

Abstract
Summary: Data-dependent acquisition (DDA) is the most common method used to control the acquisition process of shotgun proteomics experiments. While novel DDA approaches have been proposed, their evaluation is made difficult by the need of programmatic control of a mass spectrometer. An alternative is in silico analysis, for which suitable software has been unavailable. To meet this need, we have developed MSAcquisitionSimulator—a collection of C ++ programs for simulating ground truth LC-MS data and the subsequent application of custom DDA algorithms. It provides an opportunity for researchers to test, refine and evaluate novel DDA algorithms prior to implementation on a mass spectrometer.
Availability and implementation: The software is freely available from its Github repository http://www.github.com/DennisGoldfarb/MSAcquisitionSimulator/ which contains further documentation and usage instructions.
Contact: weiwang@cs.ucla.edu or ben_major@med.unc.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Mass spectrometry is an analytical technique used in proteomics to identify and quantify proteins. Many of its applications share the underlying goal of uncovering the full protein complement in a biological sample. However, for complex samples this is rarely achieved partly because the number of ion populations exceeds that which contemporary instruments can individually target for sequence analysis with a tandem mass spectrum scan (MS/MS) (Michalski et al., 2011). Acquisition algorithms are necessary to control the data acquisition process and manage the limited scan speed.
Data-dependent acquisition (DDA) constitutes a major class of data acquisition methods. DDA methods perform a precursor scan to determine the mass-to-charge ratio (m/z) and abundance of ions currently entering the mass spectrometer, followed by sequence determining MS/MS scans on ions from a subset of detected peaks. The standard DDA algorithm, TopN, selects ions for MS/MS scans that contributed to peaks of greatest signal intensity from the latest precursor scan. Several other approaches, as well as adjustments to TopN, have been proposed in order to increase peptide and protein identifications (Graumann et al., 2012; Liu et al., 2012; Rudomin et al., 2009; Scherl et al., 2004; Zerck et al., 2013). However, TopN continues to be the dominant choice for acquisition control despite its bias toward abundant proteins and relatively poor reproducibility.
Currently, evaluation of novel acquisition strategies requires access to both a mass spectrometer and its application programming interface (API). Unfortunately, few instrument vendors provide an API, and therefore the pool of researchers with the necessary tools to explore this field is extremely limited. An alternative is to evaluate methods with in silico simulations. Existing simulator software for mass spectrometry proteomics has focused on generating ground truth data and realistic signals for precursor and MS/MS scans (Bielow et al., 2011; Noyce et al., 2013; Schulz-Trieglaff et al., 2008; Smith et al., 2015). However, the size of the simulations is limited due to speed, memory and/or disk space requirements. Most importantly, the simulated MS/MS spectra lead to nearly perfect peptide-spectrum-matches (PSMs) when analyzed with existing database search algorithms. The reasons for this include the absence of co-fragmentation from neighboring ions within an isolation window, the difficulty of predicting fragmentation patterns, and potentially other not yet understood phenomena. This limitation makes evaluating any acquisition strategy impractical as the metrics for success are based on the number of confident peptide and protein identifications.
Here, we present an acquisition simulator that produces PSMs with realistic peptide sequence and probability assignments for DDA strategies by foregoing fragmentation simulation and instead directly generating PSMs based on the precursor ion fractions of MS/MS scans. It builds upon previous work on LC-MS simulations and scales to larger datasets due to probabilistic models of ion generation and subsequent pruning of rare ions. This allows for an increased number of proteins, peptides and post-translational modifications that can be simulated.
2 Methods
MSAcquisitionSimulator consists of three standalone command-line programs. The first, FASTASampler, assists users in creating a FASTA file containing the proteins to be included in the simulation. Users select a protein FASTA file, the distribution of protein abundance and the number of proteins to be sampled. The output contains a random subset of proteins with each header appended with a ‘#’ followed by the protein’s abundance.
Next, GroundTruthSimulator uses the previously created FASTA file and a configuration file containing simulation parameters to simulate digestion, residue modifications, retention time, chromatographic elution profiles, ionization efficiency and charge and isotope distributions. It outputs the tab delimited ground truth data on the generated ions, which will be used for testing acquisition algorithms. In contrast to previous simulators, there are no limits on missed cleavages, enzymatic termini or dynamic modifications. A probabilistic approach is taken instead and rare peptides below a user-defined threshold are efficiently pruned. Details on the probability calculations are in the Supplementary methods.
Finally, the AcquisitionSimulator program takes as input the previously generated ground truth file and another configuration file. This program simulates a user-defined DDA algorithm on the ground truth data. It models ion accumulation, precursor scan spectra, scan time durations and database search PSMs (Supplementary Fig. S1). Currently, MS/MS fragmentation spectra are not simulated. Instead, the PSMs are generated by sampling from the list of precursors isolated in an MS/MS scan and candidate peptides from a database search. This approach leads to true positives, false positives and reverse decoy matches similar to real experiments (Supplementary Figs. S2–S4 ). Ion accumulation is simulated by numerically integrating an ion’s elution profile. Ion transmission rates are not modeled and are assumed to be 100%. The elapsed time for a scan is equal to the scan overhead time plus the larger of either the injection time or the transient time. This models the scan time for a QExactive-like instrument. The output includes an mzML file and a PSM graph file, which is used as input for the Fido protein inference algorithm (Serang et al., 2010). Speed and memory usage comparisons with existing simulation software are provided in Supplementary Figure S5.
3 Case study
To demonstrate the utility of MSAcquisitionSimulator, we evaluated three simple DDA algorithms—TopN, RandomN and WeightedN. TopN selects the most abundant precursor peaks for MS/MS scans, RandomN samples from a uniform random distribution of the observed peaks and WeightedN samples from a random distribution weighted by observed peak intensity. Dynamic exclusion was enabled, and precursor scans were de-isotoped prior to MS/MS selection decisions. FASTASampler was executed on the human proteome provided by UniProtKB using a lognormal abundance distribution and 50% of the proteins, resulting in 45 809 protein sequences. Default configuration files were used with both GroundTruthSimulator and AcquisitionSimulator. TopN resulted in the greatest number of confident protein and peptide identifications, closely followed by WeightedN, and RandomN provided far fewer protein identifications (Fig. 1). RandomN’s poor performance stemmed from the challenges in targeting low abundant ions. The wide MS/MS isolation window of 2 m/z captured neighboring abundant ions and created spectra dominated by peptides whose monoisotopic mass fell outside the small precursor mass tolerance used to simulate the database search. In addition, fewer scans were performed due to increased injection time.
Fig. 1.
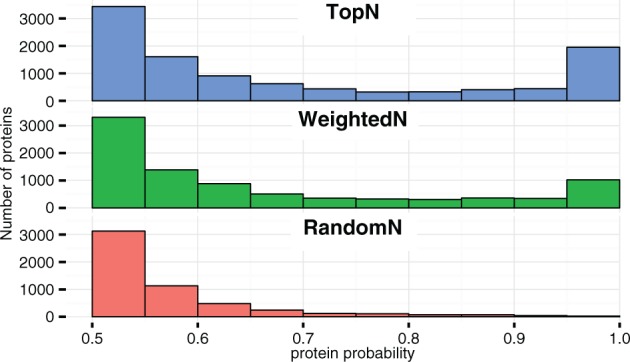
Distribution of protein probabilities using various DDA algorithms on simulated data. Fido was used for protein inference with parameters alpha = 0.1, beta = 0.01 and gamma = 0.5
4 Conclusion
DDA simulation will assist in the development and assessment of novel methods. The next generation of algorithms will likely further integrate data generation with data analysis, such as real-time peptide sequencing and protein inference. They may also become more goal-oriented, seeking to identify subsets of proteins, specific modifications or to improve quantification. Their sophistication may also come at a computational cost too great for their implementation on contemporary mass spectrometers. For such a scenario, MSAcquisitionSimulator will be a great tool for their evaluation.
Supplementary Material
Acknowledgements
We would like to thank Leonard McMillan for useful discussions and members of the Major Lab for proofreading the manuscript.
Funding
This work has been supported by the National Cancer Institute [Grant number R21-CA178760-01].
Conflict of Interest: none declared.
References
- Bielow C. et al. (2011) MSSimulator: simulation of mass spectrometry data. J. Proteome Res., 10, 2922–2929. [DOI] [PubMed] [Google Scholar]
- Graumann J. et al. (2012) A framework for intelligent data acquisition and real-time database searching for shotgun proteomics. Mol. Cell Proteomics, 11, M111.013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H. et al. (2012) Automated iterative MS/MS acquisition: a tool for improving efficiency of protein identification using a LC-MALDI MS workflow. Anal. Chem., 83, 6286–6293. [DOI] [PubMed] [Google Scholar]
- Michalski A. et al. (2011) More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC-MS/MS. J. Proteome Res., 10, 1785–1793. [DOI] [PubMed] [Google Scholar]
- Noyce A.B. et al. (2013) Mspire-Simulator: LC-MS shotgun proteomic simulator for creating realistic gold standard data. J. Proteome Res., 12, 5742–5749. [DOI] [PubMed] [Google Scholar]
- Rudomin E.L. et al. (2009) Directed sample interrogation utilizing an accurate mass exclusion-based data-dependent acquisition strategy (AMEx). J. Proteome Res., 8, 3154–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherl A. et al. (2004) Nonredundant mass spectrometry: a strategy to integrate mass spectrometry acquisition and analysis. Proteomics, 4, 917–927. [DOI] [PubMed] [Google Scholar]
- Schulz-Trieglaff O. et al. (2008) LC-MSsim–a simulation software for liquid chromatography mass spectrometry data. BMC Bioinformatics, 9, 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serang O. et al. (2010) Efficient marginalization to compute protein posterior probabilities from shotgun mass spectrometry data. J. Proteome Res., 9, 5345–5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R. et al. (2015) JAMSS: proteomics mass spectrometry simulation in Java. Bioinformatics, 31, 791–793. [DOI] [PubMed] [Google Scholar]
- Zerck A. et al. (2013) Optimal precursor ion selection for LC-MALDI MS/MS. BMC Bioinformatics, 14, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.