cells were enumerated in single cell suspensions from whole lung. cDC: conventional CD11b+ dendritic cells; Mφ, macrophages. (B–J) Time-series of mean +/− SEM of indicated lung leukocyte populations; n = 20–24 per time point per group from 4 experiments. Time 0 represents uninfected animals. (* p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, 2-way ANOVA).
To understand how the M-CSF-CSF1R axis contributes to the numbers of lung mononuclear phagocytes during Klebsiella pneumonia, we reasoned that the observed reduction in lung monocyte-derived cells, and possibly alveolar macrophages and conventional CD11b+ DC, may be attributable to impairments in monocytopoeisis, trafficking between the bone marrow, blood and lung compartments, or survival of leukocytes that have arrived in the lungs. We found that neutralization of M-CSF had no effect on the concentration of Ly6Chi and Ly6Clo monocyte numbers in bone marrow or blood (Figure 4A–B). Consistent with this, we found no difference in the proliferation of bone marrow macrophage-dendritic cell progenitor (MDP) or the recently described committed-monocyte progenitor (cMoP) (40) after neutralization of M-CSF (Figure 4C–D). These data indicate that M-CSF is dispensable for the generation of monocytes from progenitor cells in the bone marrow during bacterial pneumonia.
Figure 4. M-CSF is dispensable for maintenance of bone marrow and blood monocytes and progenitor proliferation during pneumonia.

(A–B) Time-series of mean +/− SEM of indicated leukocyte populations in the blood and bone marrow. n = 10–12 from 2 experiments. (C) Representative flow plots of CD45+ bone marrow cells, showing gating strategy of bone marrow to identify progenitor cells. (D) Time-series of mean +/− SEM of percent Ki67+ bone marrow monocyte progenitors. n = 6–12 per group per time point from 2 experiments. Time 0 represents uninfected animals.
M-CSF can be chemotactic for mononuclear phagocytes (41, 42). To assess for this possibility, we pulsed uninfected mice with the thymidine analog, BrdU, thereby labeling populations of proliferating cells that were in S-phase at the time of the pulse and then quantified the number of labeled leukocytes in various compartments after the onset of infection. As expected, we found a decreased number of BrdU-positive bone marrow neutrophils and a marked increase in labeled neutrophils in the blood and lung of infected mice compared to uninfected controls that was independent of M-CSF neutralization (Figure 5A–C). M-CSF neutralization also did not influence the numbers of labeled Ly6Chi monocytes in the bone marrow, blood, or lungs during infection (Figure 5A–C), suggesting that M-CSF is also dispensable for the recruitment of lung monocyte-derived cells during pneumonia.
Figure 5. M-CSF is dispensable to the influx of mononuclear phagocytes during pneumonia.

Mean +/− SEM of BrdU-positive leukocyte populations in the bone marrow (A) blood (B) and lungs (C). n = 6–12/group per time point from 2 experiments. (* p < 0.05, one-way ANOVA with Dunn post-test).
We next assessed the role of M-CSF on survival of mononuclear phagocytes during infection, since the M-CSF-CSF1R axis can mediate survival of mononuclear phagocytes under homeostatic conditions (43). We addressed this question by measuring the proportion of dead cells in the bronchoalveolar lavage fluid of infected animals, in order to minimize the artifact of ex vivo cell death that inevitably occurs during processing of samples. M-CSF neutralization resulted in a significant increase in the proportion of dead Ly6Chi monocytes in the lungs of infected animals (Figure 6A), potentially implicating M-CSF as a survival factor for lung monocyte-derived cells during pneumonia. To directly examine the role of M-CSF in mediating monocyte survival during infection, we also assessed the survival of bone marrow-derived macrophages co-incubated with K. pneumoniae. As compared to cells incubated with bacteria in the presence of M-CSF, monocyte-derived macrophages incubated with bacteria in the absence of M-CSF had reduced survival (Figure 6B). In addition, the surviving monocyte-derived macrophages had impaired phagocytosis of K. pneumoniae in the absence of M-CSF (Figure 6C), suggesting that M-CSF promotes the survival of mononuclear phagocytes during infection and also contributes to the antimicrobial functions of these cells independent of its effect on the number of leukocytes.
Figure 6. M-CSF is required for the survival and function of mononuclear phagocytes during pneumonia.
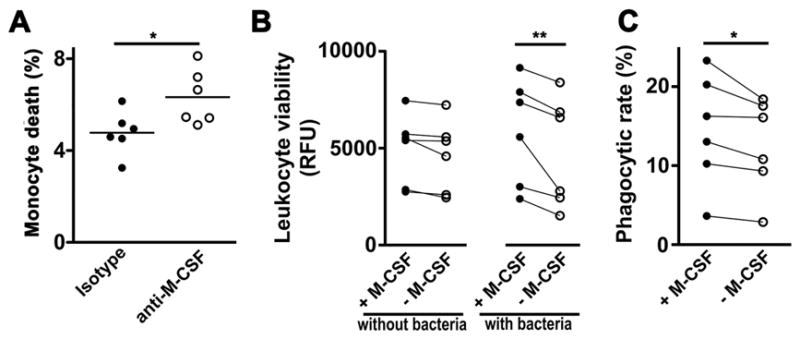
(A) Percentage of 7AAD+ lung Ly6Chi monocytes on day 3 of infection. n = 6 per group from 2 experiments (* p < 0.05, Mann-Whitney). (B) Effect of M-CSF on viability of bone marrow-derived macrophages incubated with or without Klebsiella in vitro, as measured in relative fluorescence units (RFU). Each data point represents an independent biological replicate paired to its respective control (** p < 0.01, 2-way repeated measures ANOVA). (C) Effect of M-CSF on phagocytosis of CypHer5E-labeled Klebsiella by bone marrow-derived macrophages. Each data point represents an independent biological replicate paired to its respective control (* p < 0.05, Wilcoxon matched-pairs signed rank test).
Role of M-CSF in controlling liver injury during bacterial pneumonia
In the course of the above studies, we noticed that M-CSF neutralization during pneumonia was associated with gross visual evidence of liver injury. To investigate this, we assessed the effect of M-CSF neutralization on the histology of lung and liver during experimental pneumonia. The lung histology did not differ between pneumonic mice treated with anti-M-CSF and isotype controls; in contrast, M-CSF neutralization during pneumonia resulted in large areas of sharply demarcated hepatocellular necrosis, associated with prominent hepatocyte microvesicles (Figure 7A). Consistent with this, neutralization of M-CSF resulted in elevations in plasma concentrations of aspartate transaminase and alanine transaminase during pneumonia (Figure 7B). Since M-CSF neutralization resulted in increased incidence and extent of bacteremia (Figure 1), we next assessed whether the observed hepatocellular injury was associated with increased hepatic bacterial content after the liver had been perfused to displace its blood content. We found that, in mice with pneumonia, M-CSF-neutralization resulted in increased liver bacterial burden (Figure 7C), potentially explaining the increased liver injury. To assess whether the hepatocellular injury is merely a reflection of multi-organ dysfunction in the context of sepsis, we also quantified other parameters of end-organ dysfunction. As expected, M-CSF neutralization resulted in increased lung injury, as measured by albumin concentration in the bronchoalveolar lavage fluid (Figure 8A). In contrast to its effect on the liver, however, M-CSF neutralization did not affect renal function, as measured by the concentration of blood urea nitrogen, nor cause significant end-organ hypo-perfusion, as quantified by plasma lactate concentration (Figure 8B–C), suggesting that M-CSF neutralization results in specific injury to the liver in addition to the lungs.
Figure 7. M-CSF is required for controlling bacterial dissemination and secondary liver damage during pneumonia.
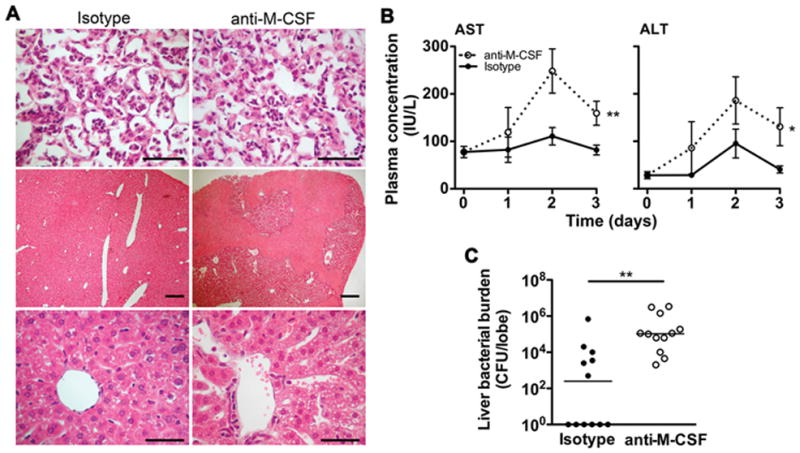
(A) H&E stain of lung (top panels) and liver sections (middle and bottom panels) on day 3 of infection in animals treated with anti-M-CSF or isotype control Ab. Top and bottom panels: scale bars 40 μm, original magnification x400; middle panels: scale bars 200 μm, original magnification x40. Representative data from 3 mice per group. Top panels show alveolar infiltration by acute inflammatory cells. Middle panels show areas of necrosis with anti-M-CSF treatment (bright pink areas). Bottom panels show central hepatic veins and surrounding lobule; the hepatocytes in mice treated with anti-M-CSF show prominent microvesicles. (B) Time-series of mean +/− SEM of plasma aspartate aminotransferase (AST) and alanine aminotransferase (ALT) during pneumonia, as measured in international units per liter (IU/L). n = 10–12 from 2 experiments. (* p < 0.05, ** p < 0.01, 2-way ANOVA). (C) Bacterial burden in the right lobe of the liver in mice treated with anti-M-CSF or isotype control Ab. Each data point represents 1 animal and horizontal lines indicate medians; samples with no recoverable bacteria are depicted as 1 colony-forming unit (CFU) on the logarithmic scale. Data from 2 experiments (** p < 0.01, Mann-Whitney).
Figure 8. M-CSF neutralization causes lung injury but not renal failure or tissue hypoperfusion during pneumonia.
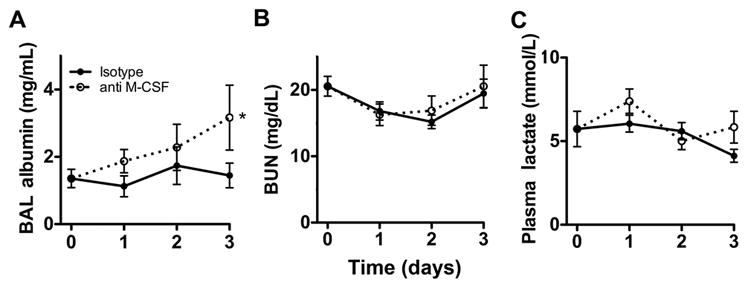
Time-series of mean +/− SEM of bronchoalveolar lavage (BAL) albumin concentration (panel A), blood urea nitrogen (BUN, panel B) and plasma lactate (panel C) in mice with pneumonia, treated with anti-M-CSF or isotype Ab. n = 8–12 per group per time point from 2 experiments. (* p < 0.05, 2-way ANOVA). Time 0 represents uninfected animals.
In order to investigate the contribution of M-CSF in the liver during bacterial pneumonia, we assessed the liver levels of M-CSF protein, since the plasma concentrations of M-CSF were undetectable during the infection. The liver contained high concentrations of M-CSF protein at baseline that did not change significantly during the infection (Figure 9A). We therefore tested the hypothesis that, similar to our findings in the lungs, liver M-CSF mediates the survival of liver mononuclear phagocytes during the infection, leading to better clearance of bacteria from the liver. Consistent with this and analogous to the lungs, we found that liver monocyte-derived macrophages and F4/80+ CD11b- resident Kupffer cells were reduced in number with M-CSF neutralization during pneumonia (Figure 9B–C). Also similar to our findings in the lungs, M-CSF neutralization resulted in increased death of liver Ly6Chi monocytes during pneumonia (Figure 9D). Taken together, these data indicate that M-CSF is also required for mononuclear phagocyte-mediated defense in the liver during bacterial pneumonia.
Figure 9. M-CSF is required for the maintenance of mononuclear phagocytes in the liver during pneumonia.

(A) Time-series of mean +/− SEM of M-CSF protein in the left liver lobe in mice with pneumonia. n = 6–16 per time point per group from 2 experiments. (B) Representative flow cytometry plots showing gating strategy for Kupffer cells. (C) Time-series of mean +/− SEM of indicated mononuclear phagocytes in the median liver lobe. Monocyte-derived macrophages were identified as depicted in Figure 3A. n = 10–12 per group per time point from 2 independent experiments. (*** p < 0.001, 2-way ANOVA). (D) 7AAD-positive Ly6Chi liver monocytes on day 3 of infection; data from 2 independent experiments (* p < 0.05, Mann-Whitney). Time 0 represents uninfected animals.
DISCUSSION
The mononuclear phagocyte system has been implicated in host defense against bacterial pneumonia (16, 17, 44, 45), but the mechanisms that generate and maintain these cells during the infection have not been defined. In the present study, we report that M-CSF, but not IL-34, is induced in the lung during the infection. M-CSF was essential to the survival of the host as a critical survival signal for mononuclear phagocytes in infected tissues, but appeared to be dispensable for the generation, recruitment and differentiation of mononuclear phagocytes during the infection. The absence of M-CSF thus resulted in reduced numbers of both monocyte-derived cells and resident macrophage populations in the lungs and the liver, resulting in increased bacterial burden and injury to these organs and increased incidence and extent of bacteremia.
M-CSF is produced by many cell types, including mononuclear phagocytes themselves and has been documented to have a multitude of effects on mononuclear phagocytes in different experimental conditions, including development during embryogenesis, monocytopoeisis in adulthood, proliferation, differentiation, chemotaxis and survival (as reviewed in (11)). M-CSF has not been studied extensively in the context of infections, but has been associated with host defense in intra-cellular infections caused by L. monocytogenes (22, 46) and influences macrophage phenotype in experimental tuberculosis and schistosomiasis (47, 48). We report that M-CSF appeared to be dispensable for the proliferation and recruitment of monocytes and their progenitors during pneumonia, similar to recent findings in sterile peritonitis (39). Our studies did not directly examine the role of M-CSF in the differentiation of mononuclear phagocytes in the lungs in vivo, but the intact recruitment of monocytes to the lungs, together with the role of M-CSF in maintaining the numbers of both lung monocytes and monocyte-derived cells during infection indicates that the differentiation of monocytes into other mononuclear phagocytes during pneumonia remained intact despite M-CSF neutralization. In contrast, M-CSF was essential in maintaining lung and liver monocytic phagocyte populations by promoting their survival. M-CSF has been shown to be critical to survival of mononuclear phagocytes during development and under homeostatic conditions (49–52) but, to our knowledge, has not been shown to mediate mononuclear phagocyte survival in the setting of infection or inflammation. In this context, macrophage survival is a known regulatory factor in the context of infection: alveolar macrophage apoptosis, for example, is both anti-microbial and anti-inflammatory during pneumococcal pneumonia (53, 54), whereas macrophage necroptosis induces lung damage during Gram-negative and S. aureus pneumonia (55, 56).
In addition, we found that M-CSF mediates enhanced phagocytosis of bacteria by surviving mononuclear phagocytes. In this context, macrophages from op/op animals have been shown to have impaired phagocytosis against bacteria but not parasites (57, 58), but it is difficult to differentiate whether any effect in op/op macrophages is attributable to developmental abnormalities of the cells or lack of exposure to M-CSF during acute infection. Similar to our findings, however, M-CSF results in enhanced phagocytosis of fungal conidia by human monocyte-derived macrophages in vitro (59). Taken together, these data suggest that locally produced M-CSF can enhance anti-microbial properties of mononuclear phagocytes in target organs.
An unexpected finding in our study was the role of M-CSF in protecting the liver from injury during experimental pneumonia. The propensity of Gram-negative bacteria, specifically Klebsiella, to infect the liver is recognized clinically (60). In the context of experimental pneumonia, the liver is responsible for a protective acute phase response, but is also susceptible to TNF-mediated injury (61–63). M-CSF has recently been shown to mediate proliferation of liver macrophages and recruitment of monocytes to the injured liver (64). Our data adds to this literature, indicating that endogenous production of M-CSF during pneumonia mediates the accumulation of both monocyte-derived macrophages and Kupffer cells and protects the liver from disseminated infection.
The present work has several implications for future research. First, our data suggest that M-CSF is required not only for maintaining the number of monocyte-derived cells, but also alveolar macrophages and CD24+ (and thus pre-DC derived (4)) CD11b+ DC in the lungs as well as Kupffer cells in the liver. This finding is reminiscent of our prior report in CCR2-deficient mice (17) and suggests either that Ly6Chi monocytes repopulate other lineages of mononuclear phagocytes in the acute setting, as has been shown previously (9, 65), or that M-CSF is necessary to the maintenance of all 3 lineages of mononuclear phagocytes in the setting of acute injury. Second, we found M-CSF to have a marked role in the accumulation of Ly6Clo monocytes in the lungs, but any role of these cells in the setting of acute infection remains undefined. Third, our work documents the critical role of M-CSF during bacterial pneumonia but does not preclude an additional contribution for IL-34, or its second receptor, protein-tyrosine phosphatase-ζ, in the context of pneumonia. Finally, our findings may be relevant to efforts to inhibit M-CSF signaling as a therapeutic strategy in cancer (66), since such strategies may be expected to predispose patients to more severe pneumonia.
Acknowledgments
Funding: NIH grants HL098329, AI117397
Footnotes
Author contributions: AB, MB, AK and BM conceived and designed experiments; AB, ZZ, KM, REC, IV, AK, MB and BM performed experiments; AB, AK and IV analyzed data; AK and IV contributed reagents and analysis tools; AB and BM wrote the paper; and all authors revised and approved the text.
References
- 1.Mehrad B, Clark NM, Zhanel GG, Lynch JPr. Antimicrobial resistance in hospital-acquired gram-negative bacterial infections. Chest. 2015;147:1413–1421. doi: 10.1378/chest.14-2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mizgerd JP. Acute lower respiratory tract infection. N Engl J Med. 2008;358:716–727. doi: 10.1056/NEJMra074111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Segura E, Tussiwand R, Yona S. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol. 2014;14:571–578. doi: 10.1038/nri3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schlitzer A, McGovern N, Teo P, Zelante T, Atarashi K, Low D, Ho AWS, See P, Shin A, Wasan PS, Hoeffel G, Malleret B, Heiseke A, Chew S, Jardine L, Purvis HA, Hilkens CMU, Tam J, Poidinger M, Stanley ER, Krug AB, Renia L, Sivasankar B, Ng LG, Collin M, Ricciardi-Castagnoli P, Honda K, Haniffa M, Ginhoux F. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity. 2013;38:970–983. doi: 10.1016/j.immuni.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kopf M, Schneider C, Nobs SP. The development and function of lung-resident macrophages and dendritic cells. Nat Immunol. 2015;16:36–44. doi: 10.1038/ni.3052. [DOI] [PubMed] [Google Scholar]
- 6.Stanley ER, Hansen G, Woodcock J, Metcalf D. Colony stimulating factor and the regulation of granulopoiesis and macrophage production. Fed Proc. 1975;34:2272–2278. [PubMed] [Google Scholar]
- 7.Stanley ER, Cifone M, Heard PM, Defendi V. Factors regulating macrophage production and growth: identity of colony-stimulating factor and macrophage growth factor. J Exp Med. 1976;143:631–647. doi: 10.1084/jem.143.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sauter KA, Pridans C, Sehgal A, Tsai YT, Bradford BM, Raza S, Moffat L, Gow DJ, Beard PM, Mabbott NA, Smith LB, Hume DA. Pleiotropic effects of extended blockade of CSF1R signaling in adult mice. J Leukoc Biol. 2014;96:265–274. doi: 10.1189/jlb.2A0114-006R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fancke B, Suter M, Hochrein H, O'Keeffe M. M-CSF: a novel plasmacytoid and conventional dendritic cell poietin. Blood. 2008;111:150–159. doi: 10.1182/blood-2007-05-089292. [DOI] [PubMed] [Google Scholar]
- 10.Mossadegh-Keller N, Sarrazin S, Kandalla PK, Espinosa L, Stanley ER, Nutt SL, Moore J, Sieweke MH. M-CSF instructs myeloid lineage fate in single haematopoietic stem cells. Nature. 2013;497:239–243. doi: 10.1038/nature12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stanley ER, Chitu V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb Perspect Biol. 2014;6 doi: 10.1101/cshperspect.a021857. pii: a021857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin H, Lee E, Hestir K, Leo C, Huang M, Bosch E, Halenbeck R, Wu G, Zhou A, Behrens D, Hollenbaugh D, Linnemann T, Qin M, Wong J, Chu K, Doberstein SK, Williams LT. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science. 2008;320:807–811. doi: 10.1126/science.1154370. [DOI] [PubMed] [Google Scholar]
- 13.Nakamichi Y, Udagawa N, Takahashi N. IL-34 and CSF-1: similarities and differences. J Bone Miner Metab. 2013;31:486–495. doi: 10.1007/s00774-013-0476-3. [DOI] [PubMed] [Google Scholar]
- 14.Baek JH, Zeng R, Weinmann-Menke J, Valerius MT, Wada Y, Ajay AK, Colonna M, Kelley VR. IL-34 mediates acute kidney injury and worsens subsequent chronic kidney disease. J Clin Invest. 2015;125:3198–3214. doi: 10.1172/JCI81166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nandi S, Cioce M, Yeung YG, Nieves E, Tesfa L, Lin H, Hsu AW, Halenbeck R, Cheng HY, Gokhan S, Mehler MF, Stanley ER. Receptor-type protein-tyrosine phosphatase zeta is a functional receptor for interleukin-34. J Biol Chem. 2013;288:21972–21986. doi: 10.1074/jbc.M112.442731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiong H, Carter RA, Leiner IM, Tang YW, Chen L, Kreiswirth BN, Pamer EG. Distinct Contributions of Neutrophils and CCR2+ Monocytes to Pulmonary Clearance of Different Klebsiella pneumoniae Strains. Infect Immun. 2015;83:3418–3427. doi: 10.1128/IAI.00678-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen L, Zhang Z, Barletta KE, Burdick MD, Mehrad B. Heterogeneity of lung mononuclear phagocytes during pneumonia: contribution of chemokine receptors. Am J Physiol Lung Cell Mol Physiol. 2013;305:L702–711. doi: 10.1152/ajplung.00194.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roger T, Delaloye J, Chanson AL, Giddey M, Le Roy D, Calandra T. Macrophage migration inhibitory factor deficiency is associated with impaired killing of gram-negative bacteria by macrophages and increased susceptibility to Klebsiella pneumoniae sepsis. J Infect Dis. 2013;207:331–339. doi: 10.1093/infdis/jis673. [DOI] [PubMed] [Google Scholar]
- 19.Pittet LA, Quinton LJ, Yamamoto K, Robson BE, Ferrari JD, Algul H, Schmid RM, Mizgerd JP. Earliest innate immune responses require macrophage RelA during pneumococcal pneumonia. Am J Respir Cell Mol Biol. 2011;45:573–581. doi: 10.1165/rcmb.2010-0210OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bailie MB, Standiford TJ, Laichalk LL, Coffey MJ, Strieter R, Peters-Golden M. Leukotriene-deficient mice manifest enhanced lethality from Klebsiella pneumonia in association with decreased alveolar macrophage phagocytic and bactericidal activities. J Immunol. 1996;157:5221–5224. [PubMed] [Google Scholar]
- 21.Bernier T, Halter R, Pau D, Rittinghausen S, Emmendorffer A. M-CSF transgenic mice: role of M-CSF in infection and autoimmunity. Exp Toxicol Pathol. 2001;53:165–173. doi: 10.1078/0940-2993-00172. [DOI] [PubMed] [Google Scholar]
- 22.Gregory SH, Wing EJ, Tweardy DJ, Shadduck RK, Lin HS. Primary listerial infections are exacerbated in mice administered neutralizing antibody to macrophage colony-stimulating factor. J Immunol. 1992;149:188–193. [PubMed] [Google Scholar]
- 23.Doyle AG, Halliday WJ, Barnett CJ, Dunn TL, Hume DA. Effect of recombinant human macrophage colony-stimulating factor 1 on immunopathology of experimental brucellosis in mice. Infect Immun. 1992;60:1465–1472. doi: 10.1128/iai.60.4.1465-1472.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kayashima S, Tsuru S, Shinomiya N, Katsura Y, Motoyoshi K, Rokutanda M, Nagata N. Effects of macrophage colony-stimulating factor on reduction of viable bacteria and survival of mice during Listeria monocytogenes infection: characteristics of monocyte subpopulations. Infect Immun. 1991;59:4677–4680. doi: 10.1128/iai.59.12.4677-4680.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cenci E, Bartocci A, Puccetti P, Mocci S, Stanley ER, Bistoni F. Macrophage colony-stimulating factor in murine candidiasis: serum and tissue levels during infection and protective effect of exogenous administration. Infect Immun. 1991;59:868–872. doi: 10.1128/iai.59.3.868-872.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barletta KE, Cagnina RE, Burdick MD, Linden J, Mehrad B. Adenosine A(2B) receptor deficiency promotes host defenses against gram-negative bacterial pneumonia. Am J Respir Crit Care Med. 2012;186:1044–1050. doi: 10.1164/rccm.201204-0622OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mehrad B, Park SJ, Akangire G, Standiford TJ, Wu T, Zhu J, Mohan C. The lupus-susceptibility locus, Sle3, mediates enhanced resistance to bacterial infections. J Immunol. 2006;176:3233–3239. doi: 10.4049/jimmunol.176.5.3233. [DOI] [PubMed] [Google Scholar]
- 28.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, Jung S. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park SJ, Wiekowski MT, Lira SA, Mehrad B. Neutrophils regulate airway responses in a model of fungal allergic airways disease. J Immunol. 2006;176:2538–2545. doi: 10.4049/jimmunol.176.4.2538. [DOI] [PubMed] [Google Scholar]
- 30.Barletta KE, Cagnina RE, Wallace KL, Ramos SI, Mehrad B, Linden J. Leukocyte compartments in the mouse lung: distinguishing between marginated, interstitial, and alveolar cells in response to injury. J Immunol Methods. 2012;375:100–110. doi: 10.1016/j.jim.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park SJ, Burdick MD, Brix WK, Stoler MH, Askew DS, Strieter RM, Mehrad B. Neutropenia enhances lung dendritic cell recruitment in response to Aspergillus via a cytokine-to-chemokine amplification loop. J Immunol. 2010;185:6190–6197. doi: 10.4049/jimmunol.1002064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Phadke AP, Akangire G, Park SJ, Lira SA, Mehrad B. The role of CC chemokine receptor 6 in host defense in a model of invasive pulmonary aspergillosis. Am J Respir Crit Care Med. 2007;175:1165–1172. doi: 10.1164/rccm.200602-256OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GRS, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol. 2013;49:503–510. doi: 10.1165/rcmb.2013-0086MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weischenfeldt J, Porse B. Bone marrow-derived macrophages (BMM): isolation and applications. Cold Spring Harbor Protocols. 2008;2008 doi: 10.1101/pdb.prot5080. pdb.prot5080. [DOI] [PubMed] [Google Scholar]
- 35.Beletskii A, Cooper M, Sriraman P, Chiriac C, Zhao L, Abbot S, Yu L. High-throughput phagocytosis assay utilizing a pH-sensitive fluorescent dye. Biotechniques. 2005;39:894–897. doi: 10.2144/000112001. [DOI] [PubMed] [Google Scholar]
- 36.Schack L, Stapulionis R, Christensen B, Kofod-Olsen E, Skov Sorensen UB, Vorup-Jensen T, Sorensen ES, Hollsberg P. Osteopontin enhances phagocytosis through a novel osteopontin receptor, the alphaXbeta2 integrin. J Immunol. 2009;182:6943–6950. doi: 10.4049/jimmunol.0900065. [DOI] [PubMed] [Google Scholar]
- 37.Shibata Y, Zsengeller Z, Otake K, Palaniyar N, Trapnell BC. Alveolar macrophage deficiency in osteopetrotic mice deficient in macrophage colony-stimulating factor is spontaneously corrected with age and associated with matrix metalloproteinase expression and emphysema. Blood. 2001;98:2845–2852. doi: 10.1182/blood.v98.9.2845. [DOI] [PubMed] [Google Scholar]
- 38.MacDonald KPA, Palmer JS, Cronau S, Seppanen E, Olver S, Raffelt NC, Kuns R, Pettit AR, Clouston A, Wainwright B, Branstetter D, Smith J, Paxton RJ, Cerretti DP, Bonham L, Hill GR, Hume DA. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood. 2010;116:3955–3963. doi: 10.1182/blood-2010-02-266296. [DOI] [PubMed] [Google Scholar]
- 39.Louis C, Cook AD, Lacey D, Fleetwood AJ, Vlahos R, Anderson GP, Hamilton JA. Specific Contributions of CSF-1 and GM-CSF to the Dynamics of the Mononuclear Phagocyte System. J Immunol. 2015;195:134–144. doi: 10.4049/jimmunol.1500369. [DOI] [PubMed] [Google Scholar]
- 40.Hettinger J, Richards DM, Hansson J, Barra MM, Joschko AC, Krijgsveld J, Feuerer M. Origin of monocytes and macrophages in a committed progenitor. Nat Immunol. 2013;14:821–830. doi: 10.1038/ni.2638. [DOI] [PubMed] [Google Scholar]
- 41.Jenkins SJ, Ruckerl D, Thomas GD, Hewitson JP, Duncan S, Brombacher F, Maizels RM, Hume DA, Allen JE. IL-4 directly signals tissue-resident macrophages to proliferate beyond homeostatic levels controlled by CSF-1. J Exp Med. 2013;210:2477–2491. doi: 10.1084/jem.20121999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tagliani E, Shi C, Nancy P, Tay CS, Pamer EG, Erlebacher A. Coordinate regulation of tissue macrophage and dendritic cell population dynamics by CSF-1. J Exp Med. 2011;208:1901–1916. doi: 10.1084/jem.20110866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tushinski RJ, I, Oliver T, Guilbert LJ, Tynan PW, Warner JR, Stanley ER. Survival of mononuclear phagocytes depends on a lineage-specific growth factor that the differentiated cells selectively destroy. Cell. 1982;28:71–81. doi: 10.1016/0092-8674(82)90376-2. [DOI] [PubMed] [Google Scholar]
- 44.Saini Y, Wilkinson KJ, Terrell KA, Burns KA, Livraghi-Butrico A, Doerschuk CM, O'Neal WK, Boucher RC. Neonatal Pulmonary Macrophage Depletion Coupled to Defective Mucus Clearance Increases Susceptibility to Pneumonia and Alters Pulmonary Immune Responses. Am J Respir Cell Mol Biol. doi: 10.1165/rcmb.2014-0111OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Broug-Holub E, Toews GB, van Iwaarden JF, Strieter RM, Kunkel SL, Paine Rr, Standiford TJ. Alveolar macrophages are required for protective pulmonary defenses in murine Klebsiella pneumonia: elimination of alveolar macrophages increases neutrophil recruitment but decreases bacterial clearance and survival. Infect Immun. 1997;65:1139–1146. doi: 10.1128/iai.65.4.1139-1146.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gregory SH, Wing EJ. Macrophage colony-stimulating factor and the enhanced migration of monocytes are essential in primary but not secondary host defenses to Listeria organisms. J Infect Dis. 1993;168:934–942. doi: 10.1093/infdis/168.4.934. [DOI] [PubMed] [Google Scholar]
- 47.Higgins DM, Sanchez-Campillo J, Rosas-Taraco AG, Higgins JR, Lee EJ, Orme IM, Gonzalez-Juarrero M. Relative levels of M-CSF and GM-CSF influence the specific generation of macrophage populations during infection with Mycobacterium tuberculosis. J Immunol. 2008;180:4892–4900. doi: 10.4049/jimmunol.180.7.4892. [DOI] [PubMed] [Google Scholar]
- 48.Nascimento M, Huang SC, Smith A, Everts B, Lam W, Bassity E, Gautier EL, Randolph GJ, Pearce EJ. Ly6Chi monocyte recruitment is responsible for Th2 associated host-protective macrophage accumulation in liver inflammation due to schistosomiasis. PLoS Pathog. 2014;10:e1004282. doi: 10.1371/journal.ppat.1004282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fuller K, Owens JM, Jagger CJ, Wilson A, Moss R, Chambers TJ. Macrophage colony-stimulating factor stimulates survival and chemotactic behavior in isolated osteoclasts. J Exp Med. 1993;178:1733–1744. doi: 10.1084/jem.178.5.1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Melcher M, Unger B, Schmidt U, Rajantie IA, Alitalo K, Ellmeier W. Essential roles for the Tec family kinases Tec and Btk in M-CSF receptor signaling pathways that regulate macrophage survival. J Immunol. 2008;180:8048–8056. doi: 10.4049/jimmunol.180.12.8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Glantschnig H, Fisher JE, Wesolowski G, Rodan GA, Reszka AA. M-CSF, TNFalpha and RANK ligand promote osteoclast survival by signaling through mTOR/S6 kinase. Cell Death Differ. 2003;10:1165–1177. doi: 10.1038/sj.cdd.4401285. [DOI] [PubMed] [Google Scholar]
- 52.Otero K, I, Turnbull R, Poliani PL, Vermi W, Cerutti E, Aoshi T, Tassi I, Takai T, Stanley SL, Miller M, Shaw AS, Colonna M. Macrophage colony-stimulating factor induces the proliferation and survival of macrophages via a pathway involving DAP12 and beta-catenin. Nat Immunol. 2009;10:734–743. doi: 10.1038/ni.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dockrell DH, Marriott HM, Prince LR, Ridger VC, Ince PG, Hellewell PG, Whyte MKB. Alveolar macrophage apoptosis contributes to pneumococcal clearance in a resolving model of pulmonary infection. J Immunol. 2003;171:5380–5388. doi: 10.4049/jimmunol.171.10.5380. [DOI] [PubMed] [Google Scholar]
- 54.Marriott HM, Hellewell PG, Cross SS, Ince PG, Whyte MKB, Dockrell DH. Decreased alveolar macrophage apoptosis is associated with increased pulmonary inflammation in a murine model of pneumococcal pneumonia. J Immunol. 2006;177:6480–6488. doi: 10.4049/jimmunol.177.9.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gonzalez-Juarbe N, Gilley RP, Hinojosa CA, Bradley KM, Kamei A, Gao G, Dube PH, Bergman MA, Orihuela CJ. Pore-Forming Toxins Induce Macrophage Necroptosis during Acute Bacterial Pneumonia. PLoS Pathog. 2015;11:e1005337. doi: 10.1371/journal.ppat.1005337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kitur K, Parker D, Nieto P, Ahn DS, Cohen TS, Chung S, Wachtel S, Bueno S, Prince A. Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage. PLoS Pathog. 2015;11:e1004820. doi: 10.1371/journal.ppat.1004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ogiku M, Kono H, Ishii K, Hosomura N, Fujii H. Role of macrophage colony-stimulating factor in polymicrobial sepsis according to studies using osteopetrotic (op/op) mice. J Surg Res. 2011;169:106–116. doi: 10.1016/j.jss.2009.10.023. [DOI] [PubMed] [Google Scholar]
- 58.Schonlau F, Schlesiger C, Ehrchen J, Grabbe S, Sorg C, Sunderkotter C. Monocyte and macrophage functions in M-CSF-deficient op/op mice during experimental leishmaniasis. J Leukoc Biol. 2003;73:564–573. doi: 10.1189/jlb.12011003. [DOI] [PubMed] [Google Scholar]
- 59.Roilides E, Sein T, Holmes A, Chanock S, Blake C, Pizzo PA, Walsh TJ. Effects of macrophage colony-stimulating factor on antifungal activity of mononuclear phagocytes against Aspergillus fumigatus. J Infect Dis. 1995;172:1028–1034. doi: 10.1093/infdis/172.4.1028. [DOI] [PubMed] [Google Scholar]
- 60.Siu LK, Yeh KM, Lin JC, Fung CP, Chang FY. Klebsiella pneumoniae liver abscess: a new invasive syndrome. Lancet Infect Dis. 2012;12:881–887. doi: 10.1016/S1473-3099(12)70205-0. [DOI] [PubMed] [Google Scholar]
- 61.Hilliard KL, Allen E, Traber KE, Yamamoto K, Stauffer NM, Wasserman GA, Jones MR, Mizgerd JP, Quinton LJ. The Lung-Liver Axis: A Requirement for Maximal Innate Immunity and Hepatoprotection during Pneumonia. Am J Respir Cell Mol Biol. 2015;53:378–390. doi: 10.1165/rcmb.2014-0195OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moore TA, Lau HY, Cogen AL, Monteleon CL, Standiford TJ. Anti-tumor necrosis factor-alpha therapy during murine Klebsiella pneumoniae bacteremia: increased mortality in the absence of liver injury. Shock. 2003;20:309–315. doi: 10.1097/01.shk.0000087203.34916.45. [DOI] [PubMed] [Google Scholar]
- 63.Moore TA, Perry ML, Getsoian AG, Monteleon CL, Cogen AL, Standiford TJ. Increased mortality and dysregulated cytokine production in tumor necrosis factor receptor 1-deficient mice following systemic Klebsiella pneumoniae infection. Infect Immun. 2003;71:4891–4900. doi: 10.1128/IAI.71.9.4891-4900.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stutchfield BM, Antoine DJ, Mackinnon AC, Gow DJ, Bain CC, Hawley CA, Hughes MJ, Francis B, Wojtacha D, Man TY, Dear JW, Devey LR, Mowat AM, Pollard JW, Park BK, Jenkins SJ, Simpson KJ, Hume DA, Wigmore SJ, Forbes SJ. CSF1 Restores Innate Immunity After Liver Injury in Mice and Serum Levels Indicate Outcomes of Patients With Acute Liver Failure. Gastroenterology. 2015;149:1896–1909.e1814. doi: 10.1053/j.gastro.2015.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 65.Taut K, Winter C, Briles DE, Paton JC, Christman JW, Maus R, Baumann R, Welte T, Maus UA. Macrophage Turnover Kinetics in the Lungs of Mice Infected with Streptococcus pneumoniae. Am J Respir Cell Mol Biol. 2008;38:105–113. doi: 10.1165/rcmb.2007-0132OC. [DOI] [PubMed] [Google Scholar]
- 66.Hume DA, MacDonald KPA. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood. 2012;119:1810–1820. doi: 10.1182/blood-2011-09-379214. [DOI] [PubMed] [Google Scholar]