

Abstract
The detection of microbes and initiation of an innate immune response occur through pattern recognition receptors (PRRs), which are critical for the production of inflammatory cytokines and activation of the cellular microbicidal machinery. In particular, the production of reactive oxygen species (ROS) by the NADPH oxidase complex is a critical component of the macrophage bactericidal machinery. We previously characterized brain-specific angiogenesis inhibitor 1 (BAI1), a member of the adhesion family of G protein (heterotrimeric guanine nucleotide–binding protein)–coupled receptors (GPCRs), as a PRR that mediates the selective phagocytic uptake of Gram-negative bacteria by macrophages. We showed that BAI1 promoted phagosomal ROS production through activation of the Rho family guanosine triphosphatase (GTPase) Rac1, thereby stimulating NADPH oxidase activity. Primary BAI1-deficient macrophages exhibited attenuated Rac GTPase activity and reduced ROS production in response to several Gram-negative bacteria, resulting in impaired microbicidal activity. Furthermore, in a peritoneal infection model, BAI1-deficient mice exhibited increased susceptibility to death by bacterial challenge because of impaired bacterial clearance. Together, these findings suggest that BAI1 mediates the clearance of Gram-negative bacteria by stimulating both phagocytosis and NADPH oxidase activation, thereby coupling bacterial detection to the cellular microbicidal machinery.
INTRODUCTION
The innate immune system relies upon the ability of the host to detect and respond to both pathogenic and nonpathogenic microbes. Detection occurs through a limited set of germ line–encoded receptors called pattern recognition receptors (PRRs) (1, 2). The coordinated actions of these innate receptors drive the activity and specificity of the host response, and loss of individual receptors can have devastating consequences on innate immunity (3–5). Macrophages and monocytes interpret the signals from PRRs to couple microbial detection to phagocytic, microbicidal, and cell signaling machinery, which results in local inflammatory responses and bacterial clearance (6, 7). Phagocytic receptors, such as the C-type lectin receptors (8) mannose receptor (9) and Dectin-1 (10) and the scavenger receptors (11) CD36 (12) and MARCO (13), mediate the internalization of microbes from the extracellular space and their delivery to highly degradative compartments within the cell, resulting in bacterial killing and antigen processing for the generation of an adaptive immune response (14). These phagocytic receptors are crucial for innate bactericidal activity and for the compartmentalization and presentation of bacterial ligands to other PRRs, such as Toll-like receptors (TLRs) (14–16).
Brain-specific angiogenesis inhibitor 1 [BAI1; also known as adhesion G protein (heterotrimeric guanine nucleotide–binding protein)–coupled receptor B1] is a member of subgroup VII of the adhesion-type G protein– coupled receptors (GPCRs), which was originally identified for a role in inhibiting angiogenesis in brain tumor models (17). BAI1 was also recognized as a phagocytic receptor for apoptotic cells, mediating apoptotic cell clearance by several cell types, including neurons, myoblasts, epithelial cells, and myeloid lineage cells (18–21). We and others reported that, in addition to recognizing apoptotic cells, BAI1 also recognizes Gram-negative bacteria (20, 22). In this context, BAI1 recognizes the core oligosaccharide of bacterial lipopolysaccharide (LPS) through a series of five type 1 thrombospondin repeats in the extracellular domain (22). Binding of either apoptotic cells or Gram-negative bacteria to the extracellular domain of BAI1 stimulates the rapid rearrangement of the actin cytoskeleton, which culminates in phagocytosis of the bound particle. In this mechanism, the cytoplasmic domain of BAI1 interacts directly with the engulfment and cell motility protein (ELMO) and Dock180, which together function as a bipartite guanine nucleotide exchange factor (GEF) that activates the Rho family guanosine triphosphatase (GTPase) Rac1 (18, 22).
In addition to its role in phagocytosis (23, 24), Rac is also a critical part of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex, a key component of the antimicrobial reactive oxygen species (ROS) response (25–27). Active, guanosine triphosphate (GTP)–bound Rac is required for the assembly of the cytosolic regulatory subunits with the transmembrane catalytic subunit gp91phox (28–30). The activation of NADPH oxidase was characterized downstream of the opsonic phagocytic receptors Fc-γ receptor (FcγR) and complement receptor (CR), but its activation in response to nonopsonized Gram-negative bacteria is poorly understood. Here, we showed that BAI1 not only mediated the capture and internalization of several species of Gram-negative bacteria by macrophages but also enhanced oxidative killing in a Rac-dependent manner. We also showed that BAI1 mediated bacterial clearance in vivo, in a mouse model of peritoneal challenge. Together, these results suggest that BAI1 functions as a critical phagocytic PRR in the host response to Gram-negative bacteria.
RESULTS
BAI1 mediates binding and uptake of Gram-negative bacteria in primary macrophages
We previously showed that BAI1 mediates the binding and uptake of Gram-negative bacteria in several cell culture model systems (22). Consistent with our earlier studies, we found that fibroblasts [LR73 Chinese hamster ovary (CHO) cells] expressing exogenous BAI internalized Escherichia coli strain DH5α more efficiently than did control, non–BAI1-expressing cells (Fig. 1A). To test the function of endogenous BAI1 in bacterial recognition, we compared primary bone marrow–derived macrophages (BMDMs) from wild-type C57BL/6 mice to cells derived from BAI1-deficient mice (19). For this purpose, bacteria were centrifuged onto monolayers of macrophages at 4°C for 5 min to enable binding, and then the cells were warmed to 37°C for an additional 30 min to enable internalization. We used an immunofluorescence-based assay to distinguish extracellular from intracellular bacteria by specifically labeling extracellular bacteria before cell permeabilization (Fig. 1B). In this assay, the total number of E. coli associated with BAI1-deficient BMDMs was reduced by about 30% relative to that associated with BAI1-expressing control macrophages (Fig. 1, C and D). We found that although the surface binding of E. coli DH5α was not statistically significantly different between wild-type and BAI1-deficient macrophages (Fig. 1, C and D, white arrowheads), internalization was reduced by ~50% in the absence of BAI1 (Fig. 1C, white arrows). This observation suggests that BAI1-mediated uptake contributes substantially to bacterial phagocytosis in primary macrophages.
Fig. 1. BAI1 mediates the binding and uptake of Gram-negative bacteria by primary macrophages.
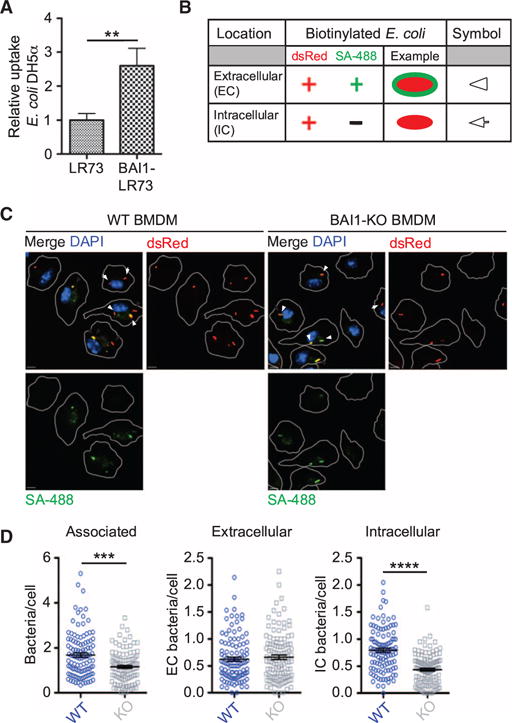
(A) The internalization of E. coli DH5α was measured in parental LR73 CHO cells and cells stably expressing exogenous BAI1 using the gentamicin protection assay as described in Materials and Methods. Data are mean fold internalization ± SEM of 10 experiments. **P < 0.01 by Mann-Whitney test. (B) Schematic of the immunofluorescence-based internalization assay. Wild-type (WT) and BAI1 knockout (BAI1-KO) BMDMs were incubated with biotinylated E. coli DH5α expressing dsRed at a multiplicity of infection (MOI) of 10 for 30 min. Cells were washed and fixed, but not permeabilized, and extracellular bacteria were labeled with Alexa Fluor 488–conjugated streptavidin (SA; green). Nuclei were labeled with 4′,6-diamidino-2-phenylindole (DAPI) (blue). In this assay, intracellular bacteria appear red (indicated by arrows), whereas extracellular bacteria appear yellow (marked by arrowheads). (C) Representative images of WT and BAI1-KO BMDMs from the immunofluorescence-based internalization assay. Scale bars, 5 μm. (D) Quantification of total cell-associated bacteria (left), extracellular bacteria (center), and intracellular bacteria (right) per cell from the experiments shown in (C). At least 125 cells per experiment were imaged, and five experiments were performed. Plots show the numbers of bacteria per cell per frame ± SEM. ***P < 0.001, ****P < 0.0001 by Mann-Whitney test.
BAI1 is recruited to sites of bacterial internalization in macrophages
We next analyzed the cellular localization of BAI1 during bacterial recognition by confocal microscopy and live-cell imaging. Because of the poor quality of existing anti-BAI1 antibodies, we used BMDMs derived from transgenic mice expressing a BAI1 construct containing an N-terminal extracellular hemagglutinin (HA) tag (31). In uninfected macrophages, BAI1 was present on the plasma membrane and in the perinuclear region in a punctate distribution, consistent with previous reports (Fig. 2A) (18, 32). Macrophages incubated with the Gram-positive pathogen Staphylococcus aureus showed very little association or enrichment with BAI1, whereas incubation with E. coli for 30 min resulted in substantial clustering of BAI1 around associated bacteria (Fig. 2, B and C, white arrows). Because these sites were also labeled with the plasma membrane marker wheat germ agglutinin (WGA), we interpret these sites to be either phagocytic cups or phagosomes.
Fig. 2. BAI1 is recruited to sites of bacterial engulfment.
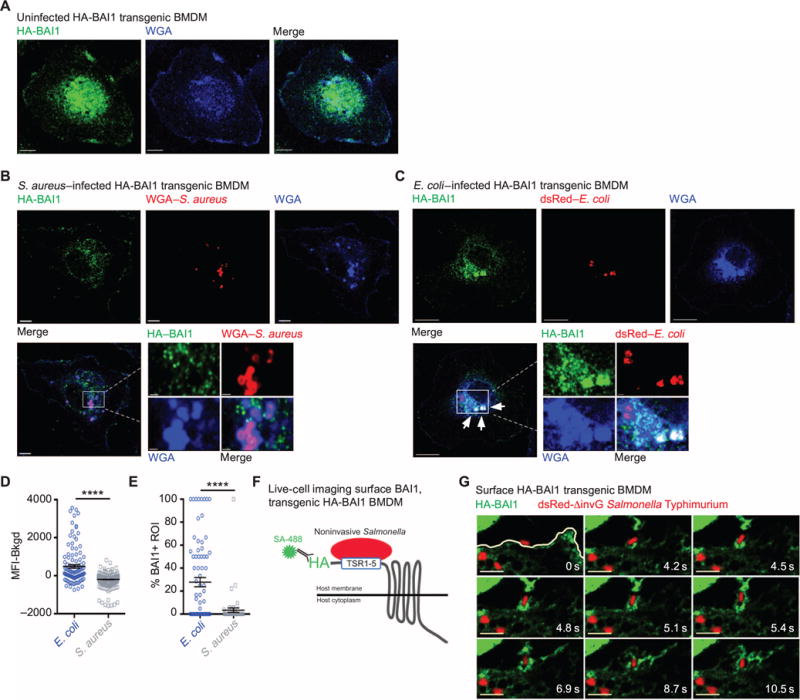
(A) Transgenic BMDMs expressing HA-BAI1 were fixed and stained with anti-HA antibody (green). The plasma membrane was labeled with WGA (blue), and cells were imaged by confocal microscopy. The representative image shows a single confocal section. Scale bars, 5 μm. (B and C) BMDMs expressing transgenic HA-BAI1 were infected for 30 min with either S. aureus (B) or E. coli (C) at an MOI of 10. The images show a single confocal section. The boxed areas of the merged images are magnified. White arrows indicate BAI1-positive bacteria. Scale bars, 5 μm; inset scale bars, 1 μm. (D) Quantification of the mean fluorescence intensity (MFI) of HA-BAI1 associated with bacteria. At least seven cells per condition per experiment were analyzed from a total of three experiments. A region of interest (ROI) was drawn around each bacterium, and the MFI was measured within the ROI (for details see Materials and Methods). Plot shows the MFI ± SEM of HA-BAI1 per ROI after subtraction of background MFI (Bkgd). ****P < 0.0001 by Mann-Whitney test. (E) Percentage of bacteria enriched for HA-BAI1. At least seven cells per condition were imaged. Plot shows the percentage of bacteria with an HA-BAI1 signal that was more than twofold greater than that of the background per cell ± SEM from three experiments. ****P < 0.0001 by Mann-Whitney test. (F) Schematic of the protocol for live-cell imaging analysis of BAI1 distribution. BMDMs expressing transgenic HA-BAI1 were incubated with fluorescently conjugated anti-HA antibody (green) to label extracellular receptors and then were incubated with noninvasive S. Typhimurium (ΔinvG) expressing dsRed. (G) Images from movie S1 are shown as a time lapse series. The white line indicates the cell periphery. Movies were generated for at least two cells from two separate experiments. Scale bars, 5 μm.
The extent of the association of BAI1 with S. aureus or E. coli was quantified in two ways. First, we determined the MFI of BAI1 at sites of bacterial association. The MFI of BAI1 associated with E. coli was statistically significantly higher than that with S. aureus (Fig. 2D). Similarly, the percentage of bacteria enriched for BAI1 was 10-fold higher for E. coli than for S. aureus (Fig. 2E). Although the overall cellular distribution of BAI1 did not change in response to infection (fig. S1), these results indicate a preferential recruitment of BAI1 to sites of interaction with Gram-negative E. coli relative to sites of interaction with the Gram-positive S. aureus. Consistent with this observation, live-cell imaging indicated that BAI1 was concentrated at sites of bacterial attachment (Fig. 2, F and G, and movie S1) and that it remained associated with bacteria during internalization. Together, these findings suggest that BAI1 preferentially recognizes Gram-negative bacteria at the macrophage plasma membrane.
BAI1 ligation stimulates cellular microbicidal activity
The route of cellular entry can markedly affect microbe survival, immune responses, and antigen processing (14, 33). Indeed, several bacterial pathogens target specific receptors during infection to alter cellular responses and compartmentalization within macrophages (34–37). Although somewhat controversial, a large body of evidence suggests that the specific subset of innate immune receptors, such as TLRs, engaged during recognition and uptake can affect phagosome maturation and particle fate (7, 38, 39). To determine whether the recognition and internalization of Gram-negative bacteria by BAI1 affected their survival, we examined intracellular microbicidal activity in primary macrophages and cell lines with a standard gentamicin protection assay. In this assay, cells were allowed to internalize bacteria for 30 min and then were chased for up to 7 hours in the presence of gentamicin, which kills extracellular, but not intracellular, bacteria. We found that BAI1-deficient BMDMs were attenuated in their ability to kill two different strains of E. coli (Fig. 3, A and B) and two Gram-negative bacterial pathogens, Salmonella Typhimurium and Pseudomonas aeruginosa (Fig. 3, C and D). Consistent with our earlier data (Fig. 2), loss of BAI1 did not affect bactericidal activity against S. aureus (Fig. 3E). Similar results were observed in peritoneal macrophages (PEMs) from wild-type and BAI1-deficient mice (Fig. 3, Fand G) and BAI1-depleted J774 cells (a macrophage cell line) (fig. S2, A to C). Although the magnitude and kinetics of bacterial killing at earlier time points were affected by the loss of BAI1, differences at later time points were not as pronounced. This presumably reflects the activity of other bactericidal machinery, including antimicrobial peptides or nitric oxide. Together, these observations suggest that BAI1 not only mediates bacterial internalization but also selectively promotes microbicidal activity against Gram-negative bacteria in infected macrophages.
Fig. 3. Intracellular killing of Gram-negative bacteria is increased by BAI1-mediated bacterial recognition.
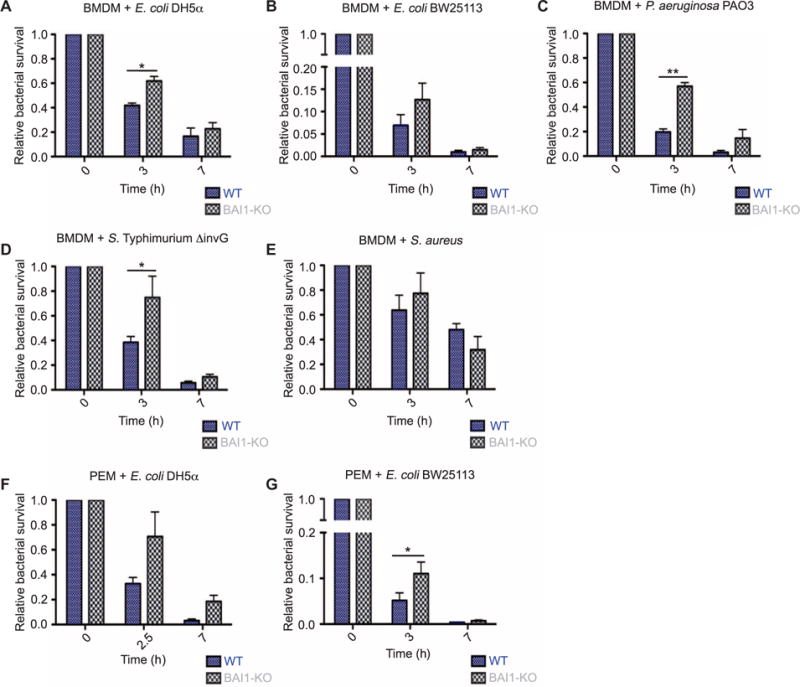
(A) WT and BAI1-KO BMDMs were incubated for 30 min with E. coli DH5α at an MOI of 25 (t = 0) and then chased in the presence of gentamicin for the indicated times to kill extracellular bacteria. Lysates were then plated to count viable intracellular bacteria. Survival is shown relative to the bacterial counts at t = 0. All graphs display relative mean ± SEM of at least three independent experiments. Data were analyzed by two-way analysis of variance (ANOVA) with Bonferroni post hoc comparisons. P values describe the source of variation in the data set (for example, cell genotype, time, or an interaction between the cell genotype and time, which can also be considered as kinetics). Statistical information in the figure shows the results from the post hoc comparison (cell, P < 0.05; time, P < 0.001; n = 3). (B to E) Intracellular bactericidal activity by BMDMs from the indicated mice against Gram-negative bacteria was measured as described in (A). These included E. coli BW25113 (time, P <0.01; n = 4) (B), P. aeruginosa (cell, P < 0.05; time, P < 0.001; n = 3) (C), noninvasive S. Typhimurium (ΔinvG) (time, P < 0.001; n = 3) (D), and the Gram-positive S. aureus (time, P < 0.05; n = 4) (E). (F and G) The survival of intracellular (F) E. coli DH5α (cell, P < 0.05; time, P < 0.01; n = 3) and (G) E. coli BW25113 (time, P < 0.001; n = 4) in PEMs from the indicated mice was measured as described in (A).
BAI1-mediated internalization of Gram-negative bacteria occurred rapidly after infection. Because the difference in bacterial survival between wild-type and BAI1-deficient cells was reduced at later time points, we hypothesized that BAI1-mediated bactericidal activity occurred earlier. To test this hypothesis, we examined microbicidal activity over a short time course in which bacteria were internalized for 15 min, washed, and then chased to 30 or 60 min. Viable associated bacteria were then quantified by colony-forming assays. BAI1-deficient BMDMs displayed statis tically significantly attenuated bactericidal activity at both 30 and 60 min against nonpathogenic E. coli (Fig. 4A). Similarly, decreased microbicidal activity in BMDMs lacking BAI1 was also observed against the pathogens P. aeruginosa and two strains of Burkholderia cenocepacia (Fig. 4, B to D). Bactericidal activity against cell-associated S. aureus at early time points was minimal and did not differ between wild-type and BAI1-deficient cells (Fig. 4E).
Fig. 4. Early microbicidal activity against Gram-negative bacteria is enhanced by BAI1 in macrophages.
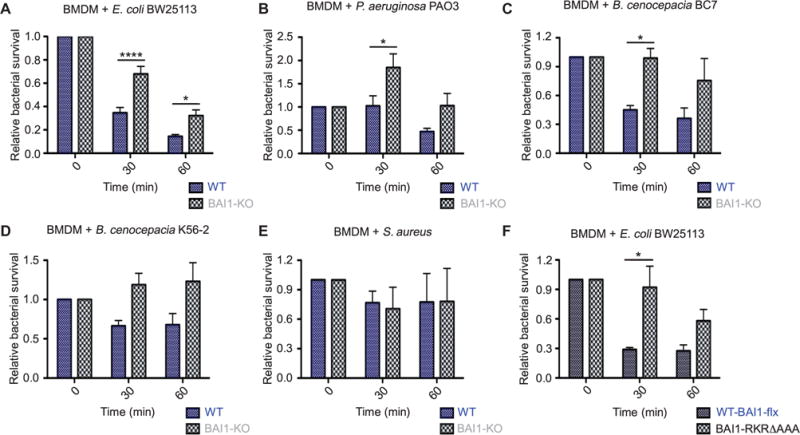
(A) WT and BAI1-KO BMDMs were incubated for 15 min with E. coli BW25113 at an MOI of 25. After extensive washing, cells were either lysed immediately (t = 0) or were chased in complete medium for 30 or 60 min. For each time point, lysates were plated on LB agar to enumerate viable bacteria. Survival is shown relative to total cell-associated bacteria at t = 0. All graphs display relative means ± SEM. Data were analyzed by two-way ANOVA with Bonferroni post hoc comparisons (cell, P < 0.0001; time, P < 0.0001; n = 8). (B to E) Cell-associated bactericidal activity of BMDMs from the indicated mice against P. aeruginosa PAO3 (cell, P < 0.01; time, P < 0.01; n = 5) (B), B. cenocepacia BC7 (cell, P < 0.01; n = 4) (C), B. cenocepacia K56-2 (cell, P < 0.01; n = 5) (D), and S. aureus (n = 5) (E) was measured as described in (A). (F) WT-flx and transgenic BAI1-RKR-AAA BMDMs were incubated with E. coli BW251113 at an MOI of 25. Bacterial killing was measured as described in (A) (cell, P < 0.01; n = 3).
We previously showed that BAI1 mediates the internalization of Gram-negative bacteria by signaling through the ELMO-Dock complex, which leads to activation of the Rho family GTPase Rac1. Macrophages depleted of either BAI1 or ELMO1 are similarly impaired in their ability to internalize noninvasive S. Typhimurium (ΔinvG), and CHO cells expressing a BAI1 mutant, BAI1-R1489KR-AAA, which is unable to couple to the ELMO-Dock complex, show impaired internalization of bacteria relative to that by cells expressing wild-type BAI1 (18, 22). To determine whether BAI1-mediated Rac1 activation contributed to the difference in bactericidal activity observed in wild-type macrophages compared to that in BAI1-deficient macrophages, we isolated BMDMs from knock-in mice expressing an HA-tagged form of this BAI1 mutant (HA–BAI1-R1489KR-AAA) (31). These cells exhibited attenuated microbicidal activity that was quantitatively similar to that of cells deficient in BAI1 (compare Fig. 4A to Fig. 4F). These results suggest that BAI1-dependent bactericidal activity is dependent on the ELMO-Dock–mediated activation of Rac1.
BAI1-mediated Rac activation is enhanced in macrophages in response to bacterial infection
We previously showed that cells overexpressing BAI1 exhibit increased Rac activity in response to the Gram-negative pathogen S. Typhimurium and that altering the ability of BAI1 to interact with the ELMO-Dock GEF complex inhibits Rac activation and phagocytosis (18, 22), as described earlier. To confirm that endogenous BAI1 was required for Rac activation in response to Gram-negative bacteria, we measured Rac activity in BMDMs with a well-characterized pull-down assay (40). Incubation of wild-type BMDMs with E. coli led to robust activation of Rac1 within 30 min (Fig. 5, A and B). In contrast, no detectable increase in Rac1 activation was observed in BMDMs lacking BAI1. Similar results were obtained with BMDMs that had been primed with interferon-γ (IFN-γ) (fig. S3, A and B). BAI1-deficient macrophages were not inherently defective in priming, because signaling in response to IFN-γ, as determined by measuring the phosphorylation of signal transducer and activator of transcription 1, was comparable between wild-type and BAI1-deficient cells (fig. S4A). These results suggest that endogenous BAI1 is required for the activation of Rac in response to Gram-negative bacteria.
Fig. 5. Loss of BAI1 impairs Rac activation in response to E. coli.
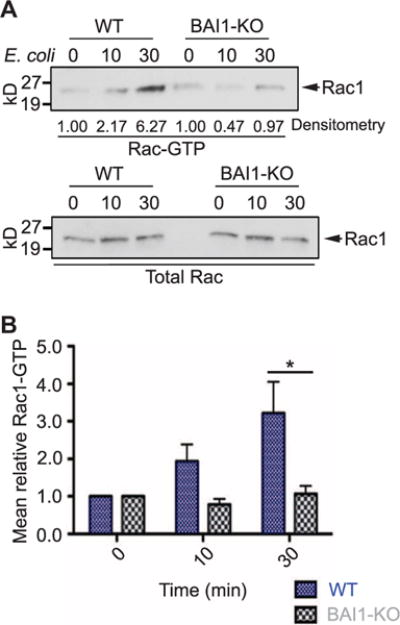
(A and B) Rac1 activation was measured by a standard pull-down assay. Unprimed BMDMs were incubated with E. coli BW25113 for 10 or 30 min. Cells were then lysed, and GTP-bound Rac was precipitated with glutathione S-transferase (GST)–p21-binding domain (PBD) beads as described in Materials and Methods. Precipitates were then analyzed by Western blotting to detect Rac1. Band intensities were quantified by densitometry. Aliquots of each cell lysate were analyzed by Western blotting for total Rac1 (bottom) to demonstrate equal total Rac1 protein in control and BAI1-KO lysates. (B) Quantitation of Western blotting data from six separate experiments. Data are mean fold changes in Rac1-GTP abundance ± SEM. Two-way ANOVA with Bonferroni post hoc comparison was used for analysis (cell, P < 0.05).
ROS production in response to Gram-negative bacteria is regulated by BAI1
As professional phagocytes, macrophages use multiple mechanisms to kill bacteria, including the production of ROS and reactive nitrogen species (RNS) (41). Macrophages use two primary systems to generate ROS for oxidative killing: mitochondria and the phagocyte NADPH oxidase (25, 26, 42–44). In the case of NADPH oxidase, upstream signaling initiates phosphorylation of the cytoplasmic regulatory subunit p47phox, which associates with two other cytosolic proteins, p67phox and p40phox (28). Assembly of this cytosolic complex on the phagosomal membrane and activation of the membrane-associated catalytic subunits gp91phox and p22phox require the activation of Rac1, Rac2, or both (30, 45). Whereas Rac2 is the predominant activating form of Rac in neutrophils (46), Rac1 is critical for ROS responses in macrophages (47–49). Our observations that BAI1 is required for Rac activation in response to Gram-negative bacteria and that microbicidal activity is reduced in BAI1-deficient cells suggested that BAI1 may stimulate ROS production upon binding to Gram-negative bacteria.
To test this hypothesis, we measured ROS production in IFN-γ–primed wild-type and BAI1-deficient BMDMs in a luminol-dependent chemiluminescence (LDCL) assay. We found that incubation of wild-type macrophages with E. coli induced the rapid and robust production of ROS, which was completely blocked by the pharmacological NADPH oxidase inhibitor diphenyleneiodonium (DPI) (Fig. 6, A and B). In contrast, ROS production was attenuated in cells lacking BAI1. Although the kinetics of activation were different, the ROS responses to two other Gram-negative bacterial pathogens, P. aeruginosa and B. cenocepacia, were attenuated in BAI1-deficient cells (Fig. 6, C to F). For comparison, no defect in ROS production was observed when macrophages were incubated with the Gram-positive bacterium S. aureus (Fig. 6, G and H) or with the phorbol ester phorbol myristate acetate (PMA) (Fig. 6, I and J). Furthermore, macrophages derived from gp91phox-deficient mice, which are completely defective in phagocyte NADPH oxidase activity, showed no detectable ROS generation in response to E. coli (Fig. 6, K and L). Similar results were observed in an in situ fluorescence assay with CellROX Green, a fluorescent ROS reporter (fig. S4B). ROS production in BAI1-deficient macrophages incubated with E. coli was reduced nearly to the level of that in gp91phox knockout cells (fig. S4, C and D). Whereas macrophage generation of ROS occurs within minutes of bacterial detection (50), generation of RNS requires the production of inducible nitric oxide synthase (iNOS), which occurs substantially later (51–53). We found that cellular iNOS protein was similarly produced in wild-type and BAI1 knockout macrophages after 6 hours of exposure to E. coli, indicating that iNOS production did not require BAI1 (fig. S4E).
Fig. 6. BAI1-deficient macrophages exhibit attenuated ROS production in response to Gram-negative bacteria.
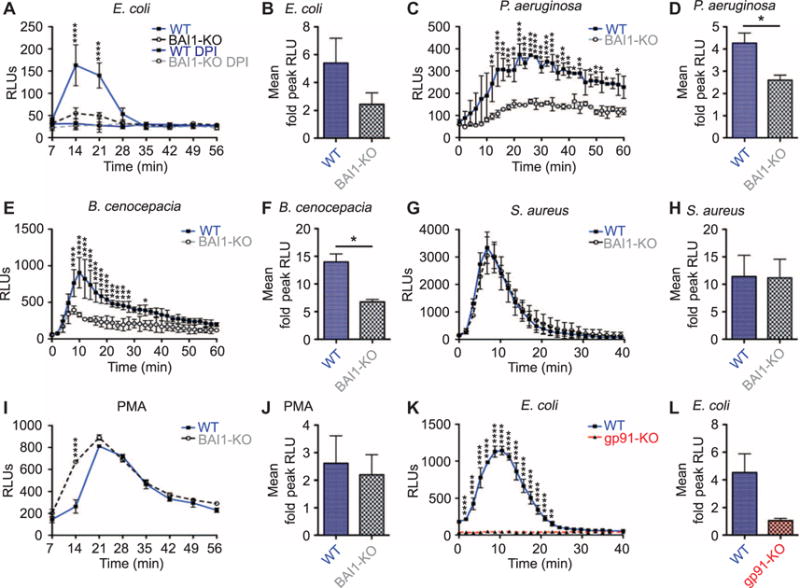
(A) LDCL assays were performed to measure ROS production by WT and BAI1-KO BMDMs after incubation with E. coli BW25113. DPI (10 μM) was added to replicate wells to inhibit NADPH oxidase activity. Graph shows a representative example of ROS activity and kinetics as mean relative light units (RLUs) ± SEM. Repeated-measures two-way ANOVA with Bonferroni post hoc comparison was used for analysis (interaction, P < 0.0001; cell, P < 0.0001; time, P < 0.0001). (B) The mean fold change in peak ROS production ± SEM of WT or BAI1-KO BMDMs treated with E. coli BW25113 from eight experiments was analyzed by Student’s t test. (C to J) BMDMs from WT or BAI1-KO mice were treated with the indicated inflammatory stimuli and analyzed as described in (A) and (B). The stimuli are listed, followed by the corresponding analysis of a representative experiment and the mean fold change in peak ROS production. (C) P. aeruginosa: interaction, P < 0.01; cell, P < 0.01; time, P <0.0001. (D) *P < 0.05; n = 5. (E) B. cenocepacia: interaction, P < 0.0001; cell, P < 0.05; time, P < 0.0001. (F) *P < 0.05; n = 2. (G) S. aureus: time, P < 0.0001. (H) n = 5. (I) PMA: interaction, P < 0.0001; cell, P < 0.01; time, P < 0.0001. (J) n = 4. (K) ROS was measured in WT or gp91phox-KO BMDMs incubated with E. coli BW25113 using LDCL and analyzed as described in (A) (interaction, P < 0.0001; cell, P < 0.0001; time, P < 0.0001). (L) The mean fold change in peak ROS production ± SEM from three experiments is shown for WT and gp91phox-KO BMDMs treated with E. coli BW25113. Data were analyzed by Student’s t test.
BAI1-mediated ROS responses result in the enhanced microbicidal activity of macrophages
To determine the extent to which BAI1-mediated bactericidal activity depended on ROS, we treated control and BAI1-deficient macrophages with the ROS scavenger N-acetylcysteine (NAC) and measured bacterial survival. Treatment of infected wild-type macrophages with NAC increased bacterial survival to an extent observed in BAI1-deficient cells (Fig. 7A). Moreover, treatment of BAI1-deficient cells with NAC did not further improve bacterial survival, confirming that the extent of ROS-derived killing at this time point in the absence of BAI1 was negligible. Similar results were observed with gp91phox-deficient macrophages, which showed defects in early microbicidal activity, but no change in bacterial killing in the presence of NAC (Fig. 7, B and C). In contrast, treatment of cells with the mitochondrial ROS inhibitor MitoTEMPO (54) had no statistically significant effect on bactericidal activity (fig. S5), indicating that most of the early microbicidal ROS was derived from the phagosomal NADPH oxidase complex.
Fig. 7. ROS-mediated microbicidal activity in BAI1-expressing macrophages.
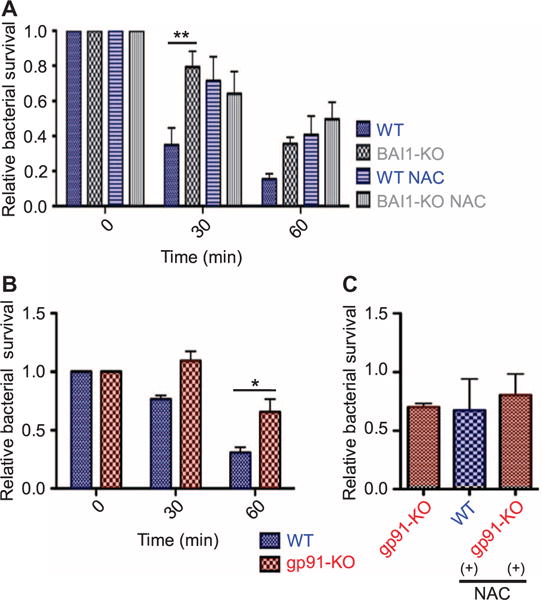
(A) BMDMs were pretreated with either vehicle or the ROS scavenger NAC before being incubated with E. coli BW25113 for the indicated times. Bacterial survival was measured as described in Fig. 3A. All graphs show mean survival ± SEM from four experiments. Data were analyzed by two-way ANOVA with Bonferroni post hoc comparisons. WT versus BAI1-KO: cell, P < 0.001; time, P < 0.001. WT versus WT-NAC: cell, P < 0.05; time, P < 0.05. (B) WT and gp91phox-KO BMDMs were infected with E. coli BW25113 for the indicated times, and the survival of the associated bacteria was measured and analyzed as described in Fig. 3A (cell, P < 0.01; time, P < 0.001; n = 6 experiments). (C) Incubation of WT cells with the ROS scavenger NAC reduces bacterial killing to the extent exhibited by gp91phox KO cells. WT and gp91phox KO BMDMs were incubated with E. coli BW25113 for 60 min in the presence or absence of NAC. Bacterial survival was measured as described in Fig. 3A. One-way ANOVA with Bonferroni post hoc comparison was used for analysis. n = 2 experiments.
BAI1 promotes bacterial clearance in vivo
Given the defect in bacterial phagocytosis and killing in BAI1-deficient primary cells, we hypothesized that BAI1 knockout animals would exhibit impaired bacterial clearance and increased susceptibility to bacterial challenge (53, 55). To test this possibility, we used a well-characterized model of bacterial peritonitis in which we infected wild-type, BAI1 knockout, and gp91phox knockout mice intraperitoneally with nonpathogenic E. coli and then analyzed several parameters of susceptibility (Fig. 8A). First, a disease score was determined for each animal based on macroscopic examination of their behavior, including posture, eye discharge, grooming, and movement at 4 hours after infection (fig. S6). BAI1-deficient animals exhibited enhanced disease activity compared to that of wild-type mice (Fig. 8B), which was comparable to that of mice lacking gp91phox. Second, BAI1 knockout animals succumbed to peritoneal infection more rapidly than did control wild-type mice (Fig. 8C). Measurement of colony-forming units (CFUs) revealed statistically significantly greater bacterial burden in the peritoneum, liver, and spleen at 4 hours after infection in BAI1 knockout mice compared to wild-type mice (Fig. 8, D to F). At 24 hours after infection, wild-type mice had almost completely cleared bacteria from the liver and spleen. In contrast, both the BAI1 knockout and gp91phox knockout animals showed persistent, viable CFUs in these tissues (Fig. 8, G to I). Furthermore, bacterial counts in the BAI1 knockout animals were similar to those in the gp91phox knockout animals, suggesting that defective ROS production contributes to increased susceptibility to bacterial infection.
Fig. 8. BAI1 mediates bacterial clearance in vivo.
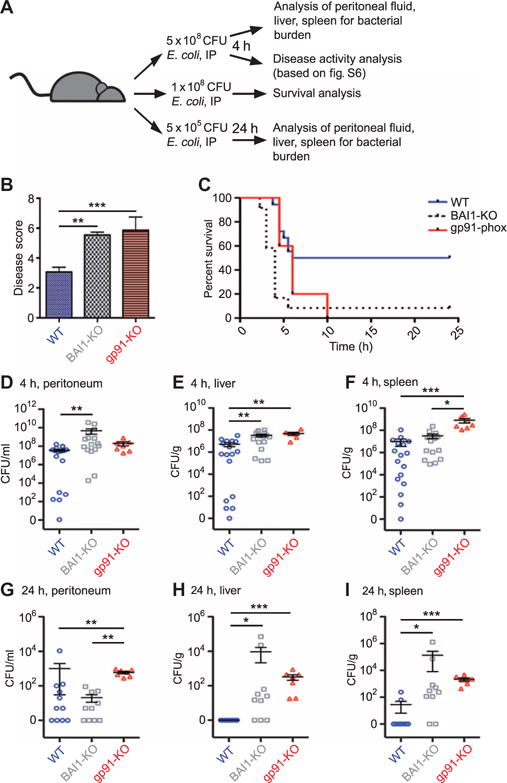
(A) WT, BAI1-KO, and gp91phox-KO mice were infected intraperitoneally with E. coli BW25113 and analyzed on the basis of several parameters of susceptibility to bacterial challenge. Bacterial dose, length of infection, and type of analysis are shown in schematic form. (B) WT, BAI1-KO, and gp91phox-KO mice were infected intraperitoneally (IP) with 5 × 108 CFU E. coli, and disease severity was analyzed 4 hours later. Graph displays mean score ± SEM of three experiments. ***P < 0.001, **P < 0.01 by one-way ANOVA Kruskal-Wallis test with Dunn’s post hoc comparisons. (C) Survival was measured in WT and BAI1-KO mice after intraperitoneal infection with 1 × 108 CFU E. coli. Survival was blindly scored on the basis of the criteria in (B). Mantel-Cox log rank was used to compare survival. **P < 0.01; n = 2 experiments. (D to F) Bacterial burden 4 hours after infection: CFUs were measured in the peritoneum (D), liver (E), and spleen (F) of the indicated mice 4 hours after challenge with 5 × 108 CFU E. coli. Each data point is representative of a single animal. Data are mean CFUs per tissue ± SEM of four experiments. Analysis was performed by one-way ANOVA Kruskal-Wallis test with Dunn’s post hoc comparison. ***P < 0.001, **P < 0.01, *P < 0.05. (G to I) Bacterial burden at 24 hours after infection: CFUs were measured in the peritoneum (G), liver (H), and spleen (I) 24 hours after challenge with 5 × 105 CFU E. coli. Data are mean CFUs per tissue ± SEM of three experiments. Analysis was performed by one-way ANOVA Kruskal-Wallis test with Dunn’s post hoc comparison. ***P < 0.001, **P < 0.01, *P < 0.05.
DISCUSSION
Innate immune cells express an array of PRRs that function in bacterial detection and phagocytosis (2, 3, 14, 56). We previously showed that BAI1 acts as a PRR for Gram-negative bacteria and that it specifically binds to the relatively invariant core oligosaccharides of bacterial LPS (22). Furthermore, this recognition mechanism is distinct from that used by TLR4, which binds to the acyl chains of LPS (57). Binding of bacteria to BAI1 stimulates their phagocytic uptake through the direct activation of the ELMO-Dock complex, which acts as a GEF for Rac (22). Here, we extend these observations to show that BAI1-mediated Rac activation not only stimulates bacterial internalization by macrophages but also is necessary for robust activation of the phagosomal NADPH oxidase complex. In vitro, primary macrophages lacking BAI1 exhibited substantially reduced bactericidal activity because of attenuated induction of ROS in response to both nonpathogenic and pathogenic Gram-negative bacteria.
The importance of the NADPH oxidase complex in the innate immune response to bacterial infection is highlighted in patients with chronic granulomatous disease (CGD), who have deficiencies in specific components of the NADPH oxidase machinery (26, 27). Consistent with the presentation of CGD in humans, mice deficient in gp91phox, the catalytic subunit of phagocyte NADPH oxidase, are highly susceptible to bacterial infections (53, 55, 58, 59). Note that patients with CGD are particularly susceptible to select bacterial pathogens, including B. cenocepacia (60, 61). Here, we showed that BAI1-deficient macrophages were similarly impaired in their ability to generate ROS in response to B. cenocepacia and several other Gram-negative pathogens, including P. aeruginosa and S. Typhimurium, which resulted in inefficient killing. Together, these data suggest that BAI1 broadly contributes to defense against Gram-negative bacteria. Although we cannot rule out other defects in the early inflammatory response to E. coli, such as defects in inflammatory signaling and cytokine production, we showed that the loss of BAI1 had an effect on susceptibility to bacterial challenge in vivo that was similar to that caused by loss of gp91phox, which suggests that BAI1-dependent ROS activity is a critical factor in early innate immunity and bacterial clearance.
The cellular mechanisms that couple nonopsonic, phagocytic receptors to cellular bactericidal machinery are not well understood (29). It is well established that Rac1 and Rac2 are critical components of the NADPH oxidase machinery in macrophages and neutrophils, respectively (47–49, 62). The recruitment and activation of Rac proteins occur through GEFs that catalyze the exchange of guanosine diphosphate for GTP (63). In neutrophils, the Rac-GEF P-Rex1 is implicated in the activation of Rac2 and NADPH oxidase by the bacterial formyl peptide fMetLeuPhe (64), whereas in both macrophages and neutrophils, the Vav family of Rho GEFs is linked to ROS and inflammatory cytokine responses downstream of TLRs (65) and FcγR (66). One study showed that deletion of the three Vav family proteins (Vav1, Vav2, and Vav3) attenuated macrophage ROS production in response to high concentrations of LPS and that the activation of Vav was dependent on the TLR adaptor protein myeloid differentiation primary response gene 88 (MyD88) (65). In that study, Vav family members mediated the activation of Rac2; however, Rac1 was not examined. In contrast, here, we observed almost complete abrogation of Rac1 activity in BAI1-deficient cells and a corresponding reduction in ROS production, suggesting that BAI1-mediated activation of these responses occurs independently of the Vav signaling pathway. Note that BAI1 does not appear to be required for phagocytosis or ROS production in response to Gram-positive bacteria, because no differences were observed between wild-type and BAI1-deficient macrophages infected with S. aureus.
BAI1 signals through several pathways that lead to Rac1 activation. These include direct binding and activation of the bipartite ELMO-Dock Rac GEF complex in response to both apoptotic cells and Gram-negative bacteria (18, 22), as well as the recruitment and activation of the Par3-Tiam1 complex during synaptogenesis (67). Rac activation during synaptogenesis requires its interaction with the Par3-Tiam1 complex but not ELMO-Dock180 (68). Here, we showed that macrophages expressing a BAI1 mutant that cannot interact with ELMO-Dock were as attenuated in bacterial killing as were cells that lacked BAI1. Although we cannot rule out an interaction between BAI1 and Tiam1 in this context, this observation suggests that the ELMO-Dock complex is the primary mediator of Rac activation in response to Gram-negative bacteria. In addition to being linked to Rac, the cytoplasmic domain of BAI1 has also been linked to the activation of RhoA and extracellular signal–regulated kinase through the G protein Gα12/13 (68).
In addition to NADPH oxidase, mitochondrial ROS has been implicated in oxidative killing in a pathway dependent on both TLR4 and MyD88 (44). Here, we found that MitoTEMPO, which selectively scavenges mitochondrial superoxide (54), had no effect on BAI1-dependent bactericidal activity, indicating that BAI1-mediated bacterial killing occurs independently of mitochondrial ROS. Moreover, in our hands, the ROS response to E. coli was completely absent in cells lacking the NADPH oxidase subunit gp91phox, which suggests that at least at the time points we examined, ROS production occurred primarily through the phagosomal NADPH oxidase. The reduced microbicidal activity of BAI1-deficient macrophages in vitro was comparable to that of gp91phox-deficient cells, and similar defects in bacterial clearance were observed in vivo in a mouse model of peritoneal infection.
Together, these results suggest that BAI1 is an innate phagocytic receptor that couples bacterial detection to the induction of oxidative killing by stimulating Rac activation in phagocytes. There are many innate immune receptors that initiate ROS production, but they do so in response to distinct stimuli. The specificity of BAI1 for nonopsonized, Gram-negative bacteria represents a previously uncharacterized mechanism for the regulation of ROS production in macrophages. This study reveals a potentially broader role for BAI1 in modulating cellular immune responses, ROS production, and inflammation not only during infection by bacterial pathogens but also under homeostatic conditions through the recognition of resident microbes at mucosal sites.
MATERIALS AND METHODS
Ethics statement
All experiments were performed in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Protocols were approved by the Institutional Animal Care and Use Committee at the University of Virginia (protocol number 3488).
Mice
Age- and sex-matched C57BL/6 mice between 6 and 10 weeks of age were used for the harvesting of primary macrophages and for peritoneal challenge experiments. BAI1 knockout mice have been described previously (19). Mice expressing transgenic wild-type BAI1 or BAI1-AAA coding sequences were generated by knocking the coding sequence for human BAI1 or its mutant into the nonessential Rosa26 locus of C57BL/6 embryonic stem (ES) cells, and generating mice with these targeted ES cells (31, 69). gp91phox knockout mice were a gift from B. Mehrad, University of Virginia (Charlottesville, VA). Mice were housed in pathogen-free conditions.
Isolation and culture of cells
Stable BAI1-depleted J774 macrophage cell lines were generated by transduction with lentiviruses encoding short hairpin RNA (shRNA) against murine BAI1 (hairpin sequence V3LHS_322807, catalog number RHS4531-NM_174991; Open Biosystems) and selection with puromycin. J774 cells were cultured in Dulbecco’s modified Eagle’s medium (4.5 g/liter glucose; Gibco) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (pen-strep). Knockdown was confirmed by quantitative reverse transcription polymerase chain reaction analysis. LR73 CHO cell lines have been described previously (18) and were cultured in a minimal essential medium (αMEM; Gibco) containing 10% FBS and 1% pen-strep. PEMs were isolated from mouse peritoneal lavage with sterile phosphate-buffered saline (PBS). To generate BMDMs, cells were seeded onto non–tissue culture–treated plastic plates and cultured in RPMI supplemented with 10% FBS, 10% L929 conditioned medium (as a source of colony-stimulating factor–1), and 1% pen-strep. BMDMs were cultured for 6 days ex vivo before use, and the culture medium was changed every 2 days. Macrophage differentiation was confirmed by flow cytometric analysis of the cell surface abundances of F4/80 (clone BM8; eBioscience) and CD11b (clone M1/70; eBioscience).
Bacterial strains and culture
All bacteria, including E. coli DH5α (18265-017; Invitrogen) or BW25113 [E. coli Genetic Stock Center Keio collection parent strain (70)], were cultured overnight in Luria-Bertani (LB) broth under aerobic conditions before use. Immunofluorescence microscopy was performed using either E. coli DH5α or noninvasive S. Typhimurium expressing dsRed (71). Δspa S. aureus Newman strain (72) was a gift from A. Criss, University of Virginia (Charlottesville, VA). P. aeruginosa PAO3 was a gift from B. Mehrad, University of Virginia (Charlottesville, VA). B. cenocepacia strains BC7 and K56-2 were gifts from C. Sifri, University of Virginia (Charlottesville, VA).
Immunofluorescence-based internalization assay
E. coli DH5α–dsRed were surface-labeled with EZ-Link Sulfo-NHS-LC-Biotin (1 mg/ml; Life Technologies) for 30 min at 4°C. BMDMs were plated on glass coverslips (Fisher) overnight before being infected for 30 min with biotinylated bacteria at an MOI of 25 in RPMI with 10% FBS. Cells were washed and then fixed with 4% paraformaldyhyde (PFA) without permeablization and then were blocked in PBS containing 3% bovine serum albumin (BSA). Extracellular bacteria were labeled with streptavidin–Alexa Fluor 488 conjugate (Life Technologies) for 30 min, after which cells were permeabilized with 0.1% Triton X-100 in PBS with 3% BSA. Cells were counterstained with DAPI to label nuclei. Images were acquired with a Nikon Eclipse E800 microscope equipped with a QImaging Retiga camera and Nikon NIS-Elements software. Test images determined optimal exposure gains, and this gain was subsequently used for all conditions within an experimental replicate. In this assay, intracellular bacteria appear red, whereas extracellular bacteria are double-positive for dsRed and Alexa Fluor 488 and appear yellow. At least 125 cells per replicate were imaged.
Immunofluoresence microscopy
Transgenic BMDMs (1 × 105) expressing HA-BAI1 were plated on fibronectin-coated coverslips (Sigma). The following day, the cells were incubated with E. coli DH5α–dsRed at an MOI of 10 for 30 min at 37°C. Cells were then fixed with 4% PFA and labeled with Alexa Fluor 647–conjugated WGA (5 mg/ml; Life Technologies) in Hanks’ balanced salt solution (HBSS) for an additional 10 min to label the plasma membrane. After washing, the cells were permeabilized for 30 min in PBS containing 3% BSA, 1% normal goat serum, FcR blocking antibody (clone 93; eBioscience), and 0.1% Triton X-100. Cells were labeled with mouse anti-HA antibody (clone 16B12; Covance) followed by Alexa Fluor 488–conjugated anti-mouse secondary antibody. ROIs for E. coli DH5α-treated cells were determined by dsRed signal. WGA signal was used to define ROIs in S. aureus conditions because the bacteria displayed substantially greater staining than did eukaryotic cell membranes. Images were captured with a Nikon C1 Plus confocal microscopewith z-stacks at 0.5μm. Analysis and processing were performed with NIS-Elements software (Nikon).
Live-cell imaging
Cells were plated on fibronectin-coated MatTek dishes (P35G-1.5-14-C) 18 hours before imaging. Imaging was performed in phenol red–free RPMI containing 10 mM Hepes (pH 7.4) and 10% heat-inactivated FBS. After blocking endogenous FcR as described earlier, surface-exposed HA-BAI1 was labeled for 30 min with Alexa Fluor 488–conjugated mouse anti-HA antibody (4 μg/ml; Life Technologies). Cells were then infected with noninvasive ΔinvG S. Typhimurium SL1344 expressing dsRed and imaged with a 100× objective fitted to a Nikon TE 2000 microscope equipped with a Yokogawa CSU 10 spinning disc and a 512×512 Hamamatsu 9100c-13 EM-BT camera. Movies were captured at a frame rate of 300 ms.
Short-course bacterial association and killing assay
BMDMs (1 × 105) were seeded onto 24-well plates 18 hours before infection with bacteria at an MOI of 25. To synchronize infections, bacteria were spun onto cells at 4°C as described earlier and then were incubated for 10 min at 37°C to enable bacterial attachment and internalization. To measure bacterial killing, cells were washed extensively with RPMI and then were placed at 37°C for the times described in the figure legends. At each time point, cells were washed and lysed and viable bacteria were enumerated as described earlier.
Gentamicin protection and intracellular bactericidal assay
The longer-course gentamicin protection assay was performed as described previously (22). Briefly, 5 × 104 CHO cells per well or 1 × 105 BMDMs per well were seeded into 24-well plates 18 hours before infection. Cells were incubated with bacteria at an MOI of 50 for 30 min at 37°C in αMEM (CHO) or RPMI (BMDMs) containing 10% heat-inactivated FBS, after spinning bacteria onto the cells at 500g for 5 min at 4°C to synchronize uptake. After 30 min of internalization, cells were treated with gentamicin (500 μg/ml; Gibco) for 30 min to kill extracellular bacteria, but leave intracellular bacteria viable. To measure bacterial killing, cells were then washed and lysed immediately or incubated with gentamicin (10 μg/ml) for the remaining times indicated in the figure legends. Cells were lysed in HBSS containing 0.5% saponin with calcium and magnesium, and viable intracellular CFUs were determined by plating cell lysates on LB agar.
Rac activation assay
The precipitation of active, GTP-bound Rac was performed as described previously (22). BMDMs were serum-starved for 2 hours in RPMI and then infected with E. coli K-12 BW25113 at an MOI of 100 for 10 or 30 min at 37°C. Cells were lysed in 50 mM tris-HCl (pH 7.5), 10 mM MgCl2, 100 mM NaCl, 10% glycerol, 0.5% NaDOC, and 1% Triton X-100. GTP-bound Rac was precipitated with a GST fusion containing the PBD of PAK immobilized on glutathione sepharose beads for 30 min. Precipitates were resolved by SDS–polyacrylamide gel electrophoresis and then analyzed by Western blotting with a Rac1-specific antibody (Millipore). Rac-GTP was quantified as a percentage of the total amount of Rac in cell lysates.
Detection of ROS
For LDCL assays, 3.5 × 105 macrophages were plated in 96-well plates in 200 μl of phenol red–free RPMI (Gibco) containing 10% FBS and then were primed overnight with IFN-γ (50 ng/ml; PeproTech). Cells were incubated with 20 μM luminol (Sigma) and treated with bacteria at 37°C in phenol red–free RPMI (Gibco). Luminescence was measured with a VICTOR3 Wallac luminometer (PerkinElmer). For in situ fluorescence assays, 1 × 105 BMDMs were plated on glass coverslips (Fisher) overnight before infection for 30 min with E. coli DH5α expressing dsRed at an MOI of 25 in phenol red–free RPMI containing 1% heat-inactivated FBS. Cells were then washed and incubated for 30 min with 5 μM CellROX Green (Molecular Probes C10444). The cells were then fixed with 4% PFA, followed by blocking and permeabilization in PBS containing 3% BSA and 0.1% Triton X-100. Cells were counterstained with DAPI (Sigma) to mark nuclei and mounted with ProLong Gold antifade (Life Technologies). Images were acquired and analyzed with a Nikon E800 microscope as described earlier. Test images determined the optimal exposure gain, which was subsequently used for all conditions within an experimental replicate. DAPI was used to select nuclei as ROIs to measure the MFI of CellROX Green. At least 300 cells were imaged per replicate.
Peritoneal infection model
Age- and sex-matched mice between 6 and 8 weeks of age were infected by peritoneal injection with 5 × 105, 1 × 108, or 5 × 108 CFUs of E. coli K-12 BW25113 in 0.2 ml of sterile Dulbecco’s PBS. Mice were monitored for disease state and severity. Disease state was determined for each animal on the basis of macroscopic examination of behavior, including posture, eye discharge, grooming, and movement. Mice were euthanized at either 4 or 24 hours after infection. Bacterial loads in the peritoneum, liver, and spleen were determined by plating the lysates of homogenized tissues on LB agar.
Statistical analysis
Statistical analysis was performed with GraphPad Prism 5 software. Statistical significance was set at the 5% standard. Data that did not match the assumptions for parametric analysis (normality, equal variance, and normalization) were analyzed with nonparametric analysis as indicated in the figure legends. All analysis was two-tailed. Graphs show means ± SEM. When appropriate, two-way ANOVA with Bonferroni post hoc comparisons was used for analysis. Information represented in the figure legend indicates the analysis regarding the two independent variables (for example, time and cell type) and whether there is an interaction between them. Statistical information represented on the graph refers to the post hoc comparison. In all data sets, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. The number of independent experimental replicates is indicated by n.
Supplementary Material
Fig. S1. BAI1 localizes to the cell periphery, the perinuclear region, and the sites of bacterial cell association.
Fig. S2. BAI1 promotes cellular microbicidal activity in J774 macrophages.
Fig. S3. Loss of BAI1 impairs Rac activation in response to E. coli in IFN-γ–primed BMDMs.
Fig. S4. Loss of BAI1 impairs intracellular ROS generation but does not affect IFN-γ priming or iNOS production induced by E. coli.
Fig. S5. Mitochondrial ROS produced in response to Gram-negative bacteria is independent of BAI1-mediated recognition and signaling.
Fig. S6. Measurement of disease activity analysis.
Movie S1. BAI1 is enriched at the phagocytic cup.
Acknowledgments
We are grateful to J. Brumell (Hospital for Sick Children, Toronto) for the dsRed expression vector, A. Criss [University of Virginia (UVA)] for the Δspa S. aureus Newman strain, S. Das (University of California, San Diego) for the pGIPZ–BAI1-shRNA vectors for BAI1 knockdown, B. Mehrad (UVA) for the P. aeruginosa PAO3 strain, C. Sifri (UVA) for the B. cenocepacia spp. strains, and X.-Q. Wang (UVA) for consultation on statistical methodology.
Funding: This work was supported by NIH RO1 grant AI093708 to J.E.C. E.A.B. was supported in part by the UVA Cell and Molecular Biology Training Grant (T32GM813626).
Footnotes
SUPPLEMENTARY MATERIALS
Author contributions: E.A.B. and J.E.C. designed the experiments and analyzed the data; K.A.O. contributed to the animal model; R.S.D. assisted with live-cell and confocal imaging studies; C.S.L. and K.S.R. generated the BAI1 knockout and transgenic animals used in this study; and E.A.B. and J.E.C. prepared the manuscript.
Competing interests: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. 2011;30:16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- 2.Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: A cell biological perspective. Annu Rev Immunol. 2015;33:257–290. doi: 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 4.Stuart LM, Ezekowitz RA. Phagocytosis: Elegant complexity. Immunity. 2005;22:539–550. doi: 10.1016/j.immuni.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 5.Taylor PR, Martinez-Pomares L, Stacey M, Lin HH, Brown GD, Gordon S. Macrophage receptors and immune recognition. Annu Rev Immunol. 2005;23:901–944. doi: 10.1146/annurev.immunol.23.021704.115816. [DOI] [PubMed] [Google Scholar]
- 6.Medzhitov R, Horng T. Transcriptional control of the inflammatory response. Nat Rev Immunol. 2009;9:692–703. doi: 10.1038/nri2634. [DOI] [PubMed] [Google Scholar]
- 7.Underhill DM, Gantner B. Integration of Toll-like receptor and phagocytic signaling for tailored immunity. Microbes Infect. 2004;6:1368–1373. doi: 10.1016/j.micinf.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 8.Kerrigan AM, Brown GD. C-type lectins and phagocytosis. Immunobiology. 2009;214:562–575. doi: 10.1016/j.imbio.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zamze S, Martinez-Pomares L, Jones H, Taylor PR, Stillion RJ, Gordon S, Wong SYC. Recognition of bacterial capsular polysaccharides and lipopolysaccharides by the macrophage mannose receptor. J Biol Chem. 2002;277:41613–41623. doi: 10.1074/jbc.M207057200. [DOI] [PubMed] [Google Scholar]
- 10.Underhill DM, Rossnagle E, Lowell CA, Simmons RM. Dectin-1 activates Syk tyrosine kinase in a dynamic subset of macrophages for reactive oxygen production. Blood. 2005;106:2543–2550. doi: 10.1182/blood-2005-03-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Areschoug T, Gordon S. Scavenger receptors: Role in innate immunity and microbial pathogenesis. Cell Microbiol. 2009;11:1160–1169. doi: 10.1111/j.1462-5822.2009.01326.x. [DOI] [PubMed] [Google Scholar]
- 12.Baranova IN, Kurlander R, Bocharov AV, Vishnyakova TG, Chen Z, Remaley AT, Csako G, Patterson AP, Eggerman TL. Role of human CD36 in bacterial recognition, phagocytosis, and pathogen-induced JNK-mediated signaling. J Immunol. 2008;181:7147–7156. doi: 10.4049/jimmunol.181.10.7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jing J, Yang IV, Hui L, Patel JA, Evans CM, Prikeris R, Kobzik L, O’Connor BP, Schwartz DA. Role of macrophage receptor with collagenous structure in innate immune tolerance. J Immunol. 2013;190:6360–6367. doi: 10.4049/jimmunol.1202942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Underhill DM, Goodridge HS. Information processing during phagocytosis. Nat Rev Immunol. 2012;12:492–502. doi: 10.1038/nri3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ip WKE, Sokolovska A, Charriere GM, Boyer L, Dejardin S, Cappillino MP, Yantosca LM, Takahashi K, Moore KJ, Lacy-Hulbert A, Stuart LM. Phagocytosis and phagosome acidification are required for pathogen processing and MyD88-dependent responses to Staphylococcus aureus. J Immunol. 2010;184:7071–7081. doi: 10.4049/jimmunol.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Husebye H, Aune MH, Stenvik J, Samstad E, Skjeldal F, Halaas Ø, Nilsen NJ, Stenmark H, Latz E, Lien E, Mollnes TE, Bakke O, Espevik T. The Rab11a GTPase controls Toll-like receptor 4-induced activation of interferon regulatory factor-3 on phago-somes. Immunity. 2010;33:583–596. doi: 10.1016/j.immuni.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaur B, Brat DJ, Calkins CC, Van Meir EG. Brain angiogenesis inhibitor 1 is differentially expressed in normal brain and glioblastoma independently of p53 expression. Am J Pathol. 2003;162:19–27. doi: 10.1016/S0002-9440(10)63794-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z, Klibanov AL, Mandell JW, Ravichandran KS. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007;450:430–434. doi: 10.1038/nature06329. [DOI] [PubMed] [Google Scholar]
- 19.Hochreiter-Hufford AE, Lee CS, Kinchen JM, Sokolowski JD, Arandjelovic S, Call JA, Klibanov AL, Yan Z, Mandell JW, Ravichandran KS. Phosphatidylserine receptor BAI1 and apoptotic cells as new promoters of myoblast fusion. Nature. 2013;497:263–267. doi: 10.1038/nature12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mazaheri F, Breus O, Durdu S, Haas P, Wittbrodt J, Gilmour D, Peri F. Distinct roles for BAI1 and TIM-4 in the engulfment of dying neurons by microglia. Nat Commun. 2014;5:4046. doi: 10.1038/ncomms5046. [DOI] [PubMed] [Google Scholar]
- 21.Das S, Sarkar A, Ryan KA, Fox S, Berger AH, Juncadella IJ, Bimczok D, Smythies LE, Harris PR, Ravichandran KS, Crowe SE, Smith PD, Ernst PB. Brain angiogenesis inhibitor 1 is expressed by gastric phagocytes during infection with Helicobacter pylori and mediates the recognition and engulfment of human apoptotic gastric epithelial cell. s FASEB J. 2014;28:2214–2224. doi: 10.1096/fj.13-243238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Das S, Owen KA, Ly KT, Park D, Black SG, Wilson JM, Sifri CD, Ravichandran KS, Ernst PB, Casanova JE. Brain angiogenesis inhibitor 1 (BAI1) is a pattern recognition receptor that mediates macrophage binding and engulfment of Gram-negative bacteria. Proc Natl Acad Sci USA. 2011;108:2136–2141. doi: 10.1073/pnas.1014775108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greenberg S, Grinstein S. Phagocytosis and innate immunity. Curr Opin Immunol. 2002;14:136–145. doi: 10.1016/s0952-7915(01)00309-0. [DOI] [PubMed] [Google Scholar]
- 24.Castellano F, Chavrier P, Caron E. Actin dynamics during phagocytosis. Semin Immunol. 2001;13:347–355. doi: 10.1006/smim.2001.0331. [DOI] [PubMed] [Google Scholar]
- 25.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 26.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 27.Bylund J, Brown KL, Movitz C, Dahlgren C, Karlsson A. Intracellular generation of superoxide by the phagocyte NADPH oxidase: How, where, and what for? Free Radic Biol Med. 2010;49:1834–1845. doi: 10.1016/j.freeradbiomed.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 28.Groemping Y, Rittinger K. Activation and assembly of the NADPH oxidase: A structural perspective. Biochem J. 2005;386:401–416. doi: 10.1042/BJ20041835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nunes P, Demaurex N, Dinauer MC. Regulation of the NADPH oxidase and associated ion fluxes during phagocytosis. Traffic. 2013;14:1118–1131. doi: 10.1111/tra.12115. [DOI] [PubMed] [Google Scholar]
- 30.Bokoch GM. Regulation of innate immunity by Rho GTPases. Trends Cell Biol. 2005;15:163–171. doi: 10.1016/j.tcb.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 31.Fond AM, Lee CS, Schulman IG, Kiss RS, Ravichandran KS. Apoptotic cells trigger a membrane-initiated pathway to increase ABCA1. J Clin Invest. 2015;125:2748–2758. doi: 10.1172/JCI80300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sokolowski JD, Nobles SL, Heffron DS, Park D, Ravichandran KS, Mandell JW. Brain-specific angiogenesis inhibitor-1 expression in astrocytes and neurons: Implications for its dual function as an apoptotic engulfment receptor. Brain Behav Immun. 2011;25:915–921. doi: 10.1016/j.bbi.2010.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Russell DG, VanderVen BC, Glennie S, Mwandumba H, Heyderman RS. The macrophage marches on its phagosome: Dynamic assays of phagosome function. Nat Rev Immunol. 2009;9:594–600. doi: 10.1038/nri2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geier H, Celli J. Phagocytic receptors dictate phagosomal escape and intracellular proliferation of. Francisella tularensis Infect Immun. 2011;79:2204–2214. doi: 10.1128/IAI.01382-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kang PB, Azad AK, Torrelles JB, Kaufman TM, Beharka A, Tibesar E, DesJardin LE, Schlesinger LS. The human macrophage mannose receptor directs Mycobacterium tuberculosis lipoarabinomannan-mediated phagosome biogenesis. J Exp Med. 2005;202:987–999. doi: 10.1084/jem.20051239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang P, Skurnik M, Zhang SS, Schwartz O, Kalyanasundaram R, Bulgheresi S, He JJ, Klena JD, Hinnebusch BJ, Chen T. Human dendritic cell-specific intercellular adhesion molecule-grabbing nonintegrin (CD209) is a receptor for Yersinia pestis that promotes phagocytosis by dendritic cells. Infect Immun. 2008;76:2070–2079. doi: 10.1128/IAI.01246-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Agramonte-Hevia J, González-Arenas A, Barrera D, Velasco-Velázquez M. Gram-negative bacteria and phagocytic cell interaction mediated by complement receptor 3. FEMS Immunol Med Microbiol. 2002;34:255–266. doi: 10.1111/j.1574-695X.2002.tb00640.x. [DOI] [PubMed] [Google Scholar]
- 38.Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science. 2004;304:1014–1018. doi: 10.1126/science.1096158. [DOI] [PubMed] [Google Scholar]
- 39.Blander JM, Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature. 2006;440:808–812. doi: 10.1038/nature04596. [DOI] [PubMed] [Google Scholar]
- 40.Criss AK, Casanova JE. Coordinate regulation of Salmonella enterica serovar Typhimurium invasion of epithelial cells by the Arp2/3 complex and Rho GTPases. Infect Immun. 2003;71:2885–2891. doi: 10.1128/IAI.71.5.2885-2891.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Flannagan RS, Cosío G, Grinstein S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol. 2009;7:355–366. doi: 10.1038/nrmicro2128. [DOI] [PubMed] [Google Scholar]
- 42.Bonini MG, Malik AB. Regulating the regulator of ROS production. Cell Res. 2014;24:908–909. doi: 10.1038/cr.2014.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Forman HJ, Torres M. Reactive oxygen species and cell signaling: Respiratory burst in macrophage signaling. Am J Respir Crit Care Med. 2002;166:S4–S8. doi: 10.1164/rccm.2206007. [DOI] [PubMed] [Google Scholar]
- 44.West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472:476–480. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hordijk PL. Regulation of NADPH oxidases: The role of Rac proteins. Circ Res. 2006;98:453–462. doi: 10.1161/01.RES.0000204727.46710.5e. [DOI] [PubMed] [Google Scholar]
- 46.Filippi MD, Harris CE, Meller J, Gu Y, Zheng Y, Williams DA. Localization of Rac2 via the C terminus and aspartic acid 150 specifies superoxide generation, actin polarity and chemotaxis in neutrophils. Nat Immunol. 2004;5:744–751. doi: 10.1038/ni1081. [DOI] [PubMed] [Google Scholar]
- 47.Noubade R, Wong K, Ota N, Rutz S, Eidenschenk C, Valdez PA, Ding J, Peng I, Sebrell A, Caplazi P, DeVoss J, Soriano RH, Sai T, Lu R, Modrusan Z, Hackney J, Ouyang W. NRROS negatively regulates reactive oxygen species during host defence and autoimmunity. Nature. 2014;509:235–239. doi: 10.1038/nature13152. [DOI] [PubMed] [Google Scholar]
- 48.Gorzalczany Y, Sigal N, Itan M, Lotan O, Pick E. Targeting of Rac1 to the phagocyte membrane is sufficient for the induction of NADPH oxidase assembly. J Biol Chem. 2000;275:40073–40081. doi: 10.1074/jbc.M006013200. [DOI] [PubMed] [Google Scholar]
- 49.Yamauchi A, Kim C, Li S, Marchal CC, Towe J, Atkinson SJ, Dinauer MC. Rac2-deficient murine macrophages have selective defects in superoxide production and phagocytosis of opsonized particles. J Immunol. 2004;173:5971–5979. doi: 10.4049/jimmunol.173.10.5971. [DOI] [PubMed] [Google Scholar]
- 50.VanderVen BC, Yates RM, Russell DG. Intraphagosomal measurement of the magnitude and duration of the oxidative burst. Traffic. 2009;10:372–378. doi: 10.1111/j.1600-0854.2009.00877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vazquez-Torres A, Jones-Carson J, Mastroeni P, Ischiropoulos H, Fang FC. Antimicrobial actions of the NADPH phagocyte oxidase and inducible nitric oxide synthase in experimental salmonellosis. I. Effects on microbial killing by activated peritoneal macrophages in vitro. J Exp Med. 2000;192:227–236. doi: 10.1084/jem.192.2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vazquez-Torres A, Fang FC. Oxygen-dependent anti-Salmonella activity of macrophages. Trends Microbiol. 2001;9:29–33. doi: 10.1016/s0966-842x(00)01897-7. [DOI] [PubMed] [Google Scholar]
- 53.Shiloh MU, MacMicking JD, Nicholson S, Brause JE, Potter S, Marino M, Fang F, Dinauer M, Nathan C. Phenotype of mice and macrophages deficient in both phagocyte oxidase and inducible nitric oxide synthase. Immunity. 1999;10:29–38. doi: 10.1016/s1074-7613(00)80004-7. [DOI] [PubMed] [Google Scholar]
- 54.Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res. 2010;107:106–116. doi: 10.1161/CIRCRESAHA.109.214601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mastroeni P, Vazquez-Torres A, Fang FC, Xu Y, Khan S, Hormaeche CE, Dougan G. Antimicrobial actions of the NADPH phagocyte oxidase and inducible nitric oxide synthase in experimental salmonellosis. II. Effects on microbial proliferation and host survival in vivo. J Exp Med. 2000;192:237–248. doi: 10.1084/jem.192.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Freeman SA, Grinstein S. Phagocytosis: Receptors, signal integration, and the cytoskeleton. Immunol Rev. 2014;262:193–215. doi: 10.1111/imr.12212. [DOI] [PubMed] [Google Scholar]
- 57.Park BS, Lee JO. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med. 2013;45:e66. doi: 10.1038/emm.2013.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pizzolla A, Hultqvist M, Nilson B, Grimm MJ, Eneljung T, Jonsson IM, Verdrengh M, Kelkka T, Gjertsson I, Segal BH, Holmdahl R. Reactive oxygen species produced by the NADPH oxidase 2 complex in monocytes protect mice from bacterial infections. J Immunol. 2012;188:5003–5011. doi: 10.4049/jimmunol.1103430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sousa SA, Ulrich M, Bragonzi A, Burke M, Worlitzsch D, Leitão JH, Meisner C, Eberl L, Sá-Correia I, Döring G. Virulence of Burkholderia cepacia complex strains in gp91phox−/− mice. Cell Microbiol. 2007;9:2817–2825. doi: 10.1111/j.1462-5822.2007.00998.x. [DOI] [PubMed] [Google Scholar]
- 60.Greenberg DE, Goldberg JB, Stock F, Murray PR, Holland SM, LiPuma JJ. Recurrent Burkholderia infection in patients with chronic granulomatous disease: 11-year experience at a large referral center. Clin Infect Dis. 2009;48:1577–1579. doi: 10.1086/598937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marciano BE, Spalding C, Fitzgerald A, Mann D, Brown T, Osgood S, Yockey L, Darnell DN, Barnhart L, Daub J, Boris L, Rump AP, Anderson VL, Haney C, Kuhns DB, Rosenzweig SD, Kelly C, Zelazny A, Mason T, DeRavin SS, Kang E, Gallin JI, Malech HL, Olivier KN, Uzel G, Freeman AF, Heller T, Zerbe CS, Holland SM. Common severe infections in chronic granulomatous disease. Clin Infect Dis. 2015;60:1176–1183. doi: 10.1093/cid/ciu1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Panday A, Sahoo MK, Osorio D, Batra S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell Mol Immunol. 2015;12:5–23. doi: 10.1038/cmi.2014.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Niedergang F, Chavrier P. Regulation of phagocytosis by Rho GTPases. Curr Top Microbiol Immunol. 2005;291:43–60. doi: 10.1007/3-540-27511-8_4. [DOI] [PubMed] [Google Scholar]
- 64.Dong X, Mo Z, Bokoch G, Guo C, Li Z, Wu D. P-Rex1 is a primary Rac2 guanine nucleotide exchange factor in mouse neutrophils. Curr Biol. 2005;15:1874–1879. doi: 10.1016/j.cub.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 65.Miletic AV, Graham DB, Montgrain V, Fujikawa K, Kloeppel T, Brim K, Weaver B, Schreiber R, Xavier R, Swat W. Vav proteins control MyD88-dependent oxidative burst. Blood. 2007;109:3360–3368. doi: 10.1182/blood-2006-07-033662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Utomo A, Cullere X, Glogauer M, Swat W, Mayadas TN. Vav proteins in neutrophils are required for FcγR-mediated signaling to Rac GTPases and nicotinamide adenine dinucleotide phosphate oxidase component p40(phox) J Immunol. 2006;177:6388–6397. doi: 10.4049/jimmunol.177.9.6388. [DOI] [PubMed] [Google Scholar]
- 67.Duman JG, Tzeng CP, Tu Y-K, Munjal T, Schwechter B, Ho TS-Y, Tolias KF. The adhesion-GPCR BAI1 regulates synaptogenesis by controlling the recruitment of the Par3/Tiam1 polarity complex to synaptic sites. J Neurosci. 2013;33:6964–6978. doi: 10.1523/JNEUROSCI.3978-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stephenson JR, Paavola KJ, Schaefer SA, Kaur B, Van Meir EG, Hall RA. Brain-specific angiogenesis inhibitor-1 signaling, regulation, and enrichment in the postsynaptic density. J Biol Chem. 2013;288:22248–22256. doi: 10.1074/jbc.M113.489757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee CS, Penberthy KK, Wheeler KM, Juncadella IJ, Vandenabeele P, Lysiak JJ, Ravichandran KS. Genetically modifying the levels of a phagocytic receptor influences apoptotic cell clearance and inflammation in vivo. Immunity. 2016 doi: 10.1016/j.immuni.2016.02.005. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol Syst Biol. 2006;2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ly KT, Casanova JE. Abelson tyrosine kinase facilitates Salmonella enterica serovar Typhimurium entry into epithelial cells. Infect Immun. 2009;77:60–69. doi: 10.1128/IAI.00639-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pishchany G, Dickey SE, Skaar EP. Subcellular localization of the Staphylococcus aureus heme iron transport components IsdA and IsdB. Infect Immun. 2009;77:2624–2634. doi: 10.1128/IAI.01531-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. BAI1 localizes to the cell periphery, the perinuclear region, and the sites of bacterial cell association.
Fig. S2. BAI1 promotes cellular microbicidal activity in J774 macrophages.
Fig. S3. Loss of BAI1 impairs Rac activation in response to E. coli in IFN-γ–primed BMDMs.
Fig. S4. Loss of BAI1 impairs intracellular ROS generation but does not affect IFN-γ priming or iNOS production induced by E. coli.
Fig. S5. Mitochondrial ROS produced in response to Gram-negative bacteria is independent of BAI1-mediated recognition and signaling.
Fig. S6. Measurement of disease activity analysis.
Movie S1. BAI1 is enriched at the phagocytic cup.