

Abstract
Long non-coding RNAs (lncRNAs) are potentially important mediators of genomic regulation. lncRNAs, however, remain poorly characterized in the rat model organism widely used in biomedical research. Using poly(A)-independent and strand-specific RNA-seq, we identified 1,500 to 1,800 lncRNAs expressed in each of the following tissues of Brown Norway rats: the renal cortex, renal outer medulla, liver, cardiac left ventricle, adrenal gland, and hypothalamus. Expression and the binding of histone H3K4me3 to promoter regions were confirmed for several lncRNAs. Rat lncRNA expression appeared to be more tissue-specific than mRNA. Rat lncRNAs had 4.5 times fewer exons and 29% shorter transcripts than mRNA. The median cumulative abundance of rat lncRNAs was 53% of that of mRNA. Approximately 28% of the lncRNAs identified in the renal outer medulla appeared to lack a poly(A) tail. Differential expression of 74 lncRNAs was detected in the renal outer medulla between Dahl SS rats, a model of salt-sensitive hypertension, and salt-insensitive, congenic SS.13BN26 rats fed a high-salt diet. Two of the differentially expressed lncRNAs, which were confirmed, were located within the congenic region and contained several sequence variants. The study identified genome-wide characteristics of lncRNAs in the rat model and suggested a role of lncRNAs in hypertension.
The relevance of a large group of non-protein-coding RNA, long non-coding RNA (lncRNA), to hypertension remains nearly completely unexplored. The significance of non-protein-coding RNA in general is being increasingly recognized in hypertension research1,2,3. Much of the work in this area has focused on microRNA, but not lncRNA. This is in spite of the rapid expansion of lncRNA research and the well-established functional importance for several lncRNAs such as the role of Xist in X chromosome inactivation4. An emerging model for the action of some, but not all, lncRNAs is that lncRNAs interact with ribonucleoproteins and position chromatin modification complexes to enable regulation of gene transcription at select locations of the genome5. An example of this is the human HOTAIR RNA, which serves as a scaffold that mediates the assembly of select histone modification complexes6. lncRNAs could also have functional impact in the cytoplasm7.
lncRNAs in the rat model organism are poorly characterized. In contrast, several thousand lncRNA transcripts have been identified and characterized in human, mouse, and several other organisms7. Rat is a favored model organism in several areas of biomedical research including hypertension research8. The lack of a systematic characterization of lncRNAs in the rat is a bottleneck for studying lncRNAs in hypertension.
In the present study, we characterized lncRNAs in several tissue regions and organs in the Brown Norway rat, the strain that provided the reference rat genome. We used poly(A)-independent and strand-specific RNA-seq since not all lncRNAs contain a poly(A) tail and some lncRNAs may be transcribed from the DNA strand opposite to the one encoding an mRNA. We then examined lncRNAs in the renal outer medulla, a tissue region important for long-term blood pressure control9, in a major genetic model of hypertension, the Dahl salt-sensitive SS rat10,11,12.
Methods
Animals and tissue collection
The animal experiments were performed in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animal protocols were approved by the Institutional Animal Care and Use Committee at the Medical College of Wisconsin (AUA206). Brown Norway rats were obtained from an in-house colony at weaning and fed the AIN-76A diet containing 0.4% NaCl since receipt. Four male rats at 6 weeks of age were used for collection of the kidneys, liver, free wall of cardiac left ventricle, adrenal glands, and brain. The brain tissue was embedded in Optimal Cutting Temperature compound and flash-frozen in 2-methylbutane with dry ice. The hypothalamus was identified and extracted as a 4 × 4 × 2 mm region between the optic chiasm, mammillary body, and lateral sulcus. The kidneys were further dissected to obtain the renal cortex and the renal outer medulla13.
Poly(A)-independent and strand-specific RNA-seq
Total RNA was extracted using TRIzol (Invitrogen) and RNA quality assessed using Agilent Bioanalyzer 210014. Only samples with an RNA integrity number greater than 8 were used. An equal amount of RNA was taken from each rat to make 2 pools of samples for each tissue, each pool representing 2 individual rats. rRNA was depleted from 1 μg of total RNA using Ribo-Zero (Illumina). cDNA libraries were prepared from the remaining RNA, without poly(A) selection, using the TruSeq Stranded RNA LT kit (Illumina) following the TruSeq stranded total RNA sample preparation guide provided by the vendor (RS-122-9007DOC, Ver. April 2013). The libraries underwent cluster generation using TruSeq PE Cluster Kit v3-cBot-HS and 100 cycles of paired-end sequencing using TruSeq SBS Kit v3-HS (Illumina) and an Illumina HiSeq 2000 sequencer as we described14,15.
Analysis of RNA-seq data
RNA-seq reads mapping and alignment, transcript construction, quantification of transcript abundance, and identification of differential expression were carried out using an in-house analytical pipeline that included Bowtie (http://bowtie-bio.sourceforge.net/index.shtml), Tophat v2.0.9 (http://tophat.cbcb.umd.edu/), and Cufflinks v2.1.1 (http://cufflinks.cbcb.umd.edu/), as we described previously14. Default parameters were used. Differential expression of transcripts was identified by Cuffdiff, implemented in Cufflinks. False discovery rate (FDR) < 0.05 was used to control for multiple comparisons.
Identification of lncRNAs
lncRNAs were identified using the workflow and criteria shown in Supplementary Figure S1. Briefly, transcripts containing single exon or <160 bp in length were removed following transcript reconstruction using Cufflinks. PhyloCSF16 was used to assess protein coding potentials of the remaining transcripts based on the alignment of the genomes of chimp, rhesus, mouse, guinea pig, cow, horse and dog. Transcripts were removed if their PhyloCSF scores were greater than 50, which would suggest a high possibility of encoding proteins. Furthermore, transcripts with complete branch lengths (CBL) >0 and open reading frames of >150 amino acids were removed since protein-coding open reading frames usually contain at least 100 amino acids. Transcripts with CBL scores of 0 due to poor sequence alignments were removed if they contained open reading frames of >50 amino acids. Finally, we used blastx with repeat masked to analyze the remaining transcripts and removed any transcripts with E-value less than 10−4. lncRNAs with FPKM of less than 0.1 were also removed. The identified transcripts were classified into different categories of lncRNAs according to the HUGO Gene Nomenclature Committee17. A previous set of RNA-seq data obtained from the renal outer medulla of SS rats and congenic SS.13BN26 rats14 was re-analyzed following the current procedure.
Real-time PCR analysis of lncRNA
Real-time PCR, as we described18,19, was used to quantify the abundance of specific lncRNAs. Primer sequences, designed using Lasergene (ver. 10.0.1, DNASTAR), are shown in Supplementary Table S1. Real-time PCR analysis was performed in total RNA samples from the four individual Brown Norway rats as well as individual rat samples from the previous study of SS rats and congenic SS.13BN26 rats14.
Histone H3K4me3 Chromatin Immunoprecipitation (ChIP) from rat tissues
ChIP was performed using a ChIP Tissue kit (#17-20000, Millipore) mostly following the vendor's protocol. Briefly, fresh rat tissue samples were minced on ice and crosslinked with 1% formadehyde for 10 min. Chromatins were sonicated for 300s using Covaris E210 following a sonication program that we optimized to generate fragments of 150–500 bp. Chromatin fragments were immunoprecipitated with anti-H3K4me3 antibody (#07-473, Millipore) or control IgG (#12-371B, Miiliore). The pullout DNA was purified using spin columns. Real-time PCR was performed to estimate the abundance of specific genomic segments, comparing the pullout DNA (output) to DNA samples used as the input for immunoprecipitation. Primer sequences are shown in Supplementary Table S1.
Statistics
Statistical analysis of RNA-seq data was conducted as described above. Real-time PCR data were analyzed by analysis of variance followed by least significant difference test. P<0.05 was considered significant. The data are shown in the figures as mean ± standard error.
Results
Identification of lncRNAs in Brown Norway rats
The number of RNA-seq reads, quality of the reads, and the mapping rate for each of the 12 libraries sequenced are summarized in Supplementary Table S2. Using the criteria shown in Supplementary Figure S1, 2,761 lncRNA transcripts were identified in at least one of the six tissue regions analyzed (Figure 1A; Supplementary Dataset). These lncRNA transcripts corresponded to 1,620 lncRNA gene loci. The number of detectable lncRNAs was 1,589 for the liver, 1,806 for the renal cortex, 1,640 for the cardiac left ventricle, 1,812 for the renal outer medulla, 1,748 for the adrenal glands, and 1,520 for the hypothalamus (Supplementary Dataset). The complete dataset was deposited into Gene Expression Omnibus (GSE60069).
Figure 1. Classification of the identified rat lncRNAs.

A. Classification of lncRNAs identified in at least one of the six tissues obtained from Brown Norway rats. lncRNAs were classified as antisense, intronic complete overlap (concs), intronic contained (incs), intronic partial overlap (poncs), and intergenic based on the spatial relationship between the genomic sequences encoding lncRNAs and protein-coding genes. B. Schematic illustration of the classification of lncRNA genes based on their spatial relationship with protein-coding genes. Black blocks and lines represent exons and introns, respectively, of protein-coding genes. Blocks and lines shown in color represent exons and introns, respectively, of lncRNA genes. Arrows indicate directions of transcription.
The vast majority of the identified lncRNAs, 2,630 out of 2,761 (95%), were novel transcripts (Figure 1A). The identified lncRNAs were classified as intergenic, intronic and antisense lncRNAs based on spatial relationships of their gene loci with protein-coding genes (Figure 1B). In general, antisense lncRNAs are transcripts that have either intronic or exonic overlaps with a protein coding gene on the opposite strand. lncRNAs that do not have any overlap with mRNA exons are either intergenic or intronic lncRNAs. It should be noted that lncRNAs described in the current study were transcripts and multiple lncRNAs may be located in the same gene loci. The identified lncRNAs were mostly intergenic lncRNAs (2,373 or 86%) and secondly antisense lncRNAs (223 or 8%) (Figure 1A, 1B).
A cutoff of mean + 2 × standard deviation, where mean and standard deviation were calculated from the FPKM values of an lncRNA in all tissues analyzed following quantile normalization, was used to identify lncRNAs enriched in a specific tissue. We found enrichment of 99 lncRNAs in the renal cortex, 69 in the renal outer medulla, 159 in the cardiac left ventricle, 224 in the liver, 152 in the adrenal gland, and 264 in the hypothalamus. Note that the smaller numbers of lncRNAs enriched in the renal cortex and the renal outer medulla were likely in part because the two tissues were similar. To verify the RNA-seq result, one lncRNA identified as enriched in each of the six tissues by RNA-seq was selected for real-time PCR analysis. The tissue enrichment was confirmed in nearly all cases (Figure 2A-2F).
Figure 2. Real-time PCR verification of rat lncRNA expression identified by RNA-seq.

Transcript abundance based on RNA-seq (left) and real-time PCR (right) is shown for an lncRNA identified by RNA-seq as enriched in each of the tissues. Li, liver; LV, cardiac left ventricle; C, renal cortex; M, renal outer medulla; H, hypothalamus; A, adrenal gland. n = 4 for real-time PCR. *, P<0.05 vs. the tissue in which the lncRNA was enriched based on RNA-seq.
Expression of rat lncRNAs was often correlated with active promoters
We examined the binding of histone H3K4me3 to the promoter regions of five lncRNA genes to further verify that the identified rat lncRNAs were likely encoded by transcriptionally active genes rather than detection artifacts. Binding of H3K4me3 around the transcriptional start sites is a characteristic of genes that are transcriptionally active or poised for activation20. As expected, immunoprecipitation using an H3K4me3 antibody, compared to a control antibody, resulted in substantial enrichment of the promoter region of the actively transcribed GAPDH gene in all five tissues analyzed, but no enrichment of an exon of the myoglobin gene which was used as a negative control (Figure 3A).
Figure 3. Higher transcript abundance for rat lncRNA genes was often associated with increased H3K4me3 binding to promoter regions.

A. Chromatin fragments immunoprecipitated from five rat tissues using an antibody for histone H3K4me3 were enriched for a GAPDH promoter region, but not a myoglobin exon region. Promoter regions for lncRNAs with higher transcript abundance in the liver (B), cardiac left ventricle (C), renal cortex (D), or renal outer medulla (E) were enriched in chromatin fragments immunoprecipitated from the respective tissue using an antibody for histone H3K4me3. The promoter enrichment was minimal for an lncRNA with higher transcript abundance in the hypothalamus (F). The lncRNAs for which promoter data are shown in the current figure correspond to those for which transcript abundance data are shown in Figure 2. Li, liver; LV, cardiac left ventricle; C, renal cortex; M, renal outer medulla; H, hypothalamus; A, adrenal gland.
The expression of each of the 5 lncRNAs we chose to analyze was enriched in one of the tissues examined (see Figure 2). The H3K4me3 binding to the promoter of the selected lncRNA gene was compared between the tissue in which it was enriched and one tissue where it was not enriched. The other tissues in which the lncRNA was not enriched were not analyzed because of the limited amount of immunoprecipitated DNA. For 4 of the 5 lncRNA genes examined, H3K4me3 binding was substantially increased in the tissue in which the lncRNA expression was enriched (Figure 3B to 3E). This was observed even when the expression abundance of an lncRNA was modest such as the selected lncRNAs enriched in the renal cortex and the renal outer medulla (see Figure 2). Increased H3K4me3 binding was not observed for the lncRNA enriched in the hypothalamus (Figure 3F). It is known that H3K4me3 binding occurs in many, but not all, transcriptionally active promoters21. These data support that the identified rat lncRNAs were likely real transcripts.
Expression of rat lncRNAs appeared more tissue-specific than mRNA
The expression profiles of lncRNAs were distinct between the six tissues (Figure 4A). The two biological replicates of each tissue were correctly clustered based on the lncRNA expression profiles (Figure 4A). The renal cortex and the renal outer medulla samples, both of which consist primarily of epithelial tissues, were clustered together as expected.
Figure 4. Expression of rat lncRNAs was tissue-specific.

A. A heat map of lncRNA expression in the six tissues. Each sample (i.e., each row in the heat map) was a pool of two rats. B. Proportions of lncRNA or mRNA transcripts found in one or more of the six tissues examined. Li, liver; LV, cardiac left ventricle; C, renal cortex; M, renal outer medulla; H, hypothalamus; A, adrenal gland.
Approximately 25% of the identified lncRNAs were expressed in all six tissues analyzed, compared to 45% of identified mRNAs (Figure 4B). The proportion of identified lncRNAs that were expressed in 4 or fewer of the 6 tissues analyzed was larger than that of mRNA. The numbers of lncRNAs expressed in one or more of the 6 tissues examined were summarized in Supplementary Figure S2. These data suggest the expression of rat lncRNAs might be more tissue-specific than mRNA.
Rat lncRNAs contained fewer exons and were less abundant than mRNA
Nearly all of the identified lncRNAs contained fewer than 10 exons, with a median of 2 exons (Figure 5A). In sharp contrast, mRNAs detected in these rat tissues contained a median of 9 exons (p = 0). Rat lncRNAs had a median length of 2,154 nucleotides, which was 29% shorter than the median length of rat mRNA (p = 4.8e-16) (Figure 5B).
Figure 5. Exon number, transcript length, and abundance of rat lncRNA compared to mRNA.
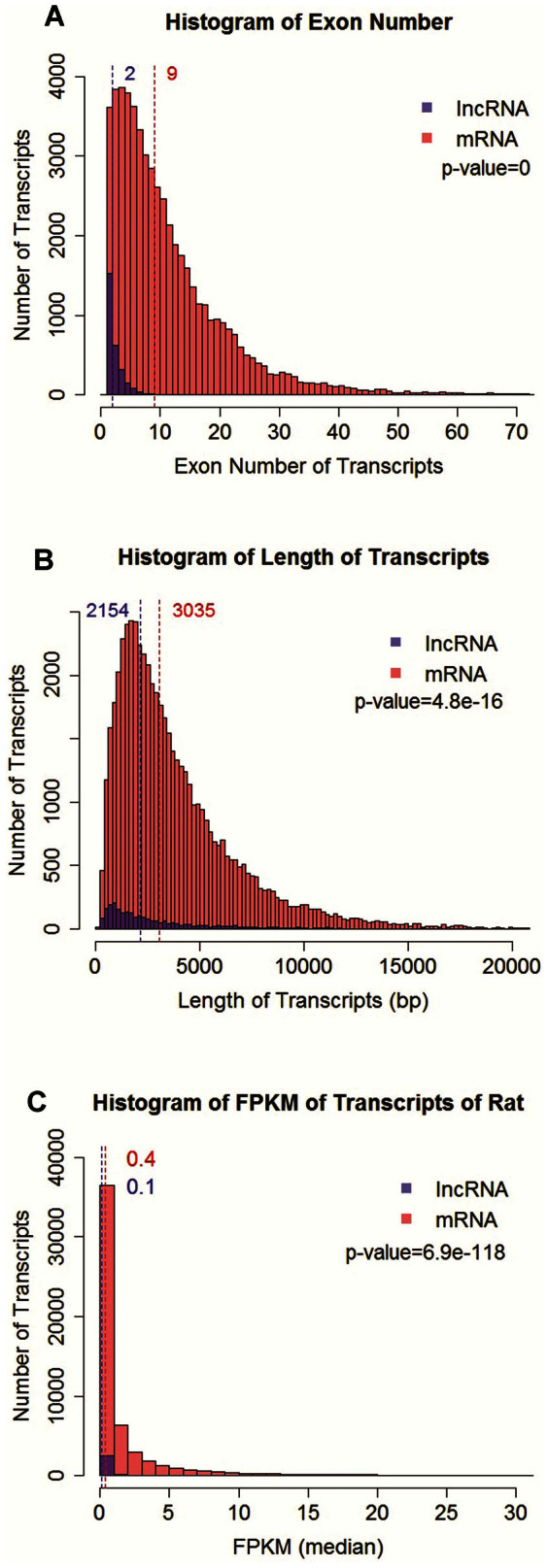
Rat lncRNA transcripts had fewer exons (A), modestly shorter lengths (B), and lower abundance (C) compared to mRNA.
The overall abundance of lncRNAs was compared to that of mRNAs by examining cumulative (sum of the 6 tissues), maximum or median abundance across the 6 tissues as well as abundance in each tissue. For all parameters examined, mRNAs were more abundant than lncRNAs by somewhere between 1.4 and 4 fold (Figure 5C and Supplementary Figure S3).
A significant fraction of Rat lncRNAs was not poly-adenylated
We compared renal outer medullary lncRNAs identified using poly(A)-independent RNA-seq in the current study to those identified in a previous study that used poly(A)-dependent RNA-seq14. lncRNAs identified as being enriched in the renal outer medulla in the current study were expressed at higher levels in the previous study compared to lncRNAs enriched in the renal cortex, supporting consistency between the two studies (Supplementary Figure S4). Of the 1,812 lncRNAs identified in the current study, only approximately 1,300 were identified in the previous study, indicating that approximately a quarter of lncRNAs in rat renal outer medulla were not poly-adenylated. This cannot be explained by differences in sequencing depth. The sequencing depth of the previous study was greater than the current study, which would increase, not decrease, the chance of detecting transcripts. It cannot be explained by different degrees of transcript fragmentation, either. The average or median number of exons per transcript was similar between the two studies. However, we cannot rule out possible roles of stranded vs. non-stranded RNA-seq or differences in rat strains in contributing to the different numbers of lncRNAs identified in the two studies.
Effects of substituting a segment of chromosome 13 and a high-salt diet on lncRNA expression in SS rats
The Dahl salt-sensitive SS rat is a widely used model of human salt-sensitive forms of hypertension and renal injury10,11,12. These disease phenotypes were significantly attenuated in congenic SS.13BN26 rats, which contain a substitution of a 12.9 Mbp segment of chromosome 13 from the Brown Norway rat genome22,23. We previously reported RNA-seq data from the renal outer medulla in SS and SS.13BN26 rats maintained on a 0.4% NaCl diet or exposed to a 4% NaCl diet for 7 days14. These RNA-seq data were re-analyzed to identify lncRNAs following the procedures described in Supplementary Figure S1.
We found 74 lncRNAs differentially expressed between SS and SS.13BN26 rats exposed to the high-salt diet (FDR<0.05) (Supplementary Table S3). No lncRNA reached the significance level when compared between SS and SS.13BN26 rats maintained on the 0.4% NaCl diet. Exposure to the high-salt diet resulted in significant differential expression of 5 lncRNAs in SS rats and 38 in SS.13BN26 rats (Supplementary Table S3 and Supplementary Figure S5).
Two of the lncRNAs that were differentially expressed between SS and SS.13BN26 rats exposed to the high-salt diet, TCONS_00028980 and TCONS_00029009, were located in the substituted genomic segment. Although these two lncRNAs did not reach significant differential expression between the two rat strains maintained on the 0.4% NaCl diet when measured by RNAseq, real-time PCR analysis confirmed the differential expression of these two lncRNAs between the rat strains on either the 0.4% or 4% NaCl diet (Figure 6A, 6B).
Figure 6. lncRNAs differentially expressed between SS and congenic SS.13BN26 rats and located on the congenic region.

Abundance levels based on RNA-seq (left) and real-time PCR (right) are shown for two lncRNAs (A, B) located in the congenic region of SS.13BN26. LS, 0.4% NaCl diet; HS, 4% NaCl diet for 7 days; L26, SS.13BN26 rat. n = 6–7 for real-time PCR. *, P<0.05 vs. SSLS; #, P<0.05 vs. SSHS. C. Genomic locations of the two lncRNAs. Nucleotides 75,226,423–77,023,203 of chromosome 13, indicated by the red square, are shown in detail. The two lncRNAs are indicated by red ovals.
The genomic segment substituted in SS.13BN26 spans nucleotides 73,664,780–86,574,314 of chromosome 13. TCONS_00028980 and TCONS_00029009 are encoded by nucleotides 75,525,885–75,529,130 and 76,635,112–76,723,739 and contain 2 and 5 exons, respectively. TCONS_00028980 is less than 10 kbp from protein-coding genes Nmnat2 and Lamc2. TCONS_00029009 is more than 100 kbp from any protein-coding gene (Figure 6C). We compared the reference genome (BN) with the genome of SS/JrHsdMcwi (SS) for these two lncRNAs24. As shown in Supplementary Table S4, 10 and 11 sequence variants were identified in the 2,000 bp genomic segment immediately upstream of the transcriptional start sites of TCONS_00028980 and TCONS_00029009, respectively. In addition, 6 and 232 sequence variants were identified in the gene bodies of TCONS_00028980 and TCONS_00029009, respectively. These data suggest genetic differences between the BN and SS alleles might contribute to the differential expression of these two lncRNAs between SS and SS.13BN26 rats.
Discussion
The present study provided a systematic characterization of lncRNAs in rat and identified several lncRNAs that might be involved in hypertension. These findings provide a new foundation for lncRNA research in hypertension and other diseases that utilize rat models.
Previous studies have reported catalogs of lncRNAs in the liver of Brown Norway rats25, the pineal gland and the colon of Sprague-Dawley rats26,27, and several whole organs in Fischer 344 rats28. Unique features of the current study compared to some or all of the previous studies included analysis of several specific tissue regions in Brown Norway rats, use of poly(A)-independent and strand-specific RNA-seq, de novo identification of lncRNAs, and, importantly, systematic examination of genome-wide characteristics of rat lncRNAs.
Some of the characteristics of rat lncRNAs appear different than lncRNAs in other species, while some characteristics are in common with other species. The number of rat lncRNAs we identified is similar to those identified in mice using methods close to ours7. The smaller number of exons and the higher degree of tissue specificity we observed in rat lncRNAs compared to mRNA have also been observed in other species7. Median abundance of lncRNAs has been reported to be as low as a tenth of mRNA and lncRNA genes are generally shorter than protein-coding genes in other species7. Our results indicate that median cumulative (sum of the 6 tissues) abundance of rat lncRNAs is about one half of that of mRNA and the median transcript length of rat lncRNAs is only 29% shorter than mRNA despite a much smaller number of exons.
lncRNAs remain nearly completely unexplored in hypertension research. However, emerging evidence indicates lncRNAs may have functional significance in the development, physiology or diseases of the heart and the vasculature29,30. Angiotensin II, a potent regulator of arterial blood pressure, has been reported to alter the expression of several lncRNAs in rat vascular smooth muscle cells, including an lncRNA that hosts two microRNAs implicated in cell proliferation31. The role of lncRNAs in the physiology and non-tumor diseases of the kidney, an organ critical for long-term control of arterial blood pressure32, is not known. Several lncRNAs were found to be differentially expressed in kidneys of mouse models of obstructive nephropathy and glomerulonephritis33.
We found approximately 4% of all lncRNAs detectable in the renal outer medulla were differentially expressed between SS and SS.13BN26 rats on a high-salt diet. This percentage is similar to the percentage of mRNAs that were found differentially expressed14. No lncRNA reached an FDR of less than 0.05 when compared between the two strains on the 0.4% NaCl diet. The number of mRNAs that were differentially expressed on the 0.4% NaCl was also much smaller than on the high-salt diet14. As shown in Figure 6, lncRNAs that did not reach the FDR cutoff based on RNA-seq could in fact be differentially expressed based on real-time PCR.
The two lncRNAs that we identified as differentially expressed between SS and SS.13BN26 rats and located in the congenic region may be controlled by sequence differences in the regulatory regions of the genes between the SS and Brown Norway alleles. It would be valuable to investigate whether and how these or other lncRNAs functionally contribute to the development of salt-sensitive hypertension.
Several lncRNAs have been found to serve as scaffolds that bring together chromatin modification complexes at select genome locations5. lncRNA-mediated assembly of chromatin modification complexes can occur locally in a cis acting manner just as lncRNAs are transcribed. Alternatively, lncRNAs can regulate the assembly of chromatin modification complex in a trans acting manner34. The result would be changes in DNA methylation, histone modification, or other chromatin characteristics at specific genomic locations, leading to changes in the expression of genes encoded by those genomic segments. We recently identified differential methylation in several hundred specific CpG islands in the renal outer medulla of SS and SS.13BN26 rats14. Several of the differentially methylated regions were located in the transcriptional start site or intergenic regions of differentially expressed protein-coding genes. DNA methylation or demethylation enzymes per se do not have the capacity to select specific sites of the genome for modification. It would be important to determine if lncRNAs contribute to the observed site-specific differential methylation in SS rats.
Epigenomic modifications and transcriptional regulation in general likely play an important role in the development and inheritance of hypertension35. Understanding of rat lncRNAs opens a novel venue for studying the role of transcriptional and other molecular control mechanisms in the development of hypertension and other diseases in rat models.
Supplementary Material
Supplementary information
Supplementary Dataset 1
Acknowledgments
This work was supported by US National Institutes of Health grants HL111580 (ML), HL121233 (ML), HL082798 (AWC, ML), and Advancing a Healthier Wisconsin Fund FP1701 and FP1703 (PL).
Footnotes
Author Contributions F.W. performed experiments. L.L., H.X. and P.L. analyzed the RNA-seq data. Y.L. and C.Y. assisted with the experiments. A.W.C., N.W. participated in study design and data interpretation. M.L. conceived and designed the study and drafted the manuscript. All authors provided input to the writing of the manuscript.
References
- Liang M. et al. MicroRNA: a new frontier in kidney and blood pressure research. Am. J. Physiol. Renal Physiol. 297, F553–558 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heggermont W. A. & Heymans S. MicroRNAs are involved in end-organ damage during hypertension. Hypertension 60, 1088–93 (2012). [DOI] [PubMed] [Google Scholar]
- Wilson F. H. et al. A cluster of metabolic defects caused by mutation in a mitochondrial tRNA. Science 306, 1190–4 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penny G. D., Kay G. F., Sheardown S. A., Rastan S. & Brockdorff N. Requirement for Xist in X chromosome inactivation. Nature 379, 131–7 (1996). [DOI] [PubMed] [Google Scholar]
- Rinn J. L. & Chang H. Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 81, 145–66 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai M. C. et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science 329, 689–93 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I. & Bartel D. P. lincRNAs: genomics, evolution, and mechanisms. Cell 154, 26–46 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob H. J., Lazar J., Dwinell M. R., Moreno C. & Geurts A. M. Gene targeting in the rat: advances and opportunities. Trends Genet. 26, 510–8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley A. W. Jr, Mattson D. L., Lu S. & Roman R. J. The renal medulla and hypertension. Hypertension 25, 663–73 (1995). [DOI] [PubMed] [Google Scholar]
- Cowley A. W. Jr The genetic dissection of essential hypertension. Nat. Rev. Genet. 7, 829–40 (2006). [DOI] [PubMed] [Google Scholar]
- Gopalakrishnan K. et al. Targeted disruption of Adamts16 gene in a rat genetic model of hypertension. Proc. Natl. Acad. Sci. U. S. A. 109, 20555–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapp J. P. Genetic analysis of inherited hypertension in the rat. Physiol. Rev. 80, 135–72 (2000). [DOI] [PubMed] [Google Scholar]
- Tian Z., Greene A. S., Pietrusz J. L., Matus I. R. & Liang M. MicroRNA-target pairs in the rat kidney identified by microRNA microarray, proteomic, and bioinformatic analysis. Genome Res. 18, 404–11 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Liu P., Yang C., Cowley A. W. Jr & Liang M. Base-resolution maps of 5-methylcytosine and 5-hydroxymethylcytosine in Dahl S rats: effect of salt and genomic sequence. Hypertension 63, 827–38 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegel A. J. et al. Characteristics of microRNAs enriched in specific cell types and primary tissue types in solid organs. Physiol. Genomics 45, 1144–56 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M. F., Jungreis I. & Kellis M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics 27, i275–282 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright M. W. A short guide to long non-coding RNA gene nomenclature. Hum. Genomics 8, 7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. et al. Renal medullary microRNAs in Dahl salt-sensitive rats: miR-29b regulates several collagens and related genes. Hypertension 55, 974–82 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mladinov D., Liu Y., Mattson D. L. & Liang M. MicroRNAs contribute to the maintenance of cell-type-specific physiological characteristics: miR-192 targets Na+/K+-ATPase beta1. Nucleic Acids Res. 41, 1273–83 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A. et al. High-resolution profiling of histone methylations in the human genome. Cell 129, 823–37 (2007). [DOI] [PubMed] [Google Scholar]
- Clouaire T. et al. Cfp1 integrates both CpG content and gene activity for accurate H3K4me3 deposition in embryonic stem cells. Genes Dev. 26, 1714–28 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng D. et al. Increased expression of NAD(P)H oxidase subunit p67(phox) in the renal medulla contributes to excess oxidative stress and salt-sensitive hypertension. Cell Metab. 15, 201–8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L. et al. Dynamic convergence and divergence of renal genomic and biological pathways in protection from Dahl salt-sensitive hypertension. Physiol. Genomics 41, 63–70 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanur S. S. et al. Genome sequencing reveals loci under artificial selection that underlie disease phenotypes in the laboratory rat. Cell 154, 691–703 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutter C. et al. Rapid turnover of long noncoding RNAs and the evolution of gene expression. PLoS Genet. 8, 26 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coon S. L. et al. Circadian changes in long noncoding RNAs in the pineal gland. Proc. Natl. Acad. Sci. U. S. A. 109, 13319–24 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triff K. et al. Genome-wide analysis of the rat colon reveals proximal-distal differences in histone modifications and proto-oncogene expression. Physiol. Genomics 45, 1229–43 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y. et al. A rat RNA-Seq transcriptomic BodyMap across 11 organs and 4 developmental stages. Nat. Commun. 5, 3230 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalik K. M. et al. Long Noncoding RNA MALAT1 Regulates Endothelial Cell Function and Vessel Growth. Circ. Res. 114, 1389–97 (2014). [DOI] [PubMed] [Google Scholar]
- Schonrock N., Harvey R. P. & Mattick J. S. Long noncoding RNAs in cardiac development and pathophysiology. Circ. Res. 111, 1349–62 (2012). [DOI] [PubMed] [Google Scholar]
- Leung A. et al. Novel long noncoding RNAs are regulated by angiotensin II in vascular smooth muscle cells. Circ. Res. 113, 266–78 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley A. W. Jr Long-term control of arterial blood pressure. Physiol. Rev. 72, 231–300 (1992). [DOI] [PubMed] [Google Scholar]
- Zhou Q. et al. Identification of novel long noncoding RNAs associated with TGF-beta/Smad3-mediated renal inflammation and fibrosis by RNA sequencing. Am. J. Pathol. 184, 409–17 (2014). [DOI] [PubMed] [Google Scholar]
- Bonasio R., Tu S. & Reinberg D. Molecular signals of epigenetic states. Science 330, 612–6 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang M., Cowley A. W. Jr, Mattson D. L., Kotchen T. A. & Liu Y. Epigenomics of hypertension. Semin. Nephrol. 33, 392–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information
Supplementary Dataset 1