

Abstract
Aims/hypothesis
The molecular mechanisms by which thiazolidinediones improve insulin sensitivity in type 2 diabetes are not fully understood. We hypothesised that pioglitazone would activate the adenosine 5′-monophosphate-activated protein kinase (AMPK) pathway and increase the expression of genes involved in adiponectin signalling, NEFA oxidation and mitochondrial function in human skeletal muscle.
Methods
A randomised, double-blind, parallel study was performed in 26 drug-naive type 2 diabetes patients treated with: (1) pioglitazone (n=14) or (2) aggressive nutritional therapy (n=12) to reduce HbA1c to levels observed in the pioglitazone-treated group. Participants were assigned randomly to treatment using a table of random numbers. Before and after 6 months, patients reported to the Clinical Research Center of the Texas Diabetes Institute for a vastus lateralis muscle biopsy followed by a 180 min euglycaemic–hyperinsulinaemic (80 mU m−2 min−1) clamp.
Results
All patients in the pioglitazone (n=14) or nutritional therapy (n=12) group were included in the analysis. Pioglitazone significantly increased plasma adiponectin concentration by 79% and reduced fasting plasma NEFA by 35% (both p<0.01). Following pioglitazone, insulin-stimulated glucose disposal increased by 30% (p<0.01), and muscle AMPK and acetyl-CoA carboxylase (ACC) phosphorylation increased by 38% and 53%, respectively (p<0.05). Pioglitazone increased mRNA levels for adiponectin receptor 1 and 2 genes (ADIPOR1, ADIPOR2), peroxisome proliferator-activated receptor gamma, coactivator 1 gene (PPARGC1) and multiple genes involved in mitochondrial function and fat oxidation. Despite a similar reduction in HbA1c and similar improvement in insulin sensitivity with nutritional therapy, there were no significant changes in muscle AMPK and ACC phosphorylation, or the expression of ADIPOR1, ADIPOR2, PPARGC1 and genes involved in mitochondrial function and fat oxidation. No adverse (or unexpected) effects or side effects were reported from the study.
Conclusions/interpretations
Pioglitazone increases plasma adiponectin levels, stimulates muscle AMPK signalling and increases the expression of genes involved in adiponectin signalling, mitochondrial function and fat oxidation. These changes may represent an important cellular mechanism by which thiazolidinediones improve skeletal muscle insulin sensitivity.
Keywords: Adiponectin, AMP kinase, Insulin sensitivity, Mitochondrial, Muscle, Pioglitazone
Introduction
Skeletal muscle insulin resistance is an early and characteristic feature of type 2 diabetes mellitus [1, 2]. Recent studies have implicated mitochondrial dysfunction as an important contributing factor in skeletal muscle insulin resistance [3, 4]. We [5] and others [6] have demonstrated that insulin-resistant patients have decreased expression of nuclear-encoded mitochondrial genes, accompanied by a decrease in expression of the PPARGC1A gene encoding peroxisome proliferator-activated receptor gamma, coactivator 1 alpha, a key transcriptional factor that regulates the coordinated expression of genes that code for multiple mitochondrial proteins.
Thiazolidinediones (TZDs) represent a class of insulin-sensitising agents that have proven effective in the treatment of patients with type 2 diabetes. TZDs are high-affinity ligands for the transcription factor peroxisome proliferator-activated receptor gamma (PPARG) [7]. Activation of PPARG alters the expression of many genes [7, 8], resulting in enhanced insulin sensitivity in liver and muscle [9, 10], reduced intracellular lipid content in liver and muscle [11, 12], and increased insulin receptor signalling in type 2 diabetes [13, 14]. However, TZDs may also improve glucose tolerance via adenosine 5′-monophosphate-activated protein kinase (AMPK), a fuel gauge and key regulator of both glucose and lipid metabolism [15]. Thus, cell culture and animal studies suggest that TZDs work by increasing fat cell secretion of adiponectin, which stimulates muscle AMPK. AMPK, in turn, augments muscle glucose uptake and fatty acid oxidation, and inhibits hepatic glucose production, triacylglycerol synthesis and lipogenesis [16–18]. Despite the widespread clinical use of TZDs, the precise molecular mechanisms by which they exert their insulin-sensitising effects in man remain unclear. Because of the similarities between the metabolic effects of AMPK activation and TZDs, we hypothesised that AMPK might play a role in mediating the metabolic actions of TZDs. Therefore, the goal of the present study was to evaluate whether pioglitazone activates the AMPK pathway and increases the expression of genes involved in adiponectin signalling, NEFA oxidation and mitochondrial function in human skeletal muscle of type 2 diabetes patients.
Methods
Patients
Twenty-six drug-naive patients with type 2 diabetes participated in the study. All patients were free of cardiovascular, renal or major organ disease, as determined by medical history, physical examination, screening blood chemistries, complete blood cell count and electrocardiogram. Bodyweight was stable (±1 kg) for at least 3 months prior to enrolment and no patient participated in an excessively heavy physical activity programme. Patients were excluded from the study if they had ever received insulin, metformin, sulfonylureas, TZDs, exenatide or dipeptidyl peptidase-IV inhibitor. Four patients in the pioglitazone group and three in the nutritional therapy group had mild hypertension treated with ACE inhibitor alone or with a diuretic; two patients in each group were receiving statin therapy. All patients gave informed written consent prior to participation in the study, which was approved by the Institutional Review Board of the University of Texas Health Science Center at San Antonio.
Study design
Following a 10–12 h overnight fast, patients came to the Clinical Research Center (CRC) of the Texas Diabetes Institute at 08:00 hours for a screening visit and OGTT. Patients consumed a weight-maintaining diet containing 50% carbohydrate, 30% fat and 20% protein (wt/wt) and were instructed not to engage in any new physical activity programme during the study period. Within 3–7 days, patients returned to the CRC at 08:00 hours after an overnight fast for vastus lateralis muscle biopsy [19], followed by a 180 min euglycaemic–hyperinsulinaemic (80 mU m−2 min−1) clamp [20]. Briefly, a catheter was inserted into an antecubital vein for infusion of all test substances. A second catheter was inserted into a vein on the dorsum of the hand for blood withdrawal and the hand was placed in a thermoregulated box (70°C). At 08:00 hours (−180 min) a prime (9.25×105 Bq×[FPG/5.55])–continuous (9.25×103 Bq/min), where FPG is fasting plasma glucose (mmol/l), infusion of [3-3H]glucose (NEN-Dupont, Boston, MA, USA) was started. Blood samples for determination of plasma glucose, insulin, NEFA and adiponectin concentrations and [3H]glucose radioactivity were obtained at −30, −20, −10 and 0 min. At 11:00 hours, a prime-continuous (80 mU m−2 min−1) insulin infusion was started. Plasma glucose concentration was allowed to decrease to 5.6 mmol/l, at which level it was maintained by a variable infusion of 20% (wt/vol.) glucose. At −60 min and at the end of the insulin clamp, biopsies of the vastus lateralis muscle were obtained. One week after the euglycaemic–hyperinsulinaemic clamp, patients were randomised to receive pioglitazone, 45 mg/day (n=14), or aggressive nutritional therapy (n=12) for 6 months. Prior to randomisation, pioglitazone-treated patients received 45 min of dietary advice, consistent with ADA guidelines. The medical nutritional therapy group received 10 h of diabetes management in an ADA-certified educational programme, an additional 2 h of one-on-one nutritional advice, and met with a dietitian for 30 min on each follow-up visit, which occurred monthly. After 6 months of treatment, patients returned to the CRC for repeat euglycaemic–hyperinsulinaemic clamps with muscle biopsies.
Muscle biopsy processing
For mRNA analyses, muscle biopsy specimens were homogenised directly in RNAStat solution (Tel-Test, Friendswood, TX, USA), using a Polytron homogeniser (Brinkmann Instruments, Westbury, NY, USA). RNA pellets were stored in 75% (wt/vol.) ethanol at −80°C. Prior to use, total RNA was purified with RNeasy and DNase I treatment (Qiagen, Chatsworth, CA, USA). For immunoblot analysis, muscle samples were weighed while still frozen and homogenised in ice-cold lysis buffer (1:10, wt/vol.) containing 50 mmol/l HEPES (pH 7.6), 150 mmol/l NaCl, 20 mmol/l sodium pyrophosphate, 20 mmol/l β-glycerophosphate, 10 mmol/l sodium fluoride, 2 mmol/l sodium orthovanadate, 2 mmol/l EDTA (pH 8.0), 1% (vol./vol.) Nonidet P-40, 10% (vol./vol.) glycerol, 1 mmol/l phenylmethylsulfonylfluoride, 1 mmol/l MgCl2, 1 mmol/l CaCl2, 10 μg/ml leupeptin and 10 fig/ml aprotinin. Homogenates were incubated on ice for 20 min and then centrifuged at 15,000 g for 20 min at 4°C Supernatant fractions were collected, and protein concentrations measured by the Lowry method. Supernatant fractions were stored at −80°C until used.
Determination of malonyl-CoA
Muscle malonyl-CoA levels were measured following extraction with 10% (wt/vol.) trichloroacetic acid. After centrifugation of precipitated proteins and ether extraction, samples were lyophilised and stored at −70°C [21]. Malonyl-CoA was assayed with a radioactive method using fatty acid synthase [21].
Quantitative real-time PCR
Muscle expression of various genes was determined using one-step quantitative real-time PCR (Q-RT-PCR) from total RNA. Q-RT-PCR was performed on an ABI PRISM 7900HT sequence detection system (Applied Biosystems, Foster City, CA, USA) using TaqMan One Step RT-PCR Master Mix reagents and Assay On Demand gene expression primer pairs and probes [see Electronic supplementary material (ESM) Table 1]. Expression data were normalised by dividing the amount of the gene of interest by the amount of RN18S gene used as the internal control.
Immunoblot analysis
Proteins were transferred to nitrocellulose membranes, and the membranes were probed with antibody against phospho-AMPK (Thr172) (Cell Signaling, Beverly, MA, USA), phospho-acetyl-CoA carboxylase (ACC) (Ser79) (Cell Signaling), AMPK panα (Cell Signaling) and PPARGC1A (Calbiochem, San Diego, CA, USA). ACC was detected using streptavidin (Pierce Biotech, Rockford, IL, USA). Bound antibodies were detected with anti-rabbit immunoglobulin-horseradish peroxidase-linked whole antibody and by using enhanced chemiluminescent reagents (PerkinElmer Life Science, Boston, MA, USA). The concentration used for primary and secondary antibodies was 1:1,000 and 1:2,000, respectively. The amount of protein loaded was 40 μg. The membranes were exposed to film, and band intensity was quantified using Image Tool Software (University of Texas Health Science Center at San Antonio, San Antonio, TX, USA).
Laboratory analyses
Plasma insulin (Diagnostic Products, Los Angeles, CA, USA) and adiponectin (Linco Research, St Charles, MO, USA) were measured by RIA, plasma glucose by the glucose oxidase method (Beckman Instruments, Fullerton, CA, USA), and HbA1c using a DCA2000 Analyzer (Bayer, Tarrytown, NY, USA). Plasma NEFA concentration was determined using a colorimetric method (Wako, Richmond, VA, USA).
Calculations
During the postabsorptive state, steady-state conditions prevail and the rate of endogenous glucose appearance, which equals rate of total body glucose uptake, was calculated as [3H]glucose infusion rate (dpm/min) divided by steady-state plasma [3H]glucose specific activity (dpm/mg). During the insulin clamp, non-steady-state conditions prevail, and the rate of glucose appearance (Ra) was calculated using Steele's equation [22]. The rate of endogenous glucose production (EGP) during the insulin clamp equals the tracer-derived Ra minus the exogenous glucose infusion rate. The rate of whole-body glucose disposal equals the rate of residual EGP plus exogenous glucose infusion rate during the 150–180 min time period. The basal hepatic insulin resistance index (IRI) is the product of basal EGP and fasting plasma insulin (FPI).
Statistical analysis
All data are expressed as means±SEM. Differences within a group, before vs after treatment, were determined using a paired t test. Between-group differences were determined by ANOVA. For all analyses, p<0.05 was considered statistically significant.
Results
Patient characteristics and in vivo data
Pioglitazone- and nutritional therapy-treated groups were well matched for age, body weight and BMI. Clinical, laboratory and metabolic variables are shown in Table 1. Of the 14 patients treated with pioglitazone, 11 responded, as defined by a decline in HbA1c ≥0.5%. After 6 months of pioglitazone treatment, FPG and HbA1c were reduced by 17% and 15%, respectively (both p<0.001), despite increased body weight and BMI (both p<0.05). Following pioglitazone treatment, FPI concentrations decreased by 41% (p<0.05), and insulin-stimulated glucose disposal increased by 30% (p<0.01). Pioglitazone treatment decreased plasma NEFA by 35% (p<0.01). In the nutritional therapy group there was a decline in FPG (14%), fasting plasma NEFA (19%) and HbA1c (12%, p<0.05), and insulin-stimulated glucose disposal increased by 20% (p<0.05) in association with weight loss.
Table 1. Clinical, laboratory and metabolic variables before and after pioglitazone and nutritional therapy.
| Variable | Nutritional therapy | Pioglitazone | p valuea | ||||
|---|---|---|---|---|---|---|---|
|
|
|
||||||
| Before | After | Change (%) | Before | After | Change (%) | ||
| n | 12 | – | – | 14 | – | – | |
| Sex (male/female) | 9/3 | – | – | 9/5 | – | – | NS |
| Age (years) | 50±2 | – | – | 51±2 | – | – | NS |
| Diabetes duration (years) | 6.9±6.0 | – | – | 7.1 ±6.6 | – | – | NS |
| Body weight (kg) | 84.4±5.4 | 82.5±5.1* | −2.2 | 89±5 | 93±5* | +5 | 0.0009 |
| BMI (kg/m2) | 30.1 ±1.4 | 29.4±1.3* | −2.1 | 31.6±1.3 | 33.4±1.4* | +6 | 0.0004 |
| FPG (mmol/l) | 9.4±0.9 | 8.0±0.8* | −14 | 10.7±0.9 | 8.9±0.8** | −17 | NS |
| FPI (pmol/l) | 76±15 | 68±9 | −11 | 90±12 | 54±12* | −41 | NS |
| HbA1c (%) | 8.1±0.4 | 7.1 ±0.5* | −12 | 8.3±0.4 | 7.1±0.4*** | −15 | NS |
| NEFA (μmol/l) | 600±33 | 489±32* | −19 | 663±61 | 433±55** | −35 | NS |
| Plasma adiponectin (μg/ml) | 4.3±0.6 | 4.5±0.6 | +4 | 5.8±0.9 | 10.4±1.8*** | +79 | 0.018 |
| Basal EGP (μmol kg−1 min−1) | 14.9± 1.7 | 13.8±2.1 | −9 | 12.9±0.9 | 11.5±0.6 | −9 | NS |
| Basal hepatic IRI (μmol kg−1 min−1 [pmol/l] −1) | 1,132±68 | 938±30* | −21 | 1,161 ±23 | 621 ±16* | −46 | NS |
| Rd (μmol kg−1 min−1) | 18.7±2.5 | 22.5±2.6* | +20 | 18.9±2.1 | 24.2±1.8* | +30 | NS |
| EGP during insulin clamp (μmol kg−1 min−1) | 2.2±1.4 | 2.1 ±0.9 | −4 | 2.3±0.8 | 1.2±0.6 | −50 | NS |
Values are means±SEM
p<0.05,
p<0.01,
p<0.001 vs pretreatment
p value (ANOVA) for the comparison between pioglitazone- and nutritional therapy-treated groups (change from baseline)
Rd, rate of whole-body glucose disposal
Effect of pioglitazone on AMPK signalling, malonyl-CoA and adiponectin receptor gene expression
Pioglitazone treatment increased AMPK phosphorylation in skeletal muscle by 38% (p<0.05 vs baseline and nutritional therapy; Fig. 1a), while there was no change in the nutritional therapy group. Consistent with activation of AMPK, pioglitazone increased ACC phosphorylation by 53% (p<0.05 vs baseline and nutritional therapy; Fig. 1b). ACC phosphorylation did not change in the nutritional therapy group. There was no difference in AMPK and ACC proteins between pioglitazone and nutritional therapy groups at baseline, and treatment had no effect on these proteins (data not shown). Plasma adiponectin concentration increased by 79% in the pioglitazone group and remained unchanged in the nutritional therapy group (Table 1). The increase in plasma adiponectin concentration and AMPK phosphorylation in muscle after pioglitazone treatment was accompanied by elevated expression of the genes ADIPOR1 (Fig. 2a) and ADIPOR2 (Fig. 2b) for adiponectin receptor 1 and 2 (both p<0.05 vs baseline and nutritional therapy). ADIPOR1 and ADIPOR2 expression did not change in the nutritional therapy group. Pioglitazone (before 2.71±0.36 vs after 2.75±0.40; p=NS) and nutritional therapy (before 2.76±0.50 vs after 2.75±0.18; p=NS) treatment had no effect on muscle malonyl-CoA content.
Fig. 1.
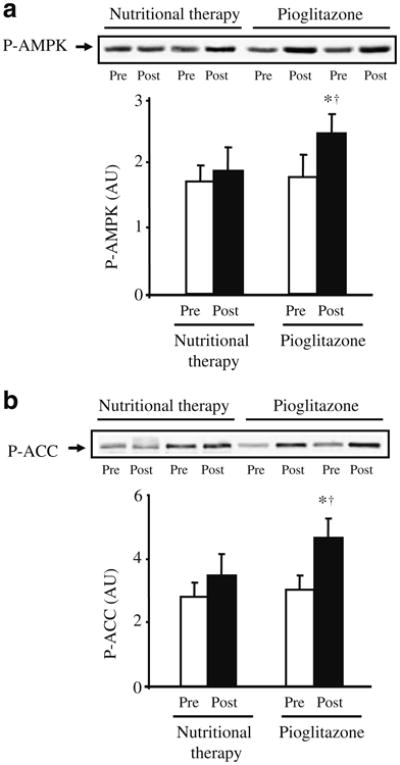
Effect of pioglitazone and nutritional therapy on AMPK (a) and ACC (b) phosphorylation (P). Data are expressed as arbitrary units (AU). Protein extracts were available for 12 pioglitazone- and nine nutritional therapy-treated patients. Representative blots for two patients are shown. *p<0.05 vs pretreatment; †p<0.05 for the comparison between pioglitazone- and nutritional therapy-treated groups (change from baseline). Means±SEM
Fig. 2.

Effect of pioglitazone and nutritional therapy on mRNA expression of ADIPOR1 (a), and ADIPOR2 (b). Expression data were normalised by dividing the amount of the gene of interest by the amount of RN18S gene used as an internal control. *p<0.05 vs pre-treatment; †p<0.05 for the comparison between pioglitazone- and nutritional therapy-treated groups (change from baseline). Means±SEM
Effect of pioglitazone on expression of PPARGC1 and genes involved in mitochondrial function
Pioglitazone significantly increased PPARGC1A protein in skeletal muscle (p<0.05 vs baseline and nutritional therapy; Fig. 3a), while there was no change in the nutritional therapy group. PPARGC1A and PPARGC1B mRNA levels were significantly increased following pioglitazone treatment (Fig. 3b,c; p<0.05 vs baseline and nutritional therapy), and were unchanged in the nutritional therapy group. Unexpectedly, NRF1, the gene for nuclear respiratory factor 1, was not affected by pioglitazone (before 0.87±0.23 vs after 0.86±0.12; p=NS). Likewise, NRF-2 was not affected by pioglitazone (before 8.28±2.13 vs after 8.54±1.78; p=NS). The mRNA levels for a number of nuclear-encoded mitochondrial genes and genes encoding for scavenging enzymes that are important in protecting mitochondria against the effects of reactive oxygen species are shown in Table 2. The genes encoding cytochrome c oxidase subunit VIc (COX6C), NADH dehydrogenase (ubiquinone) 1-alpha subcomplex, 5 (NDUFA5), cAMP-responsive element binding protein 1 (CREB1), myocyte enhancer factor 2C (MEF2C) and sirtuin 1 (SIRT1) all increased following pioglitazone treatment (all p<0.05). There were no significant changes in expression of any mRNAs in the nutritional therapy group. In addition, mRNA expression of the genes for NAD(P)H dehydrogenase, quinone 1 (NQO1) and superoxide dismutase 2 (SOD2) increased following pioglitazone treatment, but did not change in the nutritional therapy group.
Fig. 3.
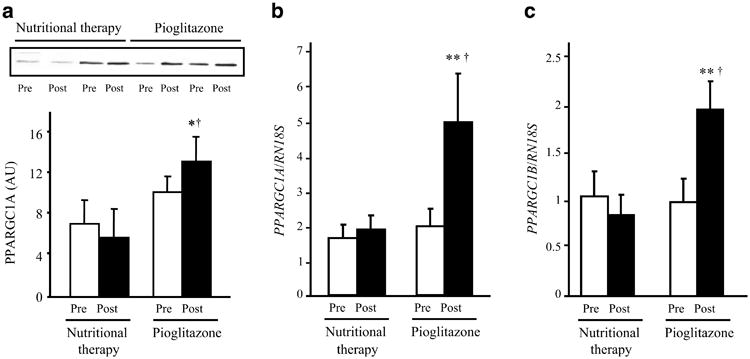
a Effect of pioglitazone and nutritional therapy on PPARGC1A protein levels. Data are expressed as arbitrary units (AU). Protein extracts were available for 12 pioglitazone- and nine nutritional therapy-treated patients. Representative blots for two patients are shown. b, c Effect of pioglitazone and nutritional therapy on mRNA expression of PPARGC1A (b) and PPARGC1B (c). Expression data were normalised by dividing the amount of the gene of interest by the amount of RN18S gene used as an internal control. *p<0.05 and **p<0.01 vs pretreatment; †p<0.05 for the comparison between pioglitazone- and nutritional therapy-treated groups (change from baseline). Means±SEM
Table 2. mRNA expression determined by Q-RT-PCR for a number of nuclear-encoded mitochondrial genes and NEFA oxidation genes.
| Gene | Nutritional therapy | Pioglitazone | p valuea | ||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||
| Before | After | Fold change | p value | Before | After | Fold change | p value | ||
| ACADM | 3.01 ±0.83 | 3.27±1.11 | 1.08 | NS | 4.37±1.54 | 8.22±1.88 | 1.88 | 0.0067 | 0.022 |
| HADH | 0.39±0.08 | 0.39±0.09 | 1.01 | NS | 0.39±0.10 | 0.84±0.11 | 2.18 | 0.000023 | 0.0041 |
| COX6C | 1.26±0.27 | 1.44±0.39 | 1.14 | NS | 1.40±0.45 | 2.18±0.47 | 1.56 | 0.000038 | NS |
| CPT1B | 0.31±0.05 | 0.32±0.08 | 1.00 | NS | 0.43±0.13 | 0.77±0.19 | 1.76 | 0.012 | 0.040 |
| CREB1 | 0.30±0.06 | 0.38±0.09 | 1.26 | NS | 0.33±0.10 | 0.54±0.09 | 1.63 | 0.011 | NS |
| IDH3B | 1.25±0.29 | 1.29±0.46 | 1.04 | NS | 1.26±0.39 | 1.88±0.33 | 1.48 | 0.0073 | NS |
| MEF2C | 0.68±0.24 | 0.69±0.20 | 1.02 | NS | 0.91 ±0.21 | 1.18±0.18 | 1.30 | 0.020 | NS |
| NDUFA5 | 1.09±0.20 | 1.02±0.27 | 0.93 | NS | 1.46±0.43 | 2.28±0.50 | 1.56 | 0.00040 | 0.0017 |
| NQO1 | 0.87±0.20 | 0.90±0.25 | 1.03 | NS | 0.51±0.23 | 2.77±1.16 | 5.4 | NS | 0.018 |
| PPARA | 0.99±0.24 | 0.64±0.13 | 0.65 | NS | 0.81±0.25 | 1.16±0.18 | 1.43 | 0.00096 | 0.019 |
| PPARD | 1.15±0.07 | 1.19±0.31 | 1.03 | NS | 0.81 ±0.27 | 1.02±0.22 | 1.26 | NS | NS |
| PPARG | 0.24±0.05 | 0.31 ±0.11 | 1.27 | NS | 0.32±0.09 | 0.47±0.08 | 1.56 | 0.0020 | NS |
| SIRT1 | 0.35±0.09 | 0.28±0.06 | 0.80 | NS | 0.26±0.08 | 0.36±0.06 | 1.36 | 0.0032 | NS |
| SOD2 | 0.039±0.016 | 0.027±0.009 | 0.69 | NS | 0.022±0.006 | 0.054±0.009 | 2.4 | 0.0069 | 0.0091 |
Values are means±SEM
p value (ANOVA) for the comparison between pioglitazone- and nutritional therapy-treated groups (change from baseline)
Effect of pioglitazone on expression of genes involved in NEFA oxidation
Pioglitazone increased expression of genes required for fatty acid oxidation (Table 2). The CPT1B gene for muscle type carnitine palmitoyltransferase 1B, the enzyme that catalyses the initial reaction in mitochondrial import of long-chain fatty acids, was significantly increased after pioglitazone. Other mRNAs that were increased following pioglitazone treatment, but remained unchanged in the nutritional therapy group, included those for the genes for acyl-CoA dehydrogenase (ACADM), hydroxyacyl-CoA dehydrogenase (HADH), isocitrate dehydrogenase 3 (NAD+) beta (IDH3B), peroxisome proliferator-activated receptor alpha (PPARA) and PPARG.
Discussion
Pioglitazone treatment for 6 months significantly improved glycaemic control (FPG and HbA1c) and insulin-stimulated whole-body (primarily reflecting muscle) glucose disposal in type 2 diabetes patients, consistent with previous results from our laboratory [23, 24] and others [25, 26]. Pioglitazone also increased the phosphorylation of AMPK (by 38%) and ACC (by 53%), consistent with animal and cell culture studies, which demonstrate that TZDs activate AMPK in skeletal muscle [27–29]. Collectively, these observations suggest that activation of AMPK signalling is involved in the insulin-sensitising effect of the TZDs.
To exclude the possibility that improved glycaemic control could explain the increases in AMPK and ACC phosphorylation, age-, sex- and BMI-matched patients with type 2 diabetes were randomised to receive intensive medical nutritional advice. Body weight decreased significantly in this group. Despite reduction in HbA1c and improvement in insulin-stimulated glucose disposal of similar magnitude to that in the pioglitazone-treated group, no changes in phosphorylation of AMPK and ACC were observed. This excludes improved glycaemic control as a causative factor in the improved AMPK and ACC phosphorylation observed with pioglitazone.
Enhanced insulin sensitivity was accompanied by a 79% increase in plasma adiponectin levels (Table 1), consistent with other studies demonstrating that TZDs increase plasma adiponectin in insulin-resistant rodents, insulin-resistant individuals without type 2 diabetes, and individuals with type 2 diabetes [30–32]. In transgenic mice, TZD-induced stimulation of adiponectin secretion from adipocytes plays a key role in its glucose-lowering action [33]. In rodents, adiponectin stimulates AMPK activity in muscle, liver and fat [16–18], resulting in increased glucose transport in skeletal muscle and adipocytes and enhanced fatty acid oxidation in muscle and liver [16–18]. These metabolic effects of adiponectin are blocked by inhibiting AMPK activity with a dominant negative mutant [16] and with a chemical inhibitor of AMPK [18], indicating that AMPK plays a central role in mediating the metabolic effects of adiponectin. The present results are consistent with these observations in rodents, but they do not allow us to definitively establish whether the stimulatory effect of pioglitazone on AMPK activity is mediated via adiponectin, or represents a possible direct effect of the TZD on AMPK. Thus, TZDs have been documented to directly activate AMPK in various tissues [28].
The present study also allowed us to examine the in vivo effect of pioglitazone on expression of genes involved in adiponectin signalling in skeletal muscle from patients with type 2 diabetes. Insulin-resistant patients are characterised by reduced adiponectin signalling [34]. We observed an increase in mRNAs for ADIPOR1 and ADIPOR2 following pioglitazone treatment in insulin-resistant type 2 diabetes patients. Although our results are in contrast to some previous studies, it should be noted that the treatment period (6 months) was much longer than the studies by Li et al. [35] (3 weeks) and Tan et al. [36] (12 weeks).
Insulin-resistant muscle exhibits decreased expression of oxidative phosphorylation (OXPHOS) genes, including PPARGC1 [5, 6], which coordinately regulates the expression of multiple genes involved in mitochondrial bioenergetics [37, 38]. In a previous study we demonstrated that an experimental increase in plasma NEFA concentration in healthy individuals resulted in a 30–40% reduction in insulin-stimulated glucose disposal and decreased expression of PPARGC1 and nuclear-encoded mitochondrial genes [39]. In this study, we hypothesised that the pioglitazone-induced increase in peripheral glucose disposal would be associated with increased expression of PPARGC1 and genes that code for mitochondrial proteins. We also hypothesised that genes involved in fatty acid oxidation in human skeletal muscle would be upregulated by pioglitazone, since TZDs have been shown to increase expression of fatty acid oxidation genes in human subcutaneous adipose tissue in vivo [38]. As hypothesised, the present results demonstrate that pioglitazone increased the expression of multiple genes involved in mitochondrial function and NEFA oxidation in human skeletal muscle in association with an increase in AMPK activity. The causality of this association remains to be seen. Consistent with this, polycystic ovary syndrome patients treated with pioglitazone for 4 months had increased expression of oxidative phosphorylation genes [40]. In contrast, Boden et al. [41] failed to observe any effect of TZDs on expression of genes coding for proteins related to mitochondrial function and fatty acid oxidation in skeletal muscle from patients with type 2 diabetes [41]. Likewise, Schrauwen-Hinderling et al. [42] demonstrated that rosiglitazone treatment for 2 months did not change muscle mitochondrial function in patients with type 2 diabetes. However, the treatment period in these studies was much shorter (2 months) than the present study (6 months), and patients in the prior study were on a variety of other glucose-lowering drugs [41], which could plausibly explain the contrasting results. Our results also demonstrate that pioglitazone increases the expression of SOD2 and NQO1, genes for enzymes that protect cells against the deleterious effects of reactive oxygen species. A prior study also demonstrated that adiponectin and 5-aminoimidazole 1 carboxamide ribonucleoside (AICAR) increased SOD2 activity in human myotubes [37]. We expected that NRF1 mRNA would be upregulated [5], but its expression was unaltered by pioglitazone. Consistent with this, a recent study in human subcutaneous adipose tissue also failed to demonstrate any effect of pioglitazone on NRF1 expression [38]. Although we observed increases in mRNAs coding for genes involved in mitochondrial function and fatty acid metabolism, these results may not reflect changes in the abundance of these proteins following pioglitazone treatment, and further studies are warranted. Importantly, the mRNA changes observed in the present study cannot be explained by improved glycaemic control, since they were not observed in patients receiving medical nutritional therapy. In contrast, Civitarese et al. [43] demonstrated increased expression of genes encoding proteins involved in mitochondrial function such as PPARGC1A, transcription factor A, mitochondrial (TFAM), endothelial nitric oxide synthase (eNOS), SIRT1 and presenilin associated, rhomboid-like (PARL) in their energy-restricted healthy young overweight individuals. Differences in gene expression responses to weight loss in the present study vs the study of Civitarese et al. [43] may be explained by differences in the study population: diabetic vs normal glucose tolerant; severity of obesity (BMI 30.1 vs 27.8 kg/m2); differences in weight loss (1.9 vs 8.3 kg). Another mechanism by which pioglitazone could enhance gene expression is via changes in the plasma insulin concentration. However, we have previously shown that insulin infusion has no effect on the mRNA expression of genes involved in mitochondrial oxidative phosphorylation [44] and the plasma insulin concentration decreased, not increased, following TZD therapy.
ACC is a key regulator of fatty acid oxidation and catalyses the conversion of acetyl-CoA to malonyl-CoA, a potent allosteric inhibitor of CPT1B. Studies in rat muscle showed that stimulation of AMPK by contraction or chemical activation with AICAR causes phosphorylation and inactivation of ACC [45, 46]. The decline in ACC activity leads to a decrease in malonyl-CoA content, and the resultant increase in CPT1B activity augments fatty acid entry into mitochondria for oxidation. Consistent with this scenario, pioglitazone increased ACC phosphorylation and expression of CPT1B and multiple genes involved in fatty acid oxidation. However, to our surprise, muscle malonyl-CoA levels were not affected by pioglitazone. In skeletal muscle from insulin-resistant obese and type 2 diabetic patients, Bandyopadhyay et al. [47] demonstrated increased malonyl-CoA levels, which they attributed to increased ACC and decreased AMPK activity. In these same patients, rosiglitazone treatment for 3 months normalised basal phosphorylated ACC levels, AMPK activity and malonyl-CoA [47]. A study in mice demonstrated that rosiglitazone treatment for 10 days led to the activation of AMPK and ACC, resulting in a significant reduction in both hepatic and muscle malonyl-CoA content [33]. Contrary to results of the present study, activity and protein content of AMPK was not increased following pioglitazone treatment in patients with polycystic ovary syndrome [48], suggesting that there may be other mechanisms, independent of AMPK, which mediate the effect of TZDs. Dean et al. [49] demonstrated that human muscle only has one-tenth of the amount of malonyl-CoA content of rat muscle and that acute exercise inhibited ACC activity and increased fat oxidation without a detectable reduction in muscle malonyl-CoA content. It is possible that the close association between malonyl-CoA content and ACC activity observed in rat muscle [45, 46] does not operate in the same manner in humans. Moreover, because of the very low malonyl concentration in skeletal muscle, changes would be difficult to detect. This may derive from the fact that cytosolic malonyl-CoA is the ACC-derived signal that regulates fat oxidation, although malonyl-CoA appears to be present in additional compartments, in particular the mitochondria and peroxisomes [50]. Perhaps, total malonyl-CoA in rat muscle better reflects cytosolic levels of this metabolite than in human muscle. However, it should be noted that the number of muscle biopsy samples available for malonyl-CoA measurement was relatively small and additional studies examining the effect of TZDs on muscle malonyl-CoA content are warranted.
In summary, we demonstrate that in patients with type 2 diabetes, chronic treatment with pioglitazone increases plasma adiponectin levels and expression of both ADIPOR1 and ADIPOR2, activates AMPK and ACC in muscle, and increases expression of genes involved in mitochondrial function and fat oxidation. We hypothesise that the increases in AMPK, ACC, CPT1B and expression of genes involved in mitochondrial function and fat oxidation lead to a decrease in toxic intracellular lipid metabolites (fatty acyl CoA, diacylglycerol, ceramides), which improves insulin signalling in muscle and augments insulin sensitivity (Fig. 4).
Fig. 4.

Proposed mechanism of action of pioglitazone. Pioglitazone increases plasma adiponectin concentration. AMPK and ACC activity is increased following pioglitazone, which is mediated via adiponectin or represents a possible direct effect of pioglitazone on AMPK. Pioglitazone stimulates the expression of PPARGC1A, CPT1B and a number of mRNAs involved in mitochondrial function and NEFA oxidation. P-, phosphorylated; DAG, diacylglycerol; FACoA, fatty acyl CoA
Supplementary Material
Acknowledgments
This work was supported by NIH grant DK24092 (R. A. DeFronzo), a VA Merit Award (R. A. DeFronzo), GCRC grant RR01346, an Executive Research Committee Research Award from the University of Texas Health Science Center at San Antonio (N. Musi), an American Diabetes Association Junior Faculty Award (N. Musi), an American Heart Association National Scientist Development Grant (D. K. Coletta), a grant from Takeda Pharmaceuticals North America and a grant from the Canadian Institute of Health Research (M. Prentki). We thank all the patients who participated in the studies and the Texas Diabetes Institute nurses who helped to perform the studies. We thank N. Cyr for excellent technical assistance.
R. A. DeFronzo has received research grant support from and is a consultant to Takeda Pharmaceuticals. He is a member of the speakers' bureau of Takeda Pharmaceuticals. N. Musi has an affiliation with Takeda Pharmaceuticals.
Abbreviations
- ACC
Acetyl-CoA carboxylase
- ADIPOR1
Adiponectin receptor 1
- ADIPOR2
Adiponectin receptor 2
- AICAR
5-Aminoimidazole 1 carboxamide ribonucleoside
- AMPK
Adenosine 5′-monophosphate-activated protein kinase
- CPT1B
Carnitine palmitoyltransferase 1B
- EGP
Endogenous glucose production
- FPG
Fasting plasma glucose
- FPI
Fasting plasma insulin
- IRI
Insulin resistance index
- PPARG
Peroxisome proliferator-activated receptor gamma
- PPARGC1A
Peroxisome proliferator-activated receptor gamma, coactivator 1 alpha
- Q-RT-PCR
Quantitative real-time PCR
- SIRT1
Sirtuin 1
- TZD
Thiazolidinedione
Footnotes
Trial registration: NCT 00816218
Electronic supplementary material The online version of this article (doi:10.1007/s00125-008-1256-9) contains supplementary material, which is available to authorised users.
Duality of interest: The other authors declare that there is no duality of interest associated with this manuscript.
Contributor Information
D. K. Coletta, Email: Dawn.Coletta@asu.edu, Center for Metabolic Biology, College of Liberal Arts and Sciences, Arizona State University, P.O. Box 873704, Tempe, AZ 85287-3704, USA.
A. Sriwijitkamol, Diabetes Division, University of Texas Health Science Center at San Antonio, San Antonio, TX, USA; Texas Diabetes Institute, San Antonio, TX, USA
E. Wajcberg, Diabetes Division, University of Texas Health Science Center at San Antonio, San Antonio, TX, USA; Texas Diabetes Institute, San Antonio, TX, USA
P. Tantiwong, Diabetes Division, University of Texas Health Science Center at San Antonio, San Antonio, TX, USA; Texas Diabetes Institute, San Antonio, TX, USA
M. Li, Diabetes Division, University of Texas Health Science Center at San Antonio, San Antonio, TX, USA; Texas Diabetes Institute, San Antonio, TX, USA
M. Prentki, Molecular Nutrition Unit, Departments of Nutrition and Biochemistry and the Montreal Diabetes Research Center, University of Montreal and Centre Hospitalier de l'Université de Montréal, Montreal, QC, Canada
M. Madiraju, Molecular Nutrition Unit, Departments of Nutrition and Biochemistry and the Montreal Diabetes Research Center, University of Montreal and Centre Hospitalier de l'Université de Montréal, Montreal, QC, Canada
C. P. Jenkinson, Diabetes Division, University of Texas Health Science Center at San Antonio, San Antonio, TX, USA; South Texas Veterans Health Care System, San Antonio, TX, USA
E. Cersosimo, Diabetes Division, University of Texas Health Science Center at San Antonio, San Antonio, TX, USA; Texas Diabetes Institute, San Antonio, TX, USA
N. Musi, Diabetes Division, University of Texas Health Science Center at San Antonio, San Antonio, TX, USA; Texas Diabetes Institute, San Antonio, TX, USA
R. A. DeFronzo, Diabetes Division, University of Texas Health Science Center at San Antonio, San Antonio, TX, USA; Texas Diabetes Institute, San Antonio, TX, USA; South Texas Veterans Health Care System, San Antonio, TX, USA
References
- 1.DeFronzo RA. Pathogenesis of type 2 diabetes: metabolic and molecular implications for identifying diabetes genes. Diabetes Rev. 1997;5:178–269. [Google Scholar]
- 2.Reaven GM. Banting lecture. Role of insulin resistance in human disease. Diabetes. 1988;37:595–607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 3.Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- 4.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patti ME, Butte AJ, Crunkhorn S, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1 alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 7.Spiegelman BM. PPAR-gamma: adipogenic regulator and thiazolidinedione receptor. Diabetes. 1998;47:507–514. doi: 10.2337/diabetes.47.4.507. [DOI] [PubMed] [Google Scholar]
- 8.Lambe KG, Tugwood JD. A human peroxisome-proliferator-activated receptor-gamma is activated by inducers of adipogenesis, including thiazolidinedione drugs. Eur J Biochem. 1996;239:1–7. doi: 10.1111/j.1432-1033.1996.0001u.x. [DOI] [PubMed] [Google Scholar]
- 9.Maggs DG, Buchanan TA, Burant CF, et al. Metabolic effects of troglitazone monotherapy in type 2 diabetes mellitus. A randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1998;128:176–185. doi: 10.7326/0003-4819-128-3-199802010-00002. [DOI] [PubMed] [Google Scholar]
- 10.Miyazaki Y, Mahankali A, Matsuda M, et al. Improved glycaemic control and enhanced insulin sensitivity in type 2 diabetic subjects treated with pioglitazone. Diabetes Care. 2001;24:710–719. doi: 10.2337/diacare.24.4.710. [DOI] [PubMed] [Google Scholar]
- 11.Mayerson AB, Hundal RS, Dufour S, et al. The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with type 2 diabetes. Diabetes. 2002;51:797–802. doi: 10.2337/diabetes.51.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belfort R, Harrison SA, Brown K, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med. 2006;355:2297–2307. doi: 10.1056/NEJMoa060326. [DOI] [PubMed] [Google Scholar]
- 13.Kim YB, Ciaraldi TP, Kong A, et al. Troglitazone but not metformin restores insulin-stimulated phosphoinositide 3-kinase activity and increases p110beta protein levels in skeletal muscle of type 2 diabetic subjects. Diabetes. 2002;51:443–448. doi: 10.2337/diabetes.51.2.443. [DOI] [PubMed] [Google Scholar]
- 14.Miyazaki Y, He H, Mandarino LJ, DeFronzo RA. Rosiglitazone improves downstream insulin receptor signaling in type 2 diabetic patients. Diabetes. 2003;52:1943–1950. doi: 10.2337/diabetes.52.8.1943. [DOI] [PubMed] [Google Scholar]
- 15.Ruderman N, Prentki M. AMP kinase and malonyl-CoA: targets for therapy of the metabolic syndrome. Nat Rev Drug Discov. 2004;3:340–351. doi: 10.1038/nrd1344. [DOI] [PubMed] [Google Scholar]
- 16.Yamauchi T, Kamon J, Minokoshi Y, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 17.Tomas E, Tsao TS, Saha AK, et al. Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: acetyl-CoA carboxylase inhibition and AMP-activated protein kinase activation. Proc Natl Acad Sci U S A. 2002;99:16309–16313. doi: 10.1073/pnas.222657499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu X, Motoshima H, Mahadev K, Stalker TJ, Scalia R, Goldstein BJ. Involvement of AMP-activated protein kinase in glucose uptake stimulated by the globular domain of adiponectin in primary rat adipocytes. Diabetes. 2003;52:1355–1363. doi: 10.2337/diabetes.52.6.1355. [DOI] [PubMed] [Google Scholar]
- 19.Cusi K, Maezono K, Osman A, et al. Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J Clin Invest. 2000;105:311–320. doi: 10.1172/JCI7535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeFronzo RA, Tobin R, Andres R. The glucose clamp technique. A method for quantifying insulin secretion and resistance. Am J Physiol. 1979;6:214–223. doi: 10.1152/ajpendo.1979.237.3.E214. [DOI] [PubMed] [Google Scholar]
- 21.Roduit R, Nolan C, Alarcon C, et al. A role for the malonyl-CoA/long-chain acyl-CoA pathway of lipid signaling in the regulation of insulin secretion in response to both fuel and nonfuel stimuli. Diabetes. 2004;53:1007–1019. doi: 10.2337/diabetes.53.4.1007. [DOI] [PubMed] [Google Scholar]
- 22.Steele R. Influences of glucose loading and of injected insulin on hepatic glucose output. Ann N Y Acad Sci. 1959;82:420–430. doi: 10.1111/j.1749-6632.1959.tb44923.x. [DOI] [PubMed] [Google Scholar]
- 23.Miyazaki Y, Mahankali A, Wajcberg E, Bajaj M, Mandarino LJ, DeFronzo RA. Effect of pioglitazone on circulating adipocytokine levels and insulin sensitivity in type 2 diabetic patients. J Clin Endocrinol Metab. 2004;89:4312–4319. doi: 10.1210/jc.2004-0190. [DOI] [PubMed] [Google Scholar]
- 24.Bajaj M, Suraamornkul S, Hardies LJ, Glass L, Musi N, DeFronzo RA. Effects of peroxisome proliferator-activated receptor (PPAR)-alpha and PPAR-gamma agonists on glucose and lipid metabolism in patients with type 2 diabetes mellitus. Diabetologia. 2007;50:1723–1731. doi: 10.1007/s00125-007-0698-9. [DOI] [PubMed] [Google Scholar]
- 25.Rasouli N, Raue U, Miles LM, et al. Pioglitazone improves insulin sensitivity through reduction in muscle lipid and redistribution of lipid into adipose tissue. Am J Physiol Endocrinol Metab. 2005;288:930–934. doi: 10.1152/ajpendo.00522.2004. [DOI] [PubMed] [Google Scholar]
- 26.Yu JG, Javorschi S, Hevener AL, et al. The effect of thiazolidinediones on plasma adiponectin levels in normal, obese, and type 2 diabetic subjects. Diabetes. 2002;51:2968–2974. doi: 10.2337/diabetes.51.10.2968. [DOI] [PubMed] [Google Scholar]
- 27.Konrad D, Rudich A, Bilan PJ, et al. Troglitazone causes acute mitochondrial membrane depolarisation and an AMPK-mediated increase in glucose phosphorylation in muscle cells. Diabetologia. 2005;48:954–966. doi: 10.1007/s00125-005-1713-7. [DOI] [PubMed] [Google Scholar]
- 28.LeBrasseur NK, Kelly M, Tsao TS, et al. Thiazolidinediones can rapidly activate AMP-activated protein kinase in mammalian tissues. Am J Physiol Endocrinol Metab. 2006;291:175–181. doi: 10.1152/ajpendo.00453.2005. [DOI] [PubMed] [Google Scholar]
- 29.Ye JM, Dzamko N, Hoy AJ, Iglesias MA, Kemp B, Kraegen E. Rosiglitazone treatment enhances acute AMP-activated protein kinase-mediated muscle and adipose tissue glucose uptake in high-fat-fed rats. Diabetes. 2006;55:2797–2804. doi: 10.2337/db05-1315. [DOI] [PubMed] [Google Scholar]
- 30.Tan GD, Fielding BA, Currie JM, et al. The effects of rosiglitazone on fatty acid and triglyceride metabolism in type 2 diabetes. Diabetologia. 2005;48:83–95. doi: 10.1007/s00125-004-1619-9. [DOI] [PubMed] [Google Scholar]
- 31.Kim H, Haluzik M, Gavrilova O, et al. Thiazolidinediones improve insulin sensitivity in adipose tissue and reduce the hyperlipidaemia without affecting the hyperglycaemia in a transgenic model of type 2 diabetes. Diabetologia. 2004;47:2215–2225. doi: 10.1007/s00125-004-1581-6. [DOI] [PubMed] [Google Scholar]
- 32.Hammarstedt A, Sopasakis VR, Gogg S, Jansson PA, Smith U. Improved insulin sensitivity and adipose tissue dysregulation after short-term treatment with pioglitazone in non-diabetic, insulin-resistant subjects. Diabetologia. 2005;48:96–104. doi: 10.1007/s00125-004-1612-3. [DOI] [PubMed] [Google Scholar]
- 33.Nawrocki AR, Rajala MW, Tomas E, et al. Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor gamma agonists. J Biol Chem. 2006;281:2654–2660. doi: 10.1074/jbc.M505311200. [DOI] [PubMed] [Google Scholar]
- 34.Civitarese AE, Jenkinson CP, Richardson D, et al. Adiponectin receptors gene expression and insulin sensitivity in non-diabetic Mexican Americans with or without a family history of type 2 diabetes. Diabetologia. 2004;47:816–820. doi: 10.1007/s00125-004-1359-x. [DOI] [PubMed] [Google Scholar]
- 35.Li W, Tonelli J, Kishore P, et al. Insulin-sensitizing effects of thiazolidinediones are not linked to adiponectin receptor expression in human fat or muscle. Am J Physiol Endocrinol Metab. 2007;292:1301–1307. doi: 10.1152/ajpendo.00312.2006. [DOI] [PubMed] [Google Scholar]
- 36.Tan GD, Debard C, Funahashi T, et al. Changes in adiponectin receptor expression in muscle and adipose tissue of type 2 diabetic patients during rosiglitazone therapy. Diabetologia. 2005;48:1585–1589. doi: 10.1007/s00125-005-1835-y. [DOI] [PubMed] [Google Scholar]
- 37.Civitarese AE, Ukropcova B, Carling S, et al. Role of adiponectin in human skeletal muscle bioenergetics. Cell Metabolism. 2006;4:75–87. doi: 10.1016/j.cmet.2006.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bogacka I, Xie H, Bray GA, Smith SR. Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes. 2005;54:1392–1399. doi: 10.2337/diabetes.54.5.1392. [DOI] [PubMed] [Google Scholar]
- 39.Richardson DK, Kashyap S, Bajaj M, et al. Lipid infusion decreases the expression of nuclear encoded mitochondrial genes and increases the expression of extracellular matrix genes in human skeletal muscle. J Biol Chem. 2005;280:10290–10297. doi: 10.1074/jbc.M408985200. [DOI] [PubMed] [Google Scholar]
- 40.Skov V, Glintborg D, Knudsen S, et al. Pioglitazone enhances mitochondrial biogenesis and ribosomal protein biosynthesis in skeletal muscle in polycystic ovary syndrome. PLoS ONE. 2008;3:e2466. doi: 10.1371/journal.pone.0002466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boden G, Homko C, Mozzoli M, Showe LC, Nichols C, Cheung P. Thiazolidinediones upregulate fatty acid uptake and oxidation in adipose tissue of diabetic patients. Diabetes. 2005;54:880–885. doi: 10.2337/diabetes.54.3.880. [DOI] [PubMed] [Google Scholar]
- 42.Schrauwen-Hinderling VB, Mensink M, Hesselink MK, Sels JP, Kooi ME, Schrauwen P. The insulin-sensitizing effect of rosiglitazone in type 2 diabetes mellitus patients does not require improved in vivo muscle mitochondrial function. J Clin Endocrinol Metab. 2008;93:2917–2921. doi: 10.1210/jc.2008-0267. [DOI] [PubMed] [Google Scholar]
- 43.Civitarese AE, Carling S, Heilbronn LK, et al. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 2007;4:e76. doi: 10.1371/journal.pmed.0040076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coletta DK, Balas B, Chavez AO, et al. Effect of acute physiological hyperinsulinemia on gene expression in human skeletal muscle in vivo. Am J Physiol Endocrinol Metab. 2008;294:910–917. doi: 10.1152/ajpendo.00607.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vavvas D, Apazidis A, Saha AK, et al. Contraction-induced changes in acetyl-CoA carboxylase and 5′-AMP-activated kinase in skeletal muscle. J Biol Chem. 1997;272:13255–13261. doi: 10.1074/jbc.272.20.13255. [DOI] [PubMed] [Google Scholar]
- 46.Merrill GF, Kurth EJ, Hardie DG, Winder WW. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am J Physiol. 1997;273:1107–1112. doi: 10.1152/ajpendo.1997.273.6.E1107. [DOI] [PubMed] [Google Scholar]
- 47.Bandyopadhyay GK, Yu JG, Ofrecio J, Olefsky JM. Increased malonyl-CoA levels in muscle from obese and type 2 diabetic subjects lead to decreased fatty acid oxidation and increased lipogenesis; thiazolidinedione treatment reverses these defects. Diabetes. 2006;55:2277–2285. doi: 10.2337/db06-0062. [DOI] [PubMed] [Google Scholar]
- 48.Højlund K, Glintborg D, Andersen NR, et al. Impaired insulin-stimulated phosphorylation of Akt and AS160 in skeletal muscle of women with polycystic ovary syndrome is reversed by pioglitazone treatment. Diabetes. 2008;57:357–366. doi: 10.2337/db07-0706. [DOI] [PubMed] [Google Scholar]
- 49.Dean D, Daugaard JR, Young ME, et al. Exercise diminishes the activity of acetyl-CoA carboxylase in human muscle. Diabetes. 2000;49:1295–1300. doi: 10.2337/diabetes.49.8.1295. [DOI] [PubMed] [Google Scholar]
- 50.Joly E, Bendayan M, Roduit R, Saha AK, Ruderman NB, Prentki M. Malonyl-CoA decarboxylase is present in the cytosolic, mitochondrial and peroxisomal compartments of rat hepatocytes. FEBS Lett. 2005;579:6581–6586. doi: 10.1016/j.febslet.2005.10.050. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.