

Abstract
We have generated a strain of mice lacking two DNA N-glycosylases of base excision repair (BER), NTH1 and NEIL1, homologs of bacterial Nth (endonuclease three) and Nei (endonuclease eight). Although these enzymes remove several oxidized bases from DNA, they do not remove the well-known carcinogenic oxidation product of guanine: 7,8-dihydro-8-oxoguanine (8-OH-Gua), which is removed by another DNA N-glycosylase, OGG1. The Nth1−/−Neil1−/− mice developed pulmonary and hepatocellular tumors in much higher incidence than either of the single knockouts, Nth1−/− and Neil1−/−. The pulmonary tumors contained, exclusively, activating GGT→GAT transitions in codon 12 of K-ras of their DNA. Such transitions contrast sharply with the activating GGT→GTT transversions in codon 12 of K-ras of the pathologically similar pulmonary tumors, which arose in mice lacking OGG1 and a second DNA N-glycosylase, MUTY. To characterize the biochemical phenotype of the knockout mice, the content of oxidative DNA base damage was analyzed from three tissues isolated from control, single and double knockout mice. The content of 8-OH-Gua was indistinguishable among all genotypes. In contrast, the content of 4,6-diamino-5-formamidopyrimidine (FapyAde) and 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyGua) derived from adenine and guanine, respectively, were increased in some but not all tissues of Neil1−/− and Neil1−/−Nth1−/− mice. The high incidence of tumors in our Nth1−/−Neil1−/− mice together with the nature of the activating mutation in the K-ras gene of their pulmonary tumors, reveal for the first time, the existence of mutagenic and carcinogenic oxidative damage to DNA which is not 8-OH-Gua.
Keywords: Mouse, Cancer, Base excision repair, NTH1, NEIL1, K-ras
1. Introduction
The removal of promutagenic and/or cytotoxic oxidized bases from genomic and mitochondrial DNA is primarily carried out by the DNA N-glycosylases of base excision repair (BER). The relationship of BER of oxidized bases to human cancer was made evident through characterization of a subset of patients who displayed the phenotype of adenomatous polyposis of the colon (APC) but did not generate affected offspring. These individuals proved not to have a defect in either germline APC tumor suppressor gene, but instead contained inactivating GC→TA transversions in both copies of the somatic APC genes of the colonic cancers [1].
Such inactivating mutations resulted from the persistence of a mispair, between the well-known product of oxidation of guanine, i.e., 7,8-dihydro-8-oxoguanine (8-OH-Gua) and adenine, which arose during DNA replication. In bacteria, such a mispair is corrected by the DNA N-glycosylase, MutY and, in eukaryotic organisms by its homolog, MUTY [2,3]. MUTY-mediated excision of the mismatched adenine initiates BER, which inserts a canonical cytosine opposite the 8-OH-Gua. Once paired with cytosine, 8-OH-Gua becomes a substrate for excision by the DNA N-glycosylase OGG1 [4–6]. The excision of 8-OH-Gua is followed by BER-mediated insertion of a normal guanine residue opposite the cytosine, thereby restoring the integrity of the oxidatively-damaged DNA. The human subpopulation, which displayed the APC phenotype, was shown to lack MUTYH (the human homolog of MUTY) activity in their tissues due to compound biallelic inactivation of their germline MUTYH genes [1,7–10].
The elucidation of the pathogenesis of this syndrome established a definitive link between endogenously-derived oxidative damage to the bases of DNA, failure to repair the consequences of such damage via BER and the development of human cancer, in this case colonic.
Mice with targeted deletion of MutY did not develop tumors. However, when coupled with targeted deletion of Ogg1, mice displayed a phenotype of increased spontaneous tumorigenesis particularly lung and lymphoma [11]. Furthermore, codon 12 of the K-ras gene of the lung tumors in MutY−/−Ogg1−/− mice contained an activating GGT→GAT transversion characteristic of MUTY deficiency. A more recent study reported that MutY−/− mice did indeed display an increase in spontaneous intestinal tumorigenesis [12]. Sakamoto et al. attributed the difference in their results from those of the earlier study [11] to the fact that their mice were allowed to age, affording the intestinal tumors more time to appear. Additional evidence pointing to oxidative damage as tumorigenic in MutY−/− mice was a further increase in the incidence of intestinal tumorigenesis when an oxidizing agent was added to their drinking water [12].
There are other oxidation products of purines and pyrimidines, which are also subject to repair via BER but by enzymes other than MUTY and OGG1. The carcinogenicity of such modified bases has not yet been demonstrated. In bacteria and eukaryotic organisms, cytotoxic and, when derived from oxidation of 5-methylcytosine [13], potentially mutagenic 5,6-dihydro-5,6-dihydroxythymine (thymine glycol) and mutagenic 5-hydroxycytosine (5-OH-Cyt) and the product of its deamination, 5-hydroxyuracil (5-OH-Ura) are removed by Escherichia coli endonuclease III (Nth) and its mammalian homologs (NTH1, NTHL1) [9,14]. The NTH enzymes are members of the Helix–Hairpin–Helix DNA N-glycosylase superfamily to which MUTY and OGG1 belong [9].
Removal of oxidized pyrimidines from DNA is also effected in E. coli by endonuclease VIII (Nei) and in eukaryotic species by its homologs, the endonuclease VIII-like (NEIL) enzymes NEIL1 and NEIL2 [14–17]. The NEIL enzymes are members of the FPG/MutM superfamily, which is characterized by a zinc finger motif, although the NEIL enzymes contain no zinc [9]. NEIL1 primarily catalyzes the release of the imidazole ring-open purine derivatives 4,6-diamino-5-formamidopyrimidine (FapyAde) and 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyGua) from the DNA backbone [14–17]. More recently, the hydantoins, guanidinohydantoin (Gh) and the two diastereomers of spiroiminodihydantoin (Sp1 and Sp2) derived from further oxidation of 8-OH-Gua, were reported to be efficiently excised in vitro by NEIL1 and to a more limited extent by NEIL2 [18,19].
To study the in vivo effects of NTH1 deficiency, we developed mice harboring a targeted deletion of Nth1. The mice displayed a normal phenotype at 12–13 months age [20]. Subsequently, Neil1−/− mice were created in the laboratory of Dr. R. Stephen Lloyd and displayed a phenotype in some but not all males, of obesity and fatty liver, which was attributed to unrepaired oxidative damage to mitochondrial DNA [21]. Dr. Lloyd sent several homozygous and heterozygous mice to NYU to breed a double knockout. Surprisingly, none of the Neil1−/− colony bred at NYU has displayed an obese phenotype after several years of continuous breeding. The variable phenotype of the Neil1−/− mice was described and commented upon in the paper initially describing the Neil1−/− mice [21] and phenotypic variability continues to be observed. However, the biological basis of phenotype variability in Neil1−/− mice is not currently known and is still under active investigation in Dr. Lloyd's laboratory.
Here we report that Nth1−/−Neil1−/− mice are viable and fertile and displayed no abnormal phenotype after 1 year of life. However, during the 2nd year, Nth1−/−Neil1−/− mice displayed a high incidence of pulmonary and hepatic tumors in comparison to Nth1−/− or Neil1−/− mice. Since Nth1−/−Neil1−/− mice contain normal Ogg1 genes, the increased tumorigenesis displayed by Nth1−/−Neil1−/− mice is the first demonstration of the carcinogenicity of oxidative damage to DNA other than 8-OH-Gua. In this report, we also present molecular analysis supporting our conclusion about the nature of the carcinogenic oxidative damage and emphasize that these results underscore the importance of BER in protecting individuals from the development of cancer due to endogenous oxidative stress.
2. Materials and methods
2.1. Generation of Nth1−/−Neil1−/− mice
Nth1−/− mice of mixed 129 SvJ/C57/BL6 background [20] were bred with Neil1−/− mice of C57/BL6 background [21]. Resulting Nth1+/−Neil+/− offspring were crossbred and litters from Nth1+/−Neil+/− pairings were genotyped to identify Nth1−/−Neil1−/− mice. All animals were housed under conditions in accordance to protocols of the Institutional Animal Care and Use Committee (IACUC).
2.2. Genotyping
PCR-based analysis was used to genotype mice. Cellular DNA was obtained from tail biopsies at the time of weaning as previously described [20,21].
2.3. Histopathological analysis
Necropsies were performed primarily when mice showed signs of stress, such as roughening of coat hair, weight loss, and inactivity. Upon exhibiting such signs, mice were euthanized following IACUC regulations. Tissues were washed in cold 1× PBS pH 7.6, trimmed, fixed in 10% formalin, washed in 50% ethanol for 16 h and stored in 70% ethanol prior to processing for histopathology. Sections were H&E stained. The diagnoses of liver tumors were made independently by three pathologists. Statistical analysis was carried out using the Fisher's Exact test.
2.4. Analysis of K-ras mutation in lung tumors
Cellular DNA was isolated from tumors and normal tissues that had been stored at −80 °C using Proteinase K digestion followed by phenol–chloroform extraction. Cellular DNA was also purified from small tumors and normal lung from paraffin-embedded formalin-fixed samples, which were processed onto PEN-membrane glass slides (Leica) and isolated using a Leica LMD6000 laser micro-dissecting microscope (Leica Microsystems Inc., Allendale, NJ). DNA from micro-dissected samples was purified via QIamp DNA Extraction Kit (Qiagen).
Primer pairs were designed to amplify codons 12 and 13 in Exon 2 of K-ras (forward primer F1, 5′-TCACTGAATTCGGAATATCTTAGAG-3′; reverse primer R1, 5′-GCACGCAGACTGTAGAGCAG-3′) and codon 61 in Exon 3 of K-ras (forward primer F3, 5′-CCAGACTGTGTTTCTCCCTTC-3′; reverse primer R3, 5′-GCTATCATTACTTCACATGCCAAC-3′) via PCR. The high GC base content in the 250 base pair region of Exon 2 for codons 12 and 13 necessitated the addition of dimethyl sulfoxide (DMSO) to a final concentration of 5% in the PCR reactions in order to amplify. Thermal cycler conditions for all reactions were as follows: (i) 94 °C for 2 min followed by 35 cycles of (ii) 94 °C for 1 min, 55 °C for 45 s, 72 °C for 1 min and (iii) final extension of 72 °C for 7 min. After an initial screening, reactions were expanded 4-fold to increase the amount of product for DNA sequencing. PCR products were purified via QiaQuick PCR Purification Kit (Qiagen) and sent for sequencing to Genewiz Inc. (South Plainfield, NJ) using primers F1 (for codons 12 and 13) and F3 (for codon 61) as sequencing primers. All trace files were analyzed for heterozygous mutations using the Staden program (Cambridge, UK). Samples that contained mutations were confirmed by reverse complementary sequencing using primer R1 for codons 12 and 13 or primer R3 for codon 61 as the sequencing primer.
2.5. Tissue DNA extraction
Liver, kidney and brain were harvested from wild type, Nth1−/−, Neil1−/− and Nth1−/−Neil1−/− male and female mice from two age groups, 9–22 months old representing aged mice and 3–4 months old representing young mice. We were unable to obtain the DNA of young Neil1−/− mice because age-matched young Neil1−/− mice were not available at the time of DNA extraction. The Neil1−/− mice stopped breeding at an earlier age than did the Nth1−/− and Nth1−/−Neil1−/− mice, a phenomenon that was described in the initial characterization of Neil1−/− mice, but which we do not yet fully understand [21].
A literature search to find an optimized DNA purification procedure for the large amounts of DNA needed for this analysis led us to employ the protocol of Hofer et al. [22], which was modified by the addition of 10mM diethylentriaminepentaacetic acid (DTPA) pH 7.5 as a second iron chelator together with deferoxamine mesylate (DFOM) from the original protocol, to minimize the oxidation of DNA which occurs during extraction and purification [23].
The DNA extracted by this method contained 1/10 the background oxidative base damage of what had been previously reported [15], thereby, greatly increasing the sensitivity of detection of endogenous oxidative damage, which had occurred in vivo. RNAase A was added to digest RNA that co-precipitated with DNA. The DNA was re-precipitated with 200mL of 3M sodium acetate and 10mL ethanol at −20 °C. Samples were centrifuged at 12,000×g for 10min at 4 °C to pellet DNA in a Sorvall RC5B centrifuge using a SS-34 rotor. The DNA pellets were washed, stored in 2mL 70% ethanol and sent on dry ice to the lab of Dr. Miral Dizdaroglu of the National Institute of Standards and Technology for analysis of content of oxidized bases.
2.6. Analysis by gas chromatography/mass spectrometry
Isolated DNA samples were dissolved in water for 24 h at 4 °C. The UV spectrum of each DNA sample was recorded by absorption spectrophotometry between 200 nm and 350 nm to ascertain DNA quality and concentration. The absorbance at 260nm was used to measure the DNA concentration of each sample (absorbance of 1=50μg of double stranded DNA/mL). Aliquots (50μg) of DNA samples were dried in a SpeedVac under vacuum. Gas chromatography/mass spectrometry (GC/MS) with isotope-dilution was used to identify and quantify FapyAde, FapyGua and 8-OH-Gua in DNA samples. The DNA samples were incubated with E. coli Fpg protein, a DNA N-glycosylase, which efficiently excises FapyAde, FapyGua and 8-OH-Gua as free bases from DNA [24]. This methodology prevents any artifactual formation of oxidized bases from normal DNA bases during sample preparation because no intact adenine or guanine is released from DNA by Fpg [25]. Fpg protein was purified as described [26]. Stable isotope-labeled analogues of FapyAde and FapyGua, i.e., FapyAde-13C,15N2 and FapyGua-13C,15N2, respectively, were purchased from Cambridge Isotope Laboratories (Cambridge, MA). The stable isotope-labeled analogue of 8-OH-Gua, i.e., 8-OH-Gua-13C,15N3 was obtained as described [27]. Aliquots (50μg) of DNA were supplemented with aliquots of the internal standards FapyGua-13C,15N2, FapyAde-13C,15N2 and 8-OH-Gua-13C,15N3, and then dried in a SpeedVac under vacuum. DNA samples were dissolved in 50μL of buffer (50mM Na2HPO4 (pH 7.4), 100mM KCl, 1mM EDTA and 0.1mM dithiothreitol), and hydrolyzed with 2μg of Fpg protein at 37 °C for 1h [26]. After incubation, 150μL of cold ethanol (−20 °C) were added. The samples were kept at −20 °C for 30min and then centrifuged at 15,000×g at 4 °C for 30 min. Supernatants and DNA pellets were separated. Supernatant fractions were freed from ethanol in a SpeedVac under vacuum, frozen in liquid nitrogen and lyophilized for 18 h. Dried samples were trimethylsilylated and analyzed by GC/MS as described [26]. For identification and quantification, selected-ion monitoring was used to monitor the characteristic ions of the trimethylsilyl derivatives of FapyAde, FapyGua and 8-OH-Gua, and their stable isotope-labeled analogues [28].
Under identical conditions, we used E. coli Nth instead of Fpg to release 5-OH-5-MeHyd, thymine glycol, 5-OH-Cyt and 5-OH-Ura from DNA. E. coli Nth is specific for removal of these lesions from DNA [14,29,30]. After hydrolysis, DNA samples were analyzed by GC/MS as described above. 13C,15N2-labeled analogues of 5-OH-5-MeHyd, thymine glycol, 5-OH-Cyt and 5-OH-Ura (purchased from Cambridge Isotope Laboratories) were used as internal standards.
2.7. Statistical analysis of spectroscopy results
Statistical analyses were performed using SPSS for Microsoft Windows 11.0 statistical program (SPSS, Chicago, IL). The statistical analysis of the significance between groups of modified DNA bases was carried out using the nonparametric Kruskal–Wallis one-way ANOVA by ranks and Mann–Whitney U-tests. A p value less than or equal to 0.05 was considered to be statistically significant.
3. Results
3.1. Generation of Nth1−/−Neil1−/− mice
The generation of Nth1−/−Neil1−/− mice was accomplished by cross-breeding Nth1−/− mice [20] of mixed 129SvJ/C57BL/6J background, with Neil1−/− mice of C57BL/6J [21] background. The resulting Nth1+/−Neil+/− offspring were intercrossed and litters from Nth1+/−Neil+/− pairings were genotyped to identify Nth1−/−Neil1−/− mice by performing two PCR reactions on the same genomic DNA sample. The first set of PCR reactions screened for targeted deletion of NTH1 using previously described conditions [20], while the second set of reactions screened for targeted deletion of NEIL1 under conditions described in Ref. [21]. Fig. 1 is a representative gel displaying a variety of genetic patterns. In particular, lane (a) displays a Nth1−/−Neil1−/− mouse, lane (b) is a Nth1+/+Neil1+/+ mouse and lane (c) is a Nth1+/−Neil1+/− mouse. The distribution of genotypes was entirely consistent with Mendelian genetics. To confirm that there was no expression from these genes, qRT-PCR analyses of the mRNA from liver, kidney, brain and thymus revealed no expression of: Nth1 in Nth1−/− mice, Neil1 in Neil1−/− mice and Nth1 or Neil1 in the double knockout (data not shown).
Fig. 1.
Electrophoretic analysis of PCR reactions to determine genotype. The upper panel screened for the targeted deletion of Nth1 while the lower panel screened for the targeted deletion of Neil1 (a) represents Nth1−/−Neil1−/− (b) represents Nth1+/+Neil1+/+and (c) represents Nth1+/−Neil1+/−.
The Nth1−/−Neil1−/− mice developed normally, displaying normal weight gain and began breeding at 3 months of age. Litter sizes were comparable to those of single knockout mice and wild type mice. Newborn mice were mothered normally. The obese phenotype reported in [21] was never observed in Neil1−/− or Nth1−/−Neil1−/− mice bred at NYU.
3.2. Histopathological analysis
During their 2nd year of life many Nth1−/−Neil1−/− mice displayed signs of stress such as roughening of their coat hair, weight loss, and inactivity. Such mice were promptly euthanized and subjected to complete necropsy. The most common finding was the presence of multiple lung tumors, averaging 4–5 per mouse, which proved to be pulmonary adenomas and carcinomas as illustrated in Fig. 2. The incidence of pulmonary tumors in all mice is presented in Table 1, with 74.4% (32 out of 43) incidence in Nth1−/−Neil1−/− males and 41.4% (12 out of 29) in Nth1−/−Neil1−/− females. The incidence of lung tumors in Nth1−/− males and females, respectively, were 1.9% and 3.7%, while the incidence in Neil1−/− mice was 12% in males and no lung tumors in females.
Fig. 2.
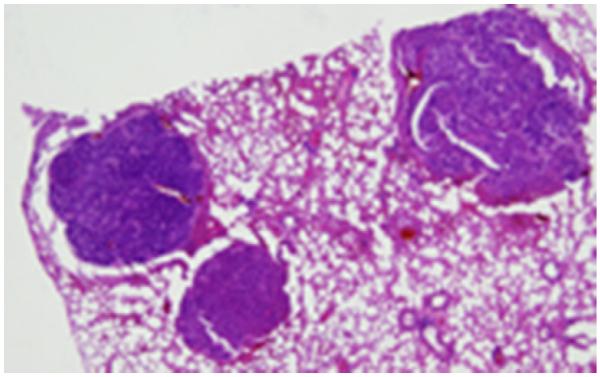
Histological sections of lung tumors in Nth1−/−Neil1−/− mice at a magnification of 4.5×. Three pulmonary adenocarcinomas are present, one extending into the bronchus.
Table 1.
Tumor incidence for Nth1−/−, Neil1−/−, and Nth1−/−Neil1−/− mice.
| Genotype | Males/females (total numbers) | Lung adenoma, adenocarcinoma (total numbers) | Liver hepatocellular carcinoma, nodular hyperplasia, severe dysplasia (total numbers) |
|---|---|---|---|
| Nth1 −/− | 52/54 | 1/2 | 8/7 |
| Neil1 −/− | 25/18 | 3/0 | 4/2 |
| Nth1 −/− Neil1 −/− | 43/29 | 32/12 | 20/5 |
Nth1−/− and Neil1−/− mice were compared to Nth1−/−Neil1−/− mice with regard to the proportion of mice with tumors using the Fisher's Exact test. A larger fraction of both Nth1−/−Neil1−/− males and females developed lung tumors (p < 0.001) than did either single knockout.
Liver tumors that were seen at necropsy proved to be hepatocellular carcinomas (Fig. 3A). Histologic examination was performed on all livers revealing nodular hyperplasia (Fig. 3B) and severe hepatocellular dysplasia (Fig. 3C). These diagnoses made by Dr. George Teebor, were confirmed independently by three pathologists. Nodular hyperplasia and severe hepatocellular dysplasia were grouped together with hepatocellular carcinomas for statistical purposes. The incidences of hepatocellular carcinomas together with the pre-malignant liver abnormalities are listed in Table 1. The incidence of liver tumors and pre-tumoral conditions in Nth1−/−Neil1−/− mice was, as with lung tumor formation, greater than either Nth1−/− or Neil1−/− mice with 46.5% (20 out of 43) in males and 17% (5 out of 29 females). Nth1−/− males had an incidence of 15.4% while females had 13%, and Neil1−/− males and females had incidences of 16% and 11.1%, respectively.
Fig. 3.

Histological sections of liver tumors and pre-malignant livers of Nth1−/−, Neil1−/− and Nth1−/−Neil1−/− mice at a magnification of 4.5×. (A) A section of a hepatocellular carcinoma. (B) Nodular hyperplasia of hepatocytes. (C) A field of severe hepatocellular dysplasia, characterized by hyperchromatic and irregularly shaped hepatocyte nuclei.
The Fisher's Exact test was applied to the liver lesions and a greater proportion of Nth1−/−Neil1−/− males developed liver abnormalities (p < 0.005) than the males from either of the single knockouts. There was no significant difference between the females of all three genotypes.
3.3. K-ras mutagenesis in lung tumors
Lung tumor formation in mice is frequently associated with activation of the K-ras oncogene, the activating hot spots being codons 12 and 13 in Exon 2 and codon 61 in Exon 3 [31,32]. Two hundred fifty base pair long regions containing codons 12 and 13 and codon 61, respectively, were amplified from cellular DNA that had been extracted from normal tissue and tumors and then sequenced to determine whether mutations had occurred in these activating hot spots. All normal tissues contained the correct wild type sequences of codons 12 and 13 (Fig. 4A), and codon 61, visualized as chromatographs with adenine fluorescing green, thymine red, guanine black, and cytosine blue. Of the 83 tumors from Nth1−/−Neil1−/− mice that were sequenced for K-ras mutations, 58 were laser capture specimens and 25 were fresh-frozen. Of the 58 laser capture samples, 54 (93%) contained a point mutation (Fig. 4B) in codon 12. Of the 25 frozen samples, 5 (20%) contained a point mutation (Fig. 4B) in codon 12. Regardless of the origin of the DNA be it from fresh-frozen, laser capture, male or female tumors, all of the point mutations in codon 12 were identical. The Staden program used to analyze the sequences will not identify a nucleotide base if a heterozygous mutation is detected. The mutation is observed as a `peak differential' (Fig. 4C) between the green adenine peak in codon 12 of the tumor sequence (Fig. 4B) compared to the black guanine peak of the wild type normal sequence (Fig. 4A). Further analysis revealed this heterozygous mutation to be a GGT→GAT transition.
Fig. 4.

Demonstration of K-ras mutagenesis in lung tumors via the Staden program for detection of heterozygous mutations. (A) DNA sequencing chromatographs of portions of amplified DNA from Exon 2 of K-ras of normal lung tissue including codons 12 and 13 containing the wild type sequence (GGT) and (GGC), respectively. Adenine fluoresces green, thymine red, guanine black and cytosine blue. (B) A sequencing chromatograph of a portion of amplified DNA extracted from a single lung tumor presenting the sequence of codon 12 as (G-T) and the sequence of codon 13 as (GGC). The Staden program does not identify a nucleotide base if there is a heterozygous mutation but instead presents it, in this case, as (G-T). (C) The trace peak differential determined by the Staden program, as the difference between the normal wild type sequence of codon 12 in (A) and the mutated sequence of codon 12 in (B) revealing it to be a GGT→GAT transition as revealed by the green adenine peak above the baseline (mutated) over the black guanine peak (wild type) below the baseline. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
No other mutations were observed in codon 13 or codon 61 in any of the specimens. The discrepancy between the incidence of mutations in fresh-frozen tissue and tissue obtained by laser capture suggests that despite our best effort to trim surrounding normal tissue, there was too much normal tissue to readily detect the heterozygous point mutation within the K-ras tumor gene. Laser capture allowed us to sample tumor within microscopically defined tumor borders thereby minimizing contamination by normal tissue. Similar findings have been reported with other genes [33,34].
K-ras was also sequenced in three lung tumors from Nth1−/− and Neil1−/− single knockout mice (Table 1) and the same GGT→GAT transition was detected.
3.4. Content of endogenous modified DNA bases
To relate the genotype of the mice to the content of oxidized bases in their cellular DNA as a function of the substrate specificity of NEIL1 and NTH1, samples of cellular DNA were extracted from liver, kidney and brain of young (average age 3 months) and old (average age 16 months) mice.
Lung DNA was not extracted because all the lung tumors arose, as is typical for such tumors, from broncho-alveolar epithelium, which represents a very small percentage of the cellular population of the lung. Thus, analysis of the content of oxidatively-modified bases of total lung DNA would not be representative of the content of modified bases in the DNA of the pre-malignant broncho-alveolar cells. This contrasts with liver where 70% of the cells are hepatocytes, the cells from which hepatocellular carcinomas arise; kidney, where the majority of cells are tubular and are the cells of origin for most renal cell carcinomas; brain where the majority of cells are glial and are the population from which most brain tumors arise.
Figs. 5 and 6 illustrate the content of oxidized purines, i.e., 8-OH-Gua, FapyAde and FapyGua in DNA extracted from tissues of old and young animals, respectively. The small differences in the content of 8-OH-Gua within tissues from each age group and genotype, were not statistically significant (p > 0.05), consistent with evidence that 8-OH-Gua is only removed from DNA by OGG1 [14–17].
Fig. 5.

Endogenous levels of modified bases in cellular DNA of old Nth1−/−, Neil1−/− and Nth1−/−Neil1−/− mice. The mean content of 8-OH-Gua, FapyAde, and FapyGua from DNA extracted from liver, kidney and brain from all three genotypes was plotted with their standard deviations. Measurements with an (*) represent values statistically significant (a p value greater than 0.05) compared to controls.
Fig. 6.

Endogenous levels of modified bases in cellular DNA of young Nth1−/−, and Nth1−/−Neil1−/− mice. The mean content of 8-OH-Gua and FapyAde, from DNA extracted from liver, kidney and brain from two genotypes was plotted with their standard deviations. Measurements with an (*) represent values statistically significant (a p value greater than 0.05) compared to controls.
In contrast, our results unequivocally show that the absence of NEIL1 resulted in the accumulation of FapyAde in cellular DNA. Old Neil1−/− mice (Fig. 5) demonstrated statistically significant accumulation of FapyAde and FapyGua in the DNA of liver (p < 0.02), kidney (p < 0.03) and brain (p < 0.02). Old Nth1−/−Neil1−/− mice also demonstrated significant accumulation of FapyAde in DNA of liver (p < 0.04), kidney (p < 0.03) and brain (p < 0.04). No increases were detectable in the DNA of most tissues from young animals. However, the liver DNA of 3–4-month-old Nth1−/− and Nth1−/−Neil1−/− mice contained significant increases in FapyAde content when compared to age-matched wild type mice. Significant accumulation of FapyGua was observed in livers of Neil1−/− and Nth1−/−Neil1−/− mice, and in the kidney of Neil1−/− mice. DNA samples from young Neil1−/− mice could not be analyzed for technical reasons. Notwithstanding that technical problem, the data of Figs. 5 and 6, demonstrate the accumulation of FapyAde with the increase of age in DNA of mice lacking NEIL1.
We attempted to measure the accumulation of 5-OH-5-MeHyd, thymine glycol, 5-OH-Cyt and 5-OH-Ura in DNA samples because these oxidized pyrimidines are substrates of Nth proteins including human NTH1 [14,29,30]. Fig. 7 demonstrates the content of 5-OH-5-MeHyd, 5-OH-Cyt and 5-OH-Ura in the liver of old mice. Despite the absence of both NEIL1 and NTH1, no significant accumulation of 5-OH-5-MeHyd, 5-OH-Cyt and 5-OH-Ura was observed in any of the samples analyzed. Thymine glycol content was below the detection level.
Fig. 7.

Endogenous levels of oxidized pyrimidines in cellular DNA of old Nth1−/−, Neil1−/− and Nth1−/−Neil1−/− mice. The mean content of 5-OH-5-MeHyd, 5-OH-Ura and 5-OH-Cyt from DNA extracted from liver from all three genotypes and wild type controls was plotted with their standard deviations. There was no statistically significant accumulation of any of the oxidized pyrimidines in any of the mice.
In summary, mice lacking both NTH1 and NEIL1 demonstrated a marked increase in pulmonary and hepatic tumorigenesis compared to mice lacking either NTH1 or NEIL1. Specifically, the lung tumors contained a point mutation in codon 12 of the K-ras oncogene which is a GGT→GAT transition, different from the transversions of codon 12 in K-ras of the pathologically similar lung tumors of mice lacking both MUTY and OGG1. The absence of NEIL1 and NTH1 resulted in the accumulation of the oxidized purine derivative, FapyAde, in all tissues measured. FapyGua was increased selectively. Most importantly, 8-OH-Gua was normal. Surprisingly, the absence of NEIL1 and NTH1 did not result in the accumulation of the oxidized pyrimidines; thymine glycol, 5-OH-5-MeHyd, 5-OH-Cyt and 5-OH-Ura.
4. Discussion
The results of this study reveal, for the first time, the existence of oxidatively-modified DNA base damage which, in the absence of NTH1 and NEIL1, is strongly carcinogenic and which is not the well-known oxidized guanine derivative 8-OH-Gua. There are several reasons for concluding that the carcinogenic damaged base(s) which accumulated in the DNA of Nth1−/−Neil1−/− mice is not 8-OH-Gua.
All knockout mice in this study contained the normal OGG1 and MUTY homologs and neither NTH1 nor NEIL1 release 8-OH-Gua or adenine to a significant degree from the DNA backbone [14–17]. Therefore, the mice retained the normal capacity to excise 8-OH-Gua when opposite cytosine and an adenine residue mispaired with 8-OH-Gua. Consistent with this conclusion was our demonstration of a normal content of 8-OH-Gua in the cellular DNA of all three genotypes. In those strains of mice lacking OGG1 activity as a consequence of targeted deletion of Ogg1, the content of 8-OH-Gua in their cellular DNA was consistently significantly elevated [12,38,39]. The accuracy of our measurement demonstrating a normal content of 8-OH-Gua in the cellular DNA of mice lacking NTH1, NEIL1 or both enzymes is supported by our simultaneous demonstration of a markedly increased content of FapyAde (Fig. 5), and to a somewhat lesser extent, of FapyGua (Fig. 5), of cellular DNA isolated from the tissues of mice lacking NEIL1. NEIL1 has been shown to be one of the primary enzymes for the removal of Fapy residues [16].
In contrast to the global accumulation of FapyAde, the content of FapyGua only increased in the liver and kidney of the old Neil1−/− mice and in the liver of the old Neil1−/−Nth1−/− mice (Fig. 5). The differences in the steady-state accumulation of FapyAde versus FapyGua can be explained by data demonstrating that FapyGua, but not FapyAde is a substrate for OGG1 [14,35,36]. Tissue-specific differences in the expression of OGG1 could account for the differential accumulation of FapyGua in the tissues of our mice [37].
Additional evidence supporting our contention that the carcinogenic damaged base is not 8-OH-Gua are the results of amplification and sequencing of the K-ras genes of the lung tumors in Nth1−/−Neil1−/− mice where like the lung tumors of mice lacking OGG1 and MUTY, most of the tumors harbored activating heterozygous mutations in codon 12. However, in contrast to the GGT→GTT transversions of codon 12 in K-ras in the lung tumors of mice lacking OGG1 and MUTY [11], which result from persistence of the mismatched adenine residue as a consequence of the absence of MUTY activity [1,11], all of the K-ras mutations in the lung tumors of Nth1−/−Neil1−/− as well as in single knockout mice proved to be GGT→GAT transitions.
Thus, for all of the preceding reasons we are confident in concluding that the high incidence of tumors in the Nth1−/−Neil1−/− mice is due to a form of endogenous oxidative damage to DNA, which is carcinogenic if not repaired by NTH1 and/or NEIL1, and which is definitely not 8-OH-Gua.
What might the carcinogenic modified bases(s) be? If we assume that the mutation in codon 12 of the K-ras gene in the lung tumors of our mice is the direct result of oxidative modification of base(s) in that codon in somatic DNA, then the promutagenic modified bases must be derived from guanine and/or cytosine since the mutation is a GC→AT transition.
It has been stated that the most common mutagenic outcome of oxidative damage to DNA is a C→T transition and that the most likely candidate for the promutagenic lesion was a deamination product of oxidized cytosine such as 5-OH-Cyt or its deaminated product 5-OH-Ura [40]. 5-OH-Cyt and 5-OH-Ura are substrates of E. coli Nth, yeast Nth and human NTH1 [14]. In addition, it was recently suggested that such cytosine-derived lesions are also removed by NEIL1 and NEIL2 [41]. Surprisingly, our measurement of the content of such oxidized bases in cellular DNA of all the knockouts did not reveal significant elevation of any of them (Fig. 7). This finding suggests that in vivo, oxidized cytosine and oxidized uracil are primarily repaired by NEIL2, which like OGG1 and MYH, is normal in all our strains. Equally surprising was the absence of accumulation of 5-OH-5-MeHyd and of thymine glycol content, which was so low as to be undetectable, both of which are repaired in vitro by NEIL1 and NTH1. This suggests, at least for thymine glycol, that nucleotide excision repair may have compensated for BER in its steady-state removal [42].
What of the possibility that the lesion is guanine-derived? Of those guanine-derived lesions measured, 8-OH-Gua content was normal and FapyGua was elevated in some tissues, but not all, consistent with the fact that FapyGua is removed by NEIL1 and is a good substrate for OGG1 [14,35,36]. However, its mutagenic properties are similar to those of 8-OH-Gua mispairing with adenine yielding transversions rather than transitions. Other guanine-derived lesions include the hydantoins (Sp1, Sp2 and Gh), which have also been shown to be substrates for NEIL1 [18,19] but whose mutagenic properties also favor transversions rather than transitions [9,19].
Most importantly, the profound increase in tumorigenicity in the double knockout compared to the single knockouts suggests we are dealing with a synergistic phenomenon, the nature of which we do not yet understand. The abnormally elevated FapyAde and FapyGua were essentially equally elevated in both Neil1−/− and Neil1−/−Nth1−/− mice, so that accumulation in and of itself, does not explain tumorigenicity.
Thus, there are at least two possibilities with respect to the nature of the novel oxidatively-modified base. The first is that the in vitro properties of one of the aforementioned oxidized bases, both its ability to be repaired and its mutagenicity, are not fully reflective of its in vivo properties. Alternatively, we may have uncovered the existence of a heretofore-uncharacterized oxidatively-modified base, whose repair is mediated by both NTH1 and NEIL1 and whose mutagenic properties lead to GC→AT transitions. It is also possible that NEIL1 and NTH1 have a relationship to modified DNA similar to OGG1 and MUTY in which one enzyme corrects a modified base and the other corrects a resulting mispair. In the absence of both enzymes the mutagenic and tumorigenic consequences of the oxidized base are greatly amplified. Once the promutagenic base has been identified, the possibility that one of the enzymes has a mismatch repair function can be verified or excluded by in vitro assay.
There is one additional clue to the nature of the lesion(s). Although both the Ogg1−/−MutY−/− mice of previous study [11] and our Nth1−/−Neil1−/− mice developed pulmonary tumors only Nth1−/−Neil1−/− mice developed liver tumors. This suggests that the lesion(s) are also sufficiently cytotoxic to induce hepatocytic death and stimulate cell division thereby permitting fixation of carcinogenic mutations. Ogg1−/− mice only developed liver tumors after treatment with peroxisome proliferators, which induced cell division. After such treatment the incidence of hepatocellular carcinomas in the Ogg1−/− mice far exceeded that of control mice treated with the same peroxisome proliferators [43].
The incidence of both lung and liver tumors was considerably greater in males than in females. For lung tumors, this gender difference has been observed in several strains but there is no mechanistic explanation currently available [44]. In liver however, the gender disparity is consistent with those of previous experiments using the chemical carcinogen ENU in which the incidence of liver tumors was far greater in males than in females [45]. This gender discrepancy was explained by the action of the hepatic cytokine IL-6, which promotes liver regeneration and whose content in Kupfer cells is regulated by testosterone.
In summary, we have shown that targeted deletion of both Nth1 and Neil1 results in a marked increase in pulmonary and hepatic tumorigenesis in mice, which repair 8-OH-Gua normally. Therefore, we have demonstrated, in the absence of repair via BER, the carcinogenic effects of an as yet uncharacterized probably oxidatively modified base(s). Our data indicates that endogenous oxidative stress causes a broader spectrum of mutagenic base modification than has heretofore been suspected and emphasizes the importance of BER in protecting the organism against the carcinogenicity of such modification(s).
Acknowledgements
We thank Dr. Frederick F. Becker, Dr. Milton J. Finegold and Dr. Timothy P. Hilbert for reviewing liver tumors and other liver lesions; Dr. Krystyna Frenkel for her assistance in modifying DNA extraction procedures to decrease exogenous damage to the bases of DNA. Certain commercial equipment or materials are identified in this paper in order to specify adequately the experimental procedure. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the materials or equipment identified are necessarily the best available for the purpose.
Footnotes
Grant support: NIH DK075974 (RSL).
Conflict of interest statement The authors declare that there are no conflicts of interest.
References
- [1].Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT, Hodges AK, Davies DR, David SS, Sampson JR, Cheadle JP. Inherited variants of MYH associated with somatic G:C→T:A mutations in colorectal tumors. Nat. Genet. 2002;30:227–232. doi: 10.1038/ng828. [DOI] [PubMed] [Google Scholar]
- [2].Michaels ML, Cruz C, Grollman AP, Miller JH. Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proc. Natl. Acad. Sci. U.S.A. 1992;89:7022–7025. doi: 10.1073/pnas.89.15.7022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tajiri T, Maki H, Sekiguchi M. Functional cooperation of MutT, MutM and MutY proteins in preventing mutations caused by spontaneous oxidation of guanine nucleotide in Escherichia coli. Mutat. Res. 1995;336:257–267. doi: 10.1016/0921-8777(94)00062-b. [DOI] [PubMed] [Google Scholar]
- [4].Aburatani H, Hippo Y, Ishida T, Takashima R, Matsuba C, Kodama T, Takao M, Yasui A, Yamamoto K, Asano M. Cloning and characterization of mammalian 8-hydroxyguanine-specific DNA glycosylase/apurinic, apyrimidinic lyase, a functional MutM homologue. Cancer Res. 1997;57:2151–2156. [PubMed] [Google Scholar]
- [5].Bjorâs M, Luna L, Johnsen B, Hoff E, Haug T, Rognes T, Seeberg E. Opposite base-dependent reactions of a human base excision repair enzyme on DNA containing 7,8-dihydro-8-oxoguanine and abasic sites. EMBO J. 1997;16:6314–6322. doi: 10.1093/emboj/16.20.6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shinmura K, Kasai H, Sasaki A, Sugimura H, Yokota J. 8-hydroxyguanine (7, 8-dihydro-8-oxoguanine) DNA glycosylase and AP lyase activities of hOGG1 protein and their substrate specificity. Mutat. Res. 1997;385:75–82. doi: 10.1016/s0921-8777(97)00041-4. [DOI] [PubMed] [Google Scholar]
- [7].Chow E, Thirlwell C, Macrae F, Lipton L. Colorectal cancer and inherited mutations in base-excision repair. Lancet Oncol. 2004;5:600–606. doi: 10.1016/S1470-2045(04)01595-5. [DOI] [PubMed] [Google Scholar]
- [8].Sampson JR, Jones S, Dolwani S, Cheadle JP. MutYH (MYH) and colorectal cancer. Biochem Soc. Trans. 2005;33:679–683. doi: 10.1042/BST0330679. [DOI] [PubMed] [Google Scholar]
- [9].David SS, O'Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kastrinos F, Syngal S. Recently identified colon cancer predispositions: MYH and MSH6 mutations. Semin. Oncol. 2007;34:418–424. doi: 10.1053/j.seminoncol.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Xie Y, Yang H, Cunanan C, Okamoto K, Shibata D, Pan J, Barnes DE, Lindahl T, McIlhatton M, Fishel R, Miller JH. Deficiencies in mouse Myh and Ogg1 result in tumor predisposition and G to T mutations in codon 12 of the K-Ras oncogene in lung tumors. Cancer Res. 2004;64:3096–3102. doi: 10.1158/0008-5472.can-03-3834. [DOI] [PubMed] [Google Scholar]
- [12].Sakamoto K, Tominaga Y, Yamauchi K, Nakatsu Y, Sakumi K, Yoshiyama K, Egashira A, Kura S, Yao T, Tsuneyoshi M, Maki H, Nakabeppu Y, Tsuzuki T. MUTYH-null mice are susceptible to spontaneous and oxidative stress induced intestinal tumorigenesis. Cancer Res. 2007;67:6599–6604. doi: 10.1158/0008-5472.CAN-06-4802. [DOI] [PubMed] [Google Scholar]
- [13].Zuo S, Boorstein RJ, Teebor GW. Oxidative damage to 5-methylcytosine in DNA. Nucleic Acids Res. 1995;23:3239–3243. doi: 10.1093/nar/23.16.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dizdaroglu M. Base-excision repair of oxidative DNA damage by DNA glycosylases. Mutat. Res. 2005;591:45–59. doi: 10.1016/j.mrfmmm.2005.01.033. [DOI] [PubMed] [Google Scholar]
- [15].Hu J, de Souza-Pinto NC, Haraguchi K, Hogue BA, Jaruga P, Greenberg MM, Dizdaroglu M, Bohr VA. Repair of formamidopyrimidines in DNA involves different glycosylases: role of the OGG1, NTH1, and NEIL1 enzymes. J. Biol. Chem. 2005;280:40544–40551. doi: 10.1074/jbc.M508772200. [DOI] [PubMed] [Google Scholar]
- [16].Jaruga P, Birinciogluc M, Rosenquist TA, Dizdaroglu M. Mouse NEIL1 protein is specific for excision of 2,6-diamino-4-hydroxy-5-formamidopyrimidine and 4, 6-diamino-5-formamidopyrimidine from oxidatively damaged DNA. Biochemistry. 2004;43:15909–15914. doi: 10.1021/bi048162l. [DOI] [PubMed] [Google Scholar]
- [17].Hazra TK, Izumi T, Boldough I, Imhoff B, Kow YW, Jaruga P, Dizdaroglu M, Mitra S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad. Sci. U.S.A. 2002;99:3523–3528. doi: 10.1073/pnas.062053799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hailer MK, Slade PG, Martin BD, Rosenquist TA, Sugden KD. Recognition of the oxidized lesions spiroiminodihydantoin and guanidinohydantoin in DNA by the mammalian base excision repair glycosylases NEIL1 and NEIL2. DNA Repair (Amst) 2005;4:41–50. doi: 10.1016/j.dnarep.2004.07.006. [DOI] [PubMed] [Google Scholar]
- [19].Krishnamurthy N, Zhao X, Burrows CJ, David SS. Superior removal of hydantoin lesions relative to other oxidized bases by the human DNA glycosylase hNEIL1. Biochemistry. 2008;47:7137–7146. doi: 10.1021/bi800160s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ocampo MT, Chaung W, Marenstein DR, Chan MK, Altamirano A, Basu AK, Boorstein RJ, Cunningham RP, Teebor GW. Targeted deletion of mNth1 reveals a novel DNA repair enzyme activity. Mol. Cell Biol. 2002;22:6111–6121. doi: 10.1128/MCB.22.17.6111-6121.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Vartanian V, Lowell B, Minko IG, Wood TG, Ceci JD, George S, Ballinger SW, Corless CL, McCullough AK, Lloyd RS. The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase. Proc. Natl. Acad. Sci. U.S.A. 2006;103:1864–1869. doi: 10.1073/pnas.0507444103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hofer T, Seo AY, Prudencio M, Leeuwenburgh C. A method to determine RNA and DNA oxidation simultaneously by HPLC-ECD: greater RNA than DNA oxidation in rat liver after doxorubicin administration. Biol. Chem. 2006;387:103–111. doi: 10.1515/BC.2006.014. [DOI] [PubMed] [Google Scholar]
- [23].Cadet J, Douki T, Gasparutto D, Ravanat JL. Oxidative damage to DNA: formation, measurement and biochemical features. Mutat. Res. 2003;531:5–23. doi: 10.1016/j.mrfmmm.2003.09.001. [DOI] [PubMed] [Google Scholar]
- [24].Boiteux S, Gajewski E, Laval J, Dizdaroglu M. Substrate specificity of the Escherichia coli Fpg protein (formamidopyrimidine-DNA glycosylase): excision of purine lesions in DNA produced by ionizing radiation or photosensitization. Biochemistry. 1992;31:106–110. doi: 10.1021/bi00116a016. [DOI] [PubMed] [Google Scholar]
- [25].Jaruga P, Kirkali G, Dizdaroglu M. Measurement of formamidopyrimidines in DNA. Free Radic. Biol. Med. 2008;45:1601–1609. doi: 10.1016/j.freeradbiomed.2008.09.019. [DOI] [PubMed] [Google Scholar]
- [26].Reddy P, Jaruga P, O'Connor T, Rodriguez H, Dizdaroglu M. Overexpression and rapid purification of Escherichia coli formamidopyrimidine-DNA glycosylase. Protein Expr. Purif. 2004;34:126–133. doi: 10.1016/j.pep.2003.11.019. [DOI] [PubMed] [Google Scholar]
- [27].Nelson RA, Boyd SJ, Ziegelstein RC, Herning R, Cadet JL, Henningfield JE, Schuster CR, Contoreggi C, Gorelick DA. Effect of rate of administration on subjective and physiological effects of intravenous cocaine in humans. Drug Alcohol Depend. 2006;82:19–24. doi: 10.1016/j.drugalcdep.2005.08.004. [DOI] [PubMed] [Google Scholar]
- [28].Dizdaroglu M. Quantitative determination of oxidative base damage in DNA by stable isotope-dilution mass spectrometry. FEBS Lett. 1993;315:1–6. doi: 10.1016/0014-5793(93)81120-o. [DOI] [PubMed] [Google Scholar]
- [29].Dizdaroglu M, Bauche C, Rodriguez H, Laval J. Novel substrates of Escherichia coli nth protein and its kinetics for excision of modified bases from DNA damaged by free radicals. Biochemistry. 2000;39:5586–5592. doi: 10.1021/bi9927787. [DOI] [PubMed] [Google Scholar]
- [30].Gasparutto D, Muller E, Boiteux S, Cadet J. Excision of the oxidatively formed 5-hydroxyhydantoin and 5-hydroxy-5-methylhydantoin pyrimidine lesions by Escherichia coli and Saccharomyces cerevisiae DNA N-glycosylases. Biochim. Biophys. Acta. 2009;1790:16–24. doi: 10.1016/j.bbagen.2008.10.001. [DOI] [PubMed] [Google Scholar]
- [31].Meuwissen R, Berns A. Mouse models for human lung cancer. Genes Dev. 2005;19:643–664. doi: 10.1101/gad.1284505. [DOI] [PubMed] [Google Scholar]
- [32].Wakamatsu N, Devereux TR, Hong HH, Sills RC. Overview of the molecular carcinogenesis of mouse lung tumor models of human lung cancer. Toxicol. Pathol. 2007;35:75–80. doi: 10.1080/01926230601059993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Esposito G. Complementary techniques: laser capture microdissection—increasing specificity of gene expression profiling of cancer specimens. Adv. Exp. Med. Biol. 2007;593:54–65. doi: 10.1007/978-0-387-39978-2_6. [DOI] [PubMed] [Google Scholar]
- [34].Budimlija ZM, Lechpammer M, Popiolek D, Fogt F, Prinz M, Bieber FR. Forensic applications of laser capture microdissection: use in DNA-based parentage testing and platform validation. Croat. Med. J. 2005;46:549–555. [PubMed] [Google Scholar]
- [35].Krishnamurthy N, Haraguchi K, Greenberg MM, David SS. Efficient removal of formamidopyrimidines by 8-oxoguanine glycosylases. Biochemistry. 2008;47:1043–1050. doi: 10.1021/bi701919u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wiederholt CJ, Delaney MO, Pope MA, David SS, Greenberg MM. Repair of DNA containing Fapy.dG and its beta-C-nucleoside analogue by formamidopyrimidine DNA glycosylase and MutY. Biochemistry. 2003;42:9755–9760. doi: 10.1021/bi034844h. [DOI] [PubMed] [Google Scholar]
- [37].Karahalil B, Hogue BA, De Souza-Pinto NC, Bohr VA. Base excision repair capacity in mitochondria and nuclei: tissue-specific variations. FASEB J. 2002;16:1895–1902. doi: 10.1096/fj.02-0463com. [DOI] [PubMed] [Google Scholar]
- [38].Russo MT, De Luca G, Degan P, Parlanti E, Dogliotti E, Barnes DE, Lindahl T, Yang H, Miller JH, Bignami M. Accumulation of the oxidative base lesion 8-hydroxyguanine in DNA of tumor-prone mice defective in both the Myh and Ogg1 DNA glycosylases. Cancer Res. 2004;64:4411–4414. doi: 10.1158/0008-5472.CAN-04-0355. [DOI] [PubMed] [Google Scholar]
- [39].Parsons JL, Elder RH. DNA N-glycosylase deficient mice: a tale of redundancy. Cancer Res. 2003;531:165–175. doi: 10.1016/j.mrfmmm.2003.05.001. [DOI] [PubMed] [Google Scholar]
- [40].Kreutzer DA, Essigmann JM. Oxidized, deaminated cytosines are a source of C→T transitions in vivo. Proc. Natl. Acad. Sci. U.S.A. 1998;95:3578–3582. doi: 10.1073/pnas.95.7.3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hazra TK, Kow YW, Hatahet Z, Imhoff B, Boldogh I, Mokkapati SK, Mitra S, Izumi T. Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J. Biol. Chem. 2002;277:30417–30420. doi: 10.1074/jbc.C200355200. [DOI] [PubMed] [Google Scholar]
- [42].Reardon JT, Bessho T, Kung HC, Bolton PH, Sancar A. In vitro repair of oxidative DNA damage by human nucleotide excision repair system: possible explanation for neurodegeneration in xeroderma pigmentosum patients. Proc. Natl. Acad. Sci. U.S.A. 1997;94:9463–9468. doi: 10.1073/pnas.94.17.9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Trapp C, Schwarz M, Epe B. The peroxisome proliferators WY-14,643 promotes hepatocarcinogenesis caused by endogenously generated oxidative DNA base modifications in repair-deficient Csbm/m/Ogg1−/− mice. Cancer Res. 2007;67:5156–5161. doi: 10.1158/0008-5472.CAN-07-0335. [DOI] [PubMed] [Google Scholar]
- [44].Hahn FF, Gigliotti A, Hutt JA. Comparative oncology of lung tumors. Toxicol. Pathol. 2007;35:130–135. doi: 10.1080/01926230601132063. [DOI] [PubMed] [Google Scholar]
- [45].Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]