

Abstract
Leishmaniasis is an important parasitic disease found in the tropics and sub-tropics. Cutaneous and visceral leishmaniasis affect an estimated 1.5 million people worldwide. Despite its human health relevance, relatively little is known about the cell death pathways that control Leishmania replication in the host. Necroptosis is a recently identified form of cell death with potent anti-viral effects. RIPK1 is a critical kinase that mediates necroptosis downstream of death receptors and toll-like receptors. Heme, a product of hemoglobin catabolism during certain intracellular pathogen infections, is also a potent inducer of macrophage necroptosis. We found that human visceral leishmaniasis patients exhibit elevated serum levels of heme. Therefore, we examined the impact of heme and necroptosis on Leishmania replication. Indeed, heme potently inhibited Leishmania replication in bone marrow derived macrophages (BMDMs). Moreover, we found that inhibition of RIPK1 kinase activity also enhanced parasite replication in the absence of heme. We further found that the mitochondrial phosphatase phosphoglycerate mutase family member 5 (PGAM5), a putative downstream effector of RIPK1, was also required for inhibition of Leishmania replication. In mouse infection, both PGAM5 and RIPK1 kinase activity are required for IL-1β expression in response to Leishmania. However, PGAM5, but not RIPK1 kinase activity, was directly responsible for Leishmania-induced IL-1β secretion and nitric oxide production in BMDMs. Collectively, these results revealed that RIPK1 and PGAM5 function independently to exert optimal control of Leishmania replication in the host.
Introduction
Leishmaniasis is a vector-borne parasitic disease caused by protozoan of the genus Leishmania. Leishmania parasites exist as extracellular flagellated promastigotes in sandflies. Upon entry into mammalian hosts, they exist as intracellular amastigotes (1). In human, disease manifestation varies depending on the Leishmania species and can range from asymptomatic infections, self-healing cutaneous lesions, to life-threatening infections involving visceral organs. For instance, Leishmania (L.) amazonensis and L. major are both known to cause cutaneous infections, while L. infantum chagasi (L. infantum) causes visceral leishmaniasis (VL). VL can be extremely debilitating, with significant morbidity and mortality if not promptly treated (2). In addition, disease progression is influenced by host immune response (3). Macrophages are widely believed to play critical roles in the control of Leishmania infection (4). Interestingly, they also serve as major reservoirs in which Leishmania amastigotes replicate.
The host immune response that controls Leishmaniasis is not fully understood. The inflammatory cytokine IL-1β is critical for innate immune defense against many different pathogens. Production of mature IL-1β in macrophages requires two signals: NF-κB-driven de novo synthesis of pro-IL-1β and inflammasome-mediated cleavage of pro-IL-1β into the mature cytokine. Mice that are deficient in the essential inflammasome sensor NLRP3, the adaptor ASC, or the IL-1β cleavage protease caspase 1 exhibited impaired control of replication of many Leishmania species (5). The protective effect of IL-1β is mediated by nitric oxide (NO) (5), which stimulates macrophage cell death. In contrast to IL-1β, NLRP3 inflammasome-mediated pro-IL-18 processing and secretion exacerbates the disease by skewing the reaction towards a Th2-biased response in susceptible BALB/c mice (6). In addition to IL-1β and IL-18, the stress-induced enzyme heme oxygenase-1 (HO-1) has been shown to facilitate Leishmania replication by limiting inflammatory cytokine expression (7). Another key function of HO-1 is to cleave heme, which results in generation of free iron, carbon monoxide and biliverdin. Biliverdin is subsequently converted to bilirubin by biliverdin reductase. Heme release is found in intracellular pathogen infections such as Plasmodium falciparum and Plasmodium vivax, the etiological agents of malaria (8, 9). Severe visceral leishmaniasis is also characterized by hematological alterations and spontaneous bleeding associated with marked inflammatory imbalance (10). These observations suggest that heme may similarly influence the outcome of Leishmania infection.
Recent studies show that free heme induces murine macrophage necroptosis (11), a recently described form of inflammatory cell death that has important protective functions in certain viral infections (12–15). Hence, HO-1 may facilitate Leishmania replication by limiting heme-induced necroptosis, thereby preserving the parasite replication reservoir. Two serine/threonine kinases, the receptor interacting protein kinase 1 (RIPK1) and RIPK3, are critical adaptors for necroptosis (16). Recently, the mitochondrial phosphatase phosphoglycerate mutase family member 5 (PGAM5) was identified as a downstream effector of necroptosis (17). In addition, PGAM5 can also function downstream of RIPK3 to control cytokine expression in NKT cells independent of necroptosis (18). However, the roles of heme, RIP kinases, PGAM5 and necroptosis have not been investigated in Leishmania infection.
In this study, we report that high levels of heme were detected in the sera of human patients with visceral leishmaniasis. In response to high doses of heme, human and mouse macrophages underwent RIPK1- and RIPK3-dependent necroptosis. High doses of heme inhibited L. infantum replication in macrophages. Although RIPK1 kinase inhibitors restored parasite replication, neither RIPK3 kinase inhibitors nor MLKL inhibitor rescued L. infantum replication in heme-treated macrophages. Importantly, RIPK1 kinase activity also inhibited Leishmania replication in the absence of heme, since enhanced Leishmania replication was detected in macrophages expressing kinase inactive RIPK1 and wild type macrophages treated with RIPK1 kinase inhibitors. In addition to RIPK1, the mitochondrial phosphatase PGAM5 was also required for optimal control of Leishmania replication in macrophages. Mechanistically, we showed that PGAM5 promotes IL-1β production, which in turn stimulates nitric oxide production. By contrast, RIPK1 regulates Leishmania replication independent of IL-1β. Collectively, our results identified RIPK1 and PGAM5 as two novel host factors that control Leishmania replication through distinct mechanisms.
Material and Methods
Determination of heme concentration in human visceral leishmaniasis
Plasma was collected between 2010 and 2012 from symptomatic individuals (N = 49) and sex-matched endemic healthy controls (N=39) in Aracaju, Sergipe, Brazil. The baseline characteristics of the study participants are shown in Luz et al., 2012. Total heme in plasma samples was estimated by a colorimetric determination at 400 nm using the QuantiChrom Heme Assay Kit (BioAssay Systems, Hayward, CA), according to the manufacturer’s protocol.
Mouse infection
All experimental procedures were approved and conducted according to the Institutional Animal Care and Use Committee of the University of the University of Massachusetts Medical School. Ripk1kd/kd (Ripk1K45A/K45A) have been described before (19). Characterization of the Pgam5−/− mice will be published in a different manuscript. For in vivo infection with Leishmania, mice were infected with 4 x 106 L. amazonensis per footpad in a volume of 20 μl. Swelling of the footpad was measured weekly by digital calipers (Fisher Scientific) and compared to the uninfected footpad. For quantification of L. amazonensis parasite load, footpads from infected mice were harvested 10 weeks post-infection, homogenized, and serially diluted (1:2) in 96-well plates in complete Schneider media. After 7 days of incubation at 26°C, the number of viable parasites was calculated from the highest dilution at which parasites were observed. For assessment of cytokine production, footpads from mice were homogenized in 1 ml PBS supplemented with protease inhibitors. The footpad extracts were analyzed for TNF and IL-1β by ELISA (BD Biosciences). For ex vivo analysis of IL-1β and TNF production, single-cell suspensions were prepared from the spleen of mice infected after 10 weeks. After red blood cell lysis, 1 x 106 cells/ml in 1 ml were plated in 24-well plates. Cells were stimulated with 50 μg/ml of L. amazonensis particulate antigens (L.A.). The supernatants were harvested after 48 hours and analyzed by ELISA.
Mouse and human cell culture
Bone marrow derived macrophages (BMDMs) were generated from femoral bone marrow cells by culture for seven days in DMEM medium supplemented with 20% L929 conditioned medium as described before (20). Human primary monocyte-derived macrophages were generated from peripheral blood mononuclear cells (PBMCs) from healthy volunteers. After Ficoll Paque (GE healthcare) density centrifugation, PBMCs were plated at 2 x 106 cells/well on 96-well plate. After 1 hour, non-adherent cells were removed, and adherent cells were cultured in RPMI 1640 supplemented with 2 mM L-glutamine and 10% FBS for 7 days. THP1 cells were stimulated with 200 nM phorbol 12-myristate 13-acetate (PMA, Sigma-Aldrich) for 3 days, followed by culture in medium without PMA for another 2 days.
NO production and Cell death assay
BMDMs (105 cells/well), PBMCs (2 x 105 cells/well), monocyte-derived macrophages (2 x 105 cells/well) and pro-monocytic THP-1 cells (2 x 105 cells/well) were cultured with 3 μM, 10 μM or 30 μM of heme. Twelve hours later, release of lactate dehydrogenase (LDH) was measured in cell-free culture supernatant by the cytotoxicity detection kit (Roche Applied Science, Germany). Where it is indicated, THP-1 cells were pre-treated with RIPK1 or RIPK3 inhibitors for 1 hour before treatment with heme. BMDMs cell viability was assessed by CytoTox96 Non-Radioactive Cytotoxicity Assay. In vitro IL-1β production by BMDMs was measured by ELISA after stimulation with 200 ng/ml ultrapure LPS (InvivoGen) for 3 hours, followed by 10 μM nigericin for another 3 hours. NO production was measured in the supernatants of L. infantum-infected BMDMs pre- treated with 200 ng/mL LPS, then stimulated with 100 U/ml IFN-γ, in some experiments 10ng/mL recombinant IL-1β was added along with IFNγ treatment. Supernatants were harvested after 48 hours and nitrite production was measured using Griess reaction.
Leishmania infection in BMDMs
L. amazonensis (strain IFLA/BR/67/PH8), L. major (strain MHOM/IL/81/Friedlin) and L. infantum (MCAN/BR/89/Ba262) promastigotes were grown in Schneider medium (Sigma-Aldrich) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin and 10% FBS. Stationary-phase promastigotes were used in all experiments. BMDMs and THP-1 cells were plated on coverslips one day before infection. Cells were infected with parasites at early stationary phase (MOI=5 for L. amazonensis and L. major, MOI=10 for L. infantum). After 4 hours, cells were washed and further cultured for 72 hours. In some experiments, 30 μM heme was added 4 hours after infection. Cells were fixed with methanol and stained with Diff-Quick (Thermo Scientific). The number of amastigotes per 100 macrophages was enumerated by counting on a light microscope. In addition, parasite load was determined by light microscopy and the production of viable promastigotes in Schneider medium. Briefly, 72 hours post-infection, cell culture medium was replaced by Schneider medium and the plates were then kept at 24°C. Seven days later, proliferating extracellular motile promastigotes were counted in a Neubauer hemocytometer. For THP-1 cells in vitro infection, PMA-differentiated cells were treated for 1 hour with GSK RIPK1 and RIPK3 inhibitors before infection with promastigotes.
Statistical analyses
All results shown are mean ± SEM. Statistical significance was determined by unpaired Student’s t test with Welch’s correction unless otherwise stated in figure legend.
Results
Elevated serum heme level is a hallmark of human visceral leishmaniasis
Heme release is a pathological consequence of hemolytic intracellular pathogen infection such as malaria (21). Heme impairs prostaglandin E2 and TGFβ production by human mononuclear cells via Cu/Zn superoxide dismutase (9). Leishmania promastigotes have been shown to degrade hemoglobin, which could lead to release of heme (22). Interestingly, HO-1, a key catabolic enzyme that neutralizes the cytotoxicity of heme (23), has been shown to promote Leishmania replication (7). We therefore asked if Leishmania infection would result in heme release. Indeed, we found that the serum of human visceral leishmaniasis patients had significant increase in heme levels compared with healthy controls (Fig. 1). This suggests that heme release is a possible contributing factor to this severe form of leishmaniasis.
Figure 1.
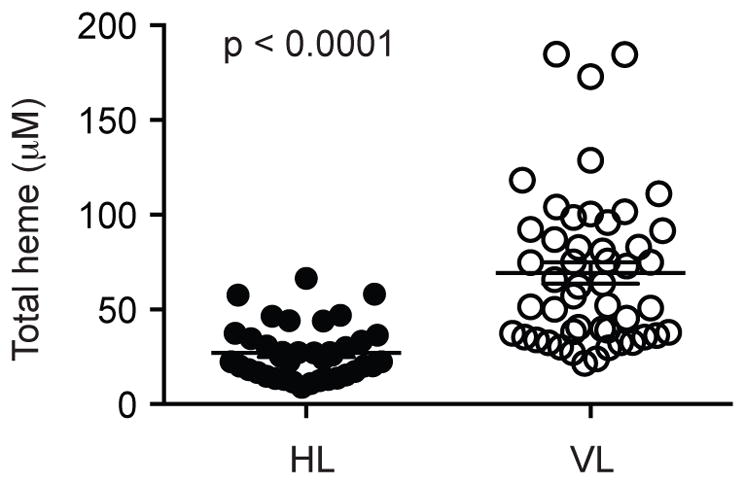
Human visceral leishmaniasis patients exhibit elevated serum concentrations of heme. Serum samples obtained from patients with VL (n=49) and healthy controls (HC, n=39) from an endemic area in the Northeast of Brazil. Total heme in plasma samples was estimated by a colorimetric determination. Mann–Whitney U test was used to verify statistical difference between HC and VL. Circles represent individual values and black bars represent median values.
Heme induces necroptosis in human macrophages
Leishmania parasites express the heme transporter LHR1 that is essential for virulence in macrophages and mice (24). This suggests that heme promotes Leishmania replication. In contrast, we previously showed that heme causes RIPK1- and RIPK3-dependent necroptosis in mouse macrophages (11). To reconcile these two observations and to determine the sensitivity of human cells to heme, we treated human peripheral blood mononuclear cells (PBMCs) from different healthy donors with different doses of heme. We found that human PBMCs were resistant to low doses of heme (3–10 μM), they underwent cell death in response to higher doses of heme (30 μM) (Fig. 2a). By contrast, human monocyte-derived macrophages were more sensitive to heme, with detectable cell death starting at 10 μM heme (Fig. 2b). The human monocytic cell line THP-1 differentiates into macrophages when stimulated with the phorbol ester PMA. Similar to primary PBMCs and macrophages, PMA-treated THP-1 cells also underwent high dose (30 μM) heme-induced cell death in a dose-dependent manner (Fig. 2c). Heme-induced cell death of THP-1 cells was inhibited by the RIPK1 kinase inhibitor necrostatin-1 (Nec-1), the RIPK3 kinase inhibitors GSK’840 and GSK’843 (25), and the MLKL inhibitor necrosulfonamide (NSA) (Fig. 2d–e). Since RIPK1, RIPK3 and MLKL are key adaptors for necroptosis (15), we conclude that high doses of heme triggered classical necroptosis in human macrophages.
Figure 2. Heme induces RIPK1-RIPK3-MLKL dependent necroptosis in human macrophages.

(a–c) Heme induced dose-dependent cell death in (a) human PBMCs, (b) primary human monocyte-derived macrophages and (c) PMA-differentiated THP-1 cells. (d–e) THP-1 cells were pre-treated with 10 μM necrostatin-1, 5 μM necrosulfonamide (NSA), or the RIPK3 kinase inhibitors GSK’840 and GSK’843 (2 μM) for 1 hour prior to treatment with 30 μM heme. Three independent experiments. Results shown are representative of mean of triplicates ± SEM. *p<0.001, ***p<0.05.
Leishmania replication is inhibited by heme and RIPK1 kinase activity
We next asked whether high dose heme-induced necroptosis might inhibit Leishmania replication. Indeed, we found that heme blunted parasite replication in a dose-dependent manner in BMDMs (Fig. 3a). Consistent with the requirement for heme for replication, low dose (3 μM) heme did not impair L. infantum replication (Fig. 3a). To determine whether the inhibition of parasite replication was due to necroptosis, we treated infected THP-1 cells with different necroptosis inhibitors. The serine/threonine kinases RIPK1 and RIPK3 are crucial for heme-induced necroptosis (11). As expected, the RIPK1 kinase inhibitors 7-Cl-O-Nec-1 (26) and GSK’963, but not the inactive enantiomer GSK’962, reversed high dose heme-induced inhibition of Leishmania replication (Fig. 3b). Surprisingly, the RIPK3 inhibitors GSK’840 and GSK’843 (25), and the MLKL inhibitor necrosulfonamide (NSA) (27), had no effects on heme-induced inhibition of Leishmania replication (Fig. 3b). These results suggest that RIPK1 kinase activity might regulate Leishmania replication independent of necroptosis. Indeed, the RIPK1 inhibitors 7-Cl-O-Nec-1 and GSK’963 strongly enhanced L. infantum replication in THP-1 cells even in the absence of heme (Fig. 3c). By contrast, the RIPK3 inhibitor GSK’843 had no effect on L. infantum replication (Fig. 3c). These results show that although high doses of heme can inhibit parasite replication through necroptosis, RIPK1 kinase activity also inhibits L. infantum replication independent of necroptosis.
Figure 3. RIPK1 kinase activity regulates Leishmania replication independent heme-induced necroptosis.

(a) High doses of heme inhibit Leishmania infantum replication. BMDMs were infected in vitro with L. infantum (multiplicity of infection 10) and treated with different doses of free heme (3, 10, 30 and 50 μM). Parasite load was counted by optical microscopy at 72 h postinfection as described in Materials and Methods. The percentage of L. infantum infected macrophages (left) and the number of amastigotes per 100 macrophages (right) are shown. Results shown are mean of triplicates ± SEM. ANOVA with Bonferroni’s post-test was used to evaluate statistical significance. *** p< 0.05. (b) RIPK1 kinase inhibitors, but not RIPK3 or MLKL inhibitors, reversed heme-induced inhibition of Leishmania infantum replication in THP-1 cells. PMA-differentiated THP-1 cells were pretreated with the indicated inhibitors for 1 hour, followed by infection with Leishmania infantum for 4 hours prior to stimulation with 30 μM heme. Parasite load was determined 24 hours post-infection. (c) RIPK1 kinase activity regulates Leishmania infantum replication in THP-1 cells independent of heme-induced necroptosis. PMA-differentiated THP-1 cells were pre-treated with the indicated inhibitors for 1 hour prior to infection with Leishmania infantum. Parasite load was determined 24 hours post-infection. Three independent experiments. Results shown are mean of triplicates ± SEM. *p<0.001, ***p<0.05.
The putative RIPK1 downstream effector PGAM5 regulates Leishmania replication
The effect of the RIPK1 kinase inhibitors on Leishmania replication prompted us to examine the underlying mechanism. Since pharmacological inhibitors can have off-target effects, we first confirmed the role of RIPK1 kinase activity in Leishmania replication using bone marrow derived macrophages (BMDMs) derived from knock-in mice expressing the kinase inactive RIPK1 mutant RIPK1-K45A (Ripk1kd/kd) (19). Similar to THP-1 cells treated with RIPK1 inhibitors, the number of L. infantum amastigotes in infected Ripk1kd/kd BMDMs was far greater than that of wild type BMDMs (Fig. 4a). Similar defects were observed in Ripk1kd/kd BMDMs infected with two other strains of Leishmania, L. amazonensis (Fig. 4b) and L. major (Fig. 4c). Importantly, no significant differences in intracellular parasite were detected 4 hours after infection (Fig. 4d), indicating that the increase in parasite replication in Ripk1kd/kd BMDMs was not due to differences in infection efficiency. Rather, these results indicate that RIPK1 kinase activity restricts Leishmania replication after entry into macrophages.
Figure 4. RIPK1 and PGAM5 control Leishmania replication in mouse BMDMs.

(a–c) Wild type (WT), Pgam5−/− and Ripk1kd/kd BMDMs were infected with (a) Leishmania infantum, (b) Leishmania amazonensis or (c) Leishmania major. Amastigotes were counted 72 hours post-infection. Representative images of amastigotes in infected BMDMs were shown on the right. (d) RIPK1 and PGAM5 do not affect infectivity. Intracellular parasites were counted 4 hours after infection with the indicated parasites. Three independent experiments. Results shown are mean of triplicates ± SEM. *p<0.001, **p<0.01, ***p<0.05.
The mitochondrial phosphatase PGAM5 has been reported to act downstream of RIPK1 and RIPK3 in necroptosis (17). Although subsequent studies have challenged the role of PGAM5 in necroptosis (28, 29), PGAM5 has been shown to mediate other RIPK1 functions (30). Indeed, we found that replication of L. infantum (Fig. 4a), L. amazonensis (Fig. 4b) and L. major (Fig. 4c) was also increased in Pgam5−/− BMDMs. The increase in parasite replication was not due to changes in infection rate, since similar number of parasites were found in WT and Pgam5−/− BMDMs 4 hours post-infection (Fig. 4d).
RIPK1 and PGAM5 controls Leishmania replication through different mechanisms
The results in BMDMs suggest that RIPK1 and PGAM5 may act along the same pathway to control Leishmania replication. To interrogate this possibility, we examined cytokine response in Ripk1kd/kd and Pgam5−/− mice in response to Leishmania infection. Because L. infantum does not cause productive infection in mice, we used L. amazonensis for the in vivo infections. A recent study demonstrated that NLRP3 inflammasome-mediated IL-1β secretion stimulates NO production and killing of Leishmania parasites (5). Since both RIPK1 and PGAM5 have been implicated to regulate cell death and pro-IL-1β cleavage (29, 30), we examined IL-1β production in mice infected with the parasites. At 4 weeks post-infection, IL-1β expression was reduced in the footpad of infected Ripk1kd/kd and Pgam5−/− mice (Fig. 5a). By contrast, TNF expression was normal in Ripk1kd/kd and Pgam5−/− mice (Fig. 5a). However, by 10 weeks post-infection, the difference in IL-1β was no longer detected in Ripk1kd/kd and Pgam5−/− mice (Fig. 4b). These results indicate that RIPK1 and PGAM5 regulate early IL-1β release during Leishmania infection.
Figure 5. RIPK1 and PGAM5 control Leishmania through distinct mechanisms.

(a–b) Production of IL-1β, but not TNF, was reduced in Leishmania-infected Pgam5−/−, but not Ripk1kd/kd mice. (a) Footpad tissue extracts from mice infected with Leishmania amazonensis for 4 weeks were stimulated with Leishmania particular antigen for 48 hours. IL-1β and TNF secretion was determined by ELISA. (b) Footpad tissue extracts from infected mice at 10 weeks were examined for IL-1β and TNF expression by ELISA. (c) Pgam5−/−, but not Ripk1kd/kd BMDMs are defective for IL-1β secretion. BMDMs were primed with LPS for 3 hours followed by 3 hours of stimulation with nigericin. IL-1β secretion was determined by ELISA. (d) Splenocytes from mice infected with Leishmania amazonensis were stimulated with Leishmania particular antigen. IL-1β secretion was measured 48 hours after stimulation. (e) WT and Pgam5−/− BMDMs were infected with L. infantum for 4 hours. Where indicated, BMDMs were also treated with LPS and the indicated cytokines. After incubation for 48 hours incubation, NO production was determined as described in materials and methods. (f) PGAM5 deficiency does not affect heme-induced necroptosis. BMDMs were stimulated with 30 μM heme and cell death was determined as described in materials and method. (g) RIPK1 and PGAM5 do not regulate BMDM survival during Leishmania infection. BMDMs were infected with the indicated Leishmania strains. Cell death was determined 24 hours post-infection. (h) WT and Ripk1kd/kd BMDMs were infected with L. infantum for 4 hours. NO production was determined 48 hours post-infection. Two independent experiments. Results shown are mean of triplicates ± SEM. *p<0.001, **p<0.01, ***p<0.05.
We next asked whether PGAM5 and RIPK1 kinase activity directly regulate IL-1β secretion. We first examined IL-1β secretion by LPS-primed macrophages stimulated with the NLRP3 inflammasome agonist nigericin. Strikingly, reduced IL-1β secretion was only observed in Pgam5−/−, but not Ripk1kd/kd BMDMs (Fig. 5c). In fact, IL-1β secretion was slightly increased in Ripk1kd/kd BMDMs compared to wild type BMDMs. To further confirm these results, we isolated splenocytes from mice infected with L. amazonensis and stimulated them with Leishmania particular antigen ex vivo. Similar to LPS-primed macrophages, IL-1β secretion was reduced in Pgam5−/−, but not Ripk1kd/kd splenocytes (Fig. 5d). A key function of IL-1β during Leishmania infection is to induce production of nitric oxide (NO), which in turn mediates killing of infected cells (5). Indeed, LPS and IFNγ-induced NO production was significantly reduced in Leishmania-infected Pgam5−/− BMDMs (Fig. 5e). Moreover, recombinant IL-1β restored NO production by Pgam5−/− BMDMs to wild type level (Fig. 5e). The defect in IL-1β secretion by Pgam5−/− BMDMs was not due to increased cell death, since Leishmania-infected wild type, Ripk1−/− and Pgam5−/− BMDMs had similar viability (Fig. 5f). Moreover, Pgam5−/− BMDMs were similarly sensitive to heme-induced cell death (Fig. 5f). This indicates that PGAM5 facilitates control of Leishmania replication through an IL-1β/NO axis. In contrast, NO production was normal in infected Ripk1kd/kd BMDMs (Fig. 5h). Hence, RIPK1 and PGAM5 inhibit Leishmania replication through distinct mechanisms.
RIPK1 and PGAM5 are required for in vivo control of Leishmania infection
We next asked how the early defect in IL-1β production by Pgam5−/− and Ripk1kd/kd mice might affect inflammation and Leishmania replication in vivo. Histological sections of the footpad revealed similar extent of neutrophil infiltration in the footpad of WT, Ripk1kd/kd and Pgam5−/− mice at 4 weeks post-infection (Fig. 6a). Importantly, an increase in the number of amastigotes in intracellular vacuoles was apparent in Ripk1kd/kd and Pgam5−/− mice. Indeed, when the numbers of parasites in the infected footpad were counted, significantly greater number of parasites was found in both Ripk1kd/kd and Pgam5−/− mice (Fig. 6b). Moreover, the lesion size in the infected footpad was significantly increased in Ripk1kd/kd mice at 10 weeks post-infection (Fig. 6c). Surprisingly, lesion size was not significantly increased in Pgam5−/− mice (Fig. 6d). These results indicate that although RIPK1 does not directly regulate pro-IL-1β processing and secretion, it has a more prominent role than PGAM5 in the control of Leishmania replication in vivo.
Figure 6. RIPK1 kinase activity and PGAM5 are required for in vivo control of Leishmania replication.

Wild type, Ripk1kd/kd or Pgam5−/− mice were infected with Leishmania amazonensis in the footpad. (a) Histology sections of infected footpad at 5 weeks post-infection were examined. Representative images were shown and revealed similar extent of inflammation marked by neutrophilic infiltration in WT, Ripk1kd/kd and Pgam5−/− mice. The boxed areas were magnified to show intracellular parasites in the vacuoles. (b) Parasite load in the footpad was determined 10 weeks after infection. (c–d) Lesion size on the footpad was monitored weekly. Two independent experiments. **p<0.01, ***p<0.05.
Discussion
Leishmania parasite causes tegumentary (cutaneous, mucocutaneous, disseminated and diffuse clinical forms) and visceral pathologies in humans and other animals. Leishmaniasis is endemic in the tropics and sub-tropics and thus is a major public health concern in the affected regions (1). Despite its importance, the relationship between host cell death pathways and immune defense against Leishmania infection is poorly understood. Certain intracellular pathogens such as Plasmodium falciparum and Plasmodium vivax elicit the release of heme (9), which can cause oxidative damage, inflammation and necroptosis (11, 21).
We recently found that heme induces RIPK1 and RIPK3-dependent necroptosis in murine macrophages (11). Inflammatory cell death of macrophages could provide a key signal that stimulates innate immune response during Leishmania infection. Damage-associated molecular patterns (DAMPs) released from necrotic cells could further stimulate expression of cytokines such as IL-1β to exacerbate tissue inflammation. Given their reported roles in inflammation and necroptosis, we speculated that RIPK1 and PGAM5 might contribute to these disease pathologies in human leishmaniasis. Indeed, we found that human patients with visceral leishmaniasis exhibited elevated serum levels of heme. High doses of heme could induce necroptosis and inhibit Leishmania replication, as we found to be the case in human macrophages. Heme-induced cytotoxicity has also been observed in Mycobacterium tuberculosis infection (31). However, low doses of heme did not induce necroptosis or inhibit Leishmania replication. This is consistent with the fact that low dose of heme is required for optimal Leishmania growth (24). Free heme is neutralized by HO-1, an enzyme that promotes Leishmania replication (7). Thus, at high doses of heme, such as that found in visceral leishmaniasis, heme-induced necroptosis may prevent excessive parasite replication by overwhelming the neutralizing effect of HO-1. Unfortunately, the lack of mouse model that mimics human visceral leishmania prevented us from directly testing this hypothesis.
RIPK1 and PGAM5 also participate in the control of parasite replication in cutaneous leishmaniasis, a more mild form of the disease. In contrast to their reported roles in apoptosis and necroptosis (17, 32), RIPK1 and PGAM5 did not regulate Leishmania replication through cell death. Rather, we found that PGAM5 facilitates optimal IL-1β and NO production in response to Leishmania infection. IL-1β has been shown to cooperate with other inflammatory cytokines such as TNF and IFNγ to enhance NO production by macrophages (5, 33). The IL-1β-NO axis causes macrophage cell death and creates an antimicrobial milieu at the tissue level, both of which are key mechanisms that limit parasite replication (34, 35). Consistent with their reported roles in regulating NLRP3 inflammasome activation (29, 30, 36), IL-1β secretion was reduced in Leishmania-infected Ripk1kd/kd and Pgam5−/− mice during the early phase of the infection. However, we found that Pgam5−/−, but not Ripk1kd/kd splenocytes from infected mice exhibited defective IL-1β secretion in response to Leishmania particular antigen. The distinct effects of RIPK1 and PGAM5 in pro-IL-1β processing are also observed in BMDMs (29). The critical role for PGAM5 in IL-1β secretion is consistent with its reported function in inflammasome polymerization (29). However, despite reduced IL-1β secretion and increased parasite load, footpad swelling was not significantly increased in Leishmania-infected Pgam5−/− mice. This is in contrast to Ripk1kd/kd mice, which showed significant increase in footpad swelling compared to wild type mice. The relatively mild phenotype of Pgam5−/− mice compared to mice lacking core inflammasome components is consistent with the fact that PGAM5 deficiency reduced, but did not abolish IL-β secretion.
Although IL-1β secretion in the footpad was reduced, IL-1β secretion by splenocytes from Leishmania-infected Ripk1kd/kd mice was normal. The mechanism by which RIPK1 kinase activity promotes IL-1β expression in vivo is unknown at present. Although the kinase activity of RIPK1 is known to promote cell death by apoptosis and necroptosis (12, 37), infected Ripk1kd/kd BMDMs did not exhibit significant difference in cell death when compared to wild type BMDMs. Besides promoting cell death, RIPK1 is a known activator of NF-κB and MAP kinase (38–42). In this regard, it is noteworthy that L. amazonensis infection in macrophages activates the p50/p50 homodimer, a NF-κB transcriptional repressor. Inhibition of NF-κB-dependent gene transcription by p50/p50 homodimer has been attributed to cause downregulation of iNOS expression in L. amazonensis infection (43, 44). It is therefore tempting to speculate that RIPK1 may promote canonical NF-κB activation to counteract this inhibitory signal from p50/p50.
The discovery that RIPK1 and PGAM5 control Leishmania replication suggests that they may have wider roles in host defense and resistance against other intracellular pathogens. In this light, Roca and Ramakrishnan have recently shown that macrophages inhibit Mycobacterium tuberculosis growth, and that this phenomenon is dependent on RIPK1- and RIPK3-mediated reactive oxygen species (ROS) production (45). Thus, RIPK1 and PGAM5 may represent preserved innate immune mechanisms against intracellular pathogens. Identifying the pathogen recognition receptors that stimulate these pathways could lead to better therapeutics against these diseases.
Acknowledgments
This work is supported by NIH grant AI119030 (F.K.M. Chan) and Fundação de Amparo à Pesquisa do Estado da Bahia-FAPESB (V. M. B.). N.F.L. receives a post-doc fellowship from Conselho Nacional de Desenvolvimento Científico e Tecnológico-CNPq. V.M.B., R.P.A., R.G. and M.T.B. are senior investigators at CNPq.
Literature cited
- 1.Alvar J, Velez ID, Bern C, Herrero M, Desjeux P, Cano J, Jannin J, den Boer M WHOLC Team. Leishmaniasis worldwide and global estimates of its incidence. Plos One. 2012;7:e35671. doi: 10.1371/journal.pone.0035671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sampaio MJ, Cavalcanti NV, Alves JG, Filho MJ, Correia JB. Risk factors for death in children with visceral leishmaniasis. PLoS neglected tropical diseases. 2010;4:e877. doi: 10.1371/journal.pntd.0000877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chappuis F, Sundar S, Hailu A, Ghalib H, Rijal S, Peeling RW, Alvar J, Boelaert M. Visceral leishmaniasis: what are the needs for diagnosis, treatment and control? Nature reviews Microbiology. 2007;5:873–882. doi: 10.1038/nrmicro1748. [DOI] [PubMed] [Google Scholar]
- 4.Zamboni DS, Lima-Junior DS. Inflammasomes in host response to protozoan parasites. Immunological reviews. 2015;265:156–171. doi: 10.1111/imr.12291. [DOI] [PubMed] [Google Scholar]
- 5.Lima-Junior DS, Costa DL, Carregaro V, Cunha LD, Silva AL, Mineo TW, Gutierrez FR, Bellio M, Bortoluci KR, Flavell RA, Bozza MT, Silva JS, Zamboni DS. Inflammasome-derived IL-1beta production induces nitric oxide-mediated resistance to Leishmania. Nature medicine. 2013;19:909–915. doi: 10.1038/nm.3221. [DOI] [PubMed] [Google Scholar]
- 6.Gurung P, Karki R, Vogel P, Watanabe M, Bix M, Lamkanfi M, Kanneganti TD. An NLRP3 inflammasome-triggered Th2-biased adaptive immune response promotes leishmaniasis. The Journal of clinical investigation. 2015;125:1329–1338. doi: 10.1172/JCI79526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luz NF, Andrade BB, Feijo DF, Araujo-Santos T, Carvalho GQ, Andrade D, Abanades DR, Melo EV, Silva AM, Brodskyn CI, Barral-Netto M, Barral A, Soares RP, Almeida RP, Bozza MT, Borges VM. Heme oxygenase-1 promotes the persistence of Leishmania chagasi infection. J Immunol. 2012;188:4460–4467. doi: 10.4049/jimmunol.1103072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sigala PA, Goldberg DE. The peculiarities and paradoxes of Plasmodium heme metabolism. Annual review of microbiology. 2014;68:259–278. doi: 10.1146/annurev-micro-091313-103537. [DOI] [PubMed] [Google Scholar]
- 9.Andrade BB, Araujo-Santos T, Luz NF, Khouri R, Bozza MT, Camargo LM, Barral A, Borges VM, Barral-Netto M. Heme impairs prostaglandin E2 and TGF-beta production by human mononuclear cells via Cu/Zn superoxide dismutase: insight into the pathogenesis of severe malaria. J Immunol. 2010;185:1196–1204. doi: 10.4049/jimmunol.0904179. [DOI] [PubMed] [Google Scholar]
- 10.Saha S, Mondal S, Banerjee A, Ghose J, Bhowmick S, Ali N. Immune responses in kala-azar. The Indian journal of medical research. 2006;123:245–266. [PubMed] [Google Scholar]
- 11.Fortes GB, Alves LS, de Oliveira R, Dutra FF, Rodrigues D, Fernandez PL, Souto-Padron T, De Rosa MJ, Kelliher M, Golenbock D, Chan FK, Bozza MT. Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood. 2012;119:2368–2375. doi: 10.1182/blood-2011-08-375303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, Orenstein J, Moss B, Lenardo MJ. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem. 2003;278:51613–51621. doi: 10.1074/jbc.M305633200. [DOI] [PubMed] [Google Scholar]
- 13.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mocarski ES, Guo H, Kaiser WJ. Necroptosis: The Trojan horse in cell autonomous antiviral host defense. Virology. 2015;479–480:160–166. doi: 10.1016/j.virol.2015.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan FK, Luz NF, Moriwaki K. Programmed necrosis in the cross talk of cell death and inflammation. Annual review of immunology. 2015;33:79–106. doi: 10.1146/annurev-immunol-032414-112248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moriwaki K, Chan FK. RIP3: a molecular switch for necrosis and inflammation. Genes & development. 2013;27:1640–1649. doi: 10.1101/gad.223321.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148:228–243. doi: 10.1016/j.cell.2011.11.030. [DOI] [PubMed] [Google Scholar]
- 18.Kang YJ, Bang BR, Han KH, Hong L, Shim EJ, Ma J, Lerner RA, Otsuka M. Regulation of NKT cell-mediated immune responses to tumours and liver inflammation by mitochondrial PGAM5-Drp1 signalling. Nature communications. 2015;6:8371. doi: 10.1038/ncomms9371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berger SB, Kasparcova V, Hoffman S, Swift B, Dare L, Schaeffer M, Capriotti C, Cook M, Finger J, Hughes-Earle A, Harris PA, Kaiser WJ, Mocarski ES, Bertin J, Gough PJ. Cutting Edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J Immunol. 2014;192:5476–5480. doi: 10.4049/jimmunol.1400499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Challa S, Woelfel M, Guildford M, Moquin D, Chan FK. Viral cell death inhibitor MC159 enhances innate immunity against vaccinia virus infection. Journal of virology. 2010;84:10467–10476. doi: 10.1128/JVI.00983-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dutra FF, Bozza MT. Heme on innate immunity and inflammation. Frontiers in pharmacology. 2014;5:115. doi: 10.3389/fphar.2014.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Graca-Souza AV, Silva-Neto MA, Oliveira PL. Urate synthesis in the blood-sucking insect rhodnius prolixus. Stimulation by hemin is mediated by protein kinase C. J Biol Chem. 1999;274:9673–9676. doi: 10.1074/jbc.274.14.9673. [DOI] [PubMed] [Google Scholar]
- 23.Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annual review of pharmacology and toxicology. 2010;50:323–354. doi: 10.1146/annurev.pharmtox.010909.105600. [DOI] [PubMed] [Google Scholar]
- 24.Huynh C, Yuan X, Miguel DC, Renberg RL, Protchenko O, Philpott CC, Hamza I, Andrews NW. Heme uptake by Leishmania amazonensis is mediated by the transmembrane protein LHR1. PLoS Pathog. 2012;8:e1002795. doi: 10.1371/journal.ppat.1002795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, Lich JD, Finger J, Kasparcova V, Votta B, Ouellette M, King BW, Wisnoski D, Lakdawala AS, DeMartino MP, Casillas LN, Haile PA, Sehon CA, Marquis RW, Upton J, Daley-Bauer LP, Roback L, Ramia N, Dovey CM, Carette JE, Chan FK, Bertin J, Gough PJ, Mocarski ES, Kaiser WJ. RIP3 Induces Apoptosis Independent of Pronecrotic Kinase Activity. Mol Cell. 2014;56:481–495. doi: 10.1016/j.molcel.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, Wang X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 28.Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, Young SN, Varghese LN, Tannahill GM, Hatchell EC, Majewski IJ, Okamoto T, Dobson RC, Hilton DJ, Babon JJ, Nicola NA, Strasser A, Silke J, Alexander WS. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39:443–453. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 29.Moriwaki K, Farias Luz N, Balaji S, De Rosa MJ, O’Donnell CL, Gough PJ, Bertin J, Welsh RM, Chan FK. The Mitochondrial Phosphatase PGAM5 Is Dispensable for Necroptosis but Promotes Inflammasome Activation in Macrophages. J Immunol. 2015 doi: 10.4049/jimmunol.1501662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity. 2013;38:27–40. doi: 10.1016/j.immuni.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 31.Silva-Gomes S, Appelberg R, Larsen R, Soares MP, Gomes MS. Heme catabolism by heme oxygenase-1 confers host resistance to Mycobacterium infection. Infect Immun. 2013;81:2536–2545. doi: 10.1128/IAI.00251-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu W, Jing L, Wang Q, Lin CC, Chen X, Diao J, Liu Y, Sun X. Bax-PGAM5L-Drp1 complex is required for intrinsic apoptosis execution. Oncotarget. 2015;6:30017–30034. doi: 10.18632/oncotarget.5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liew FY, Li Y, Millott S. Tumor necrosis factor-alpha synergizes with IFN-gamma in mediating killing of Leishmania major through the induction of nitric oxide. J Immunol. 1990;145:4306–4310. [PubMed] [Google Scholar]
- 34.Olekhnovitch R, Ryffel B, Muller AJ, Bousso P. Collective nitric oxide production provides tissue-wide immunity during Leishmania infection. The Journal of clinical investigation. 2014;124:1711–1722. doi: 10.1172/JCI72058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mauel J, Ransijn A, Buchmuller-Rouiller Y. Killing of Leishmania parasites in activated murine macrophages is based on an L-arginine-dependent process that produces nitrogen derivatives. Journal of leukocyte biology. 1991;49:73–82. doi: 10.1002/jlb.49.1.73. [DOI] [PubMed] [Google Scholar]
- 36.Kang S, Fernandes-Alnemri T, Rogers C, Mayes L, Wang Y, Dillon C, Roback L, Kaiser W, Oberst A, Sagara J, Fitzgerald KA, Green DR, Zhang J, Mocarski ES, Alnemri ES. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nature communications. 2015;6:7515. doi: 10.1038/ncomms8515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133:693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 38.Dannappel M, Vlantis K, Kumari S, Polykratis A, Kim C, Wachsmuth L, Eftychi C, Lin J, Corona T, Hermance N, Zelic M, Kirsch P, Basic M, Bleich A, Kelliher M, Pasparakis M. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature. 2014;513:90–94. doi: 10.1038/nature13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takahashi N, Vereecke L, Bertrand MJ, Duprez L, Berger SB, Divert T, Goncalves A, Sze M, Gilbert B, Kourula S, Goossens V, Lefebvre S, Gunther C, Becker C, Bertin J, Gough PJ, Declercq W, van Loo G, Vandenabeele P. RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature. 2014;513:95–99. doi: 10.1038/nature13706. [DOI] [PubMed] [Google Scholar]
- 40.Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, Komuves L, Ferrando RE, French DM, Webster J, Roose-Girma M, Warming S, Dixit VM. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343:1357–1360. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- 41.Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van TM, Lee TH, Chan FK, Pasparakis M, Kelliher MA. Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol. 2014;193:1539–1543. doi: 10.4049/jimmunol.1400590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee TH, Shank J, Cusson N, Kelliher MA. The kinase activity of Rip1 is not required for tumor necrosis factor-alpha-induced IkappaB kinase or p38 MAP kinase activation or for the ubiquitination of Rip1 by Traf2. J Biol Chem. 2004;279:33185–33191. doi: 10.1074/jbc.M404206200. [DOI] [PubMed] [Google Scholar]
- 43.Calegari-Silva TC, Pereira RM, De-Melo LD, Saraiva EM, Soares DC, Bellio M, Lopes UG. NF-kappaB-mediated repression of iNOS expression in Leishmania amazonensis macrophage infection. Immunology letters. 2009;127:19–26. doi: 10.1016/j.imlet.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 44.Calegari-Silva TC, Vivarini AC, Miqueline M, Dos Santos GR, Teixeira KL, Saliba AM, Nunes de Carvalho S, de Carvalho L, Lopes UG. The human parasite Leishmania amazonensis downregulates iNOS expression via NF-kappaB p50/p50 homodimer: role of the PI3K/Akt pathway. Open biology. 2015;5:150118. doi: 10.1098/rsob.150118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roca FJ, Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell. 2013;153:521–534. doi: 10.1016/j.cell.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]