

Abstract
Chronic viral infections (e.g., HIV, HBV, HCV) represent a significant source of morbidity and mortality with over 500 million people infected worldwide. Dendritic cells (DCs) and macrophages are key cell types for productive viral replication and persistent systemic infection. We demonstrate that the plant virus cowpea mosaic virus (CPMV) displays tropism for such antigen presenting cells in both mice and humans, thus making it an ideal candidate for targeted drug delivery toward viral infections. Furthermore, we show inhibition of a key host protein for viral infection, site-1 protease (S1P), using the small molecule PF-429242 in the model pathogen arenavirus lymphocytic choriomeningitis virus (LCMV) limits viral growth. By packaging PF-429242 in CPMV, we are able to control drug release and efficiently deliver the drug. This sets the groundwork for utilizing the natural tropism of CPMV for a therapeutic approach that specifically targets cell types most commonly subverted by chronic viruses.
Keywords: cowpea mosaic virus, nanoparticles, lymphocytic choriomeningitis virus, site-1 protease, PF-429242, tropism
Graphical abstract
Persistent viral infections1 and cancer represent two of the largest sources of morbidity and mortality, creating a significant economic and humanitarian burden across the world. It is now widely appreciated that modulation of the adaptive immune system is critical for these disease processes to continue.2–4 The control of the adaptive immune system lies largely within antigen presenting cells (APCs). This cell type presents antigens, along with costimulatory molecules and cytokines, to T cells to instruct the host’s adaptive immune response.
Although our understanding of persistent viral infections is incomplete, one unifying feature is that they persist despite an immune response that has evolved to eliminate infections. Central to this immune response are professional antigen presenting cells (pAPCs), such as dendritic cells (DCs) and macrophages; they survey the environment for pathogens and with appropriate stimuli can directly present antigens to T cells to initiate an adaptive immune response.5 Therefore, it is not surprising that many persistent infections have evolved mechanisms that affect pAPC function. Moreover, investigators have speculated that chronic viruses must subvert and exploit pAPC function to persist.1,6–8 Therefore, pAPCs present an attractive target for therapeutic approaches to pathogenic viral infections.
Toward this aim, we apply nonpathogenic viruses, which is becoming a growing field of interest and impact in the medical sector.9–11 Plant viruses are biodegradable and biocompatible materials that offer a high degree of engineerability.12 In this study, we set out to utilize plant virus-based nanocarriers targeting drugs to APCs to overcome persistent viral infections. Specifically, we investigate the 30 nm-sized icosahedral plant virus cowpea mosaic virus (CPMV), as it has been reported to have a natural tropism to APCs, at least in mouse spleens.13 The specificity of CPMV to such cell populations can then be used to partition antiviral drugs to cells critical to resolving the disease process. Although the plant virus does not infect human cells, it can deliver its cargo to the same cells that are targeted by mammalian pathogens.
CPMV is a protein-based platform that can be delivered intravenously or orally as purified nanoparticles or in edible plant tissue. Previous studies indicated that CPMV is well tolerated in animals; at dosages of up to 100 mg/kg no adverse toxic effects were observed, and the particles are removed from cells and tissues within a matter of days.14,15 The structure of CPMV is known to near atomic resolution16 and its surface chemistry (inside and out) is well established,17,18 allowing an exquisite level of functionalizability not yet achievable with synthetic nanoparticles. Additionally, CPMV can be produced at a large scale using plants, which is highly scalable and economic. Thus, CPMV provides a novel nanotechnology with potential applications in medicine.
First, we expand on findings of CPMV’s tropism to APCs in mouse spleens13 by measuring their tropism in the draining lymph node bed of mice as well as in human peripheral blood mononuclear cells (PBMCs). We analyzed the immediate systemic distribution of CPMV in mice by using particles labeled with the fluorophore Alexa Fluor 647 (AF647) through direct conjugation to exterior lysine residues (see the Supporting Information). C57BL/6J mice were injected intraperitoneally (i.p.) with 200 μg CPMV-AF647, and the lymphoid organs important for systemic infections, specifically the spleen and draining inguinal lymph nodes, were harvested 4 h post injection. The tropism of CPMV to various cell types within these organs was analyzed by flow cytometry (Figure 1A). Although there was some tropism to natural killer (NK) cells in the spleen, very few CPMV ended up in T cells, and the majority of the cells positive for CPMV were APCs, in particular B cells, plasmacytoid DCs (pDCs), conventional DCs (cDCs or mDCs), and the macrophage-monocyte family of cells. We found that splenocytes harbored the majority of the particles at this time, but we also detected significant, albeit less, acquisition of CPMV more distally in the inguinal lymph nodes.
Figure 1.
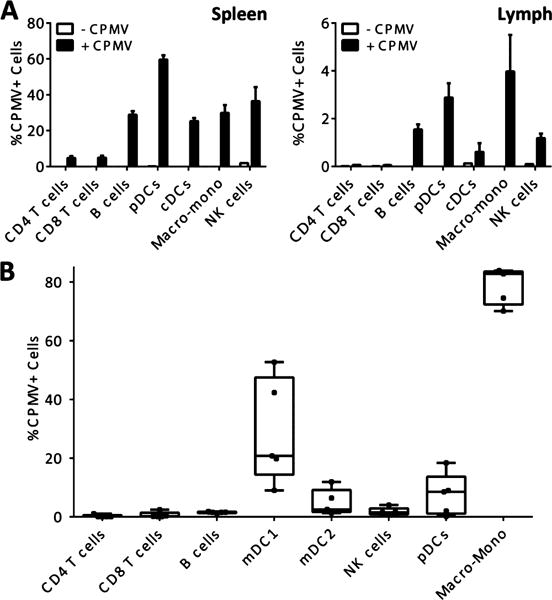
CPMV tropism in vivo and in human PBMC. (A) Tropism measured 4 h post i.p. injection in spleen and draining lymph node of wild-type mice demonstrates an antigen presenting cell preference in vivo. Spleen and lymph node were harvested from vehicle-injected animals and fluorescence was measured from the AF647 channel to define a background gate of less than 0.1%. The percentage of each cell type positive for CPMV-AF647 fluorescence is reported as the mean and SEM from 3 mice. One of 2 similar experiments is shown. (B) CPMV tropism measured 1 h post spinoculation of freshly isolated human PBMCs. PBMCs were analyzed by flow cytometry and fluorescence was measured from the AF647 channel to define a background gate of less than 0.1%. The percentage of each cell type positive for CPMV-AF647 fluorescence is reported from 5 donors using box and whisker plots and the gating strategy outlined in Figures S1 and S2. One of two similar experiments is shown.
We then set out to determine if CPMV tropism is similar in humans compared to mice. PBMCs were isolated from healthy donors, spinoculated with CPMV-AF647 for 1 h at room temperature at 300 g, then analyzed by flow cytometry (Figure 1B, Figures S1–S3). The cells capable of acquiring CPMV in vitro followed a similar distribution as found in mice, with APCs favored. Notably, B cells, which are known to have less potent APC function,19 showed less uptake of CPMV in human cells under these conditions. On the other hand, there was significant tropism observed of CPMV to DCs and the macrophage-monocyte family.
Having established the feasibility of using CPMV to target APCs, we next investigated the ability of CPMV to package and deliver antiviral drugs. In particular, we set out to target and treat cell populations infected by the prototypical arenavirus lymphocytic choriomeningitis virus (LCMV). LCMV infection can be used to address APC function, as its persistence has been linked to its specific targeting of DCs and macrophages. In particular, LCMV has been shown to inhibit expression of costimulatory and major histocompatibility complex (MHC) molecules in DCs.20,21
One requirement for the persistence of LCMV and many other pathogens is site-1 protease (S1P), a host protease used for maturation of their glycoprotein.22,23 We have shown that this event is required for the persistence of LCMV in vivo.20 In addition, others have shown that S1P is required for replication and infection of hepatitis C virus (HCV), among others.24,25 Due to the necessity of S1P for the life cycle of many viruses, S1P has become an increasingly attractive therapeutic target. Inhibition of S1P can be accomplished both genetically and pharmacologically.20,22 By specifically delivering high doses of S1P inhibitor to DCs and macrophages, the therapeutic efficacy is expected to increase while reducing potential off-target effects or undesired clearance.
To target and treat LMCV infection, we packaged and delivered the potent S1P inhibitor PF-429242, which has been shown to inhibit S1P function in vitro and in vivo and has antiviral activity against LCMV and other arenaviruses (Figure S4).26–28 Previous work has demonstrated that CPMV can be loaded with a variety of small molecules through infusion and either electrostatics or affinity associations with encapsulated nucleic acids within the particles.29 We utilized a similar method here to package PF-429242 within CPMV (Figure 2A, B). Infusion of drug into CPMV was allowed to proceed overnight, after which drug not packaged within CPMV was removed by dialysis (see the Supporting Information). We found that PF-429242 could be loaded efficiently into CPMV, with a slow release profile over time (half-life of a few days) when dialyzed or spin filter-purified against 0.1 M potassium phosphate buffer pH 7.0 (Figure 2C). The amount of drug per CPMV was quantified by high performance liquid chromatography (HPLC) using a detection wavelength of 260 nm. After drug loading, transmission electron microscopy (TEM) was used to verify that the infusion process did not grossly alter the structure of the viral nanoparticles (Figure 2D).
Figure 2.
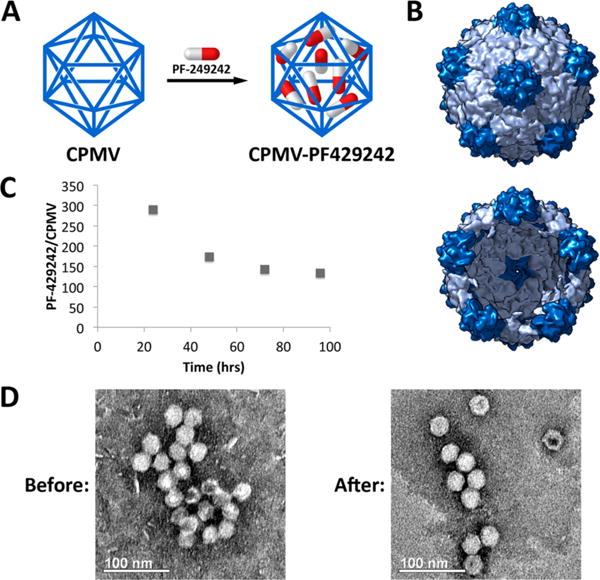
Infusion of PF-429242 into CPMV. (A) Strategy for loading PF-429242 into CPMV. (B) Exterior (top) and interior (bottom) views of the capsid with 1 nm-sized pore at its 5-fold axis. (Figure rendered with Chimera based on coordinates from PDB file 1NY7.) (C) Retention of PF-429242 within CPMV capsid over time when dialyzed against buffer. (D) TEM images of CPMV before and after loading with PF-429242 indicate the particles remain intact and the structure unaltered.
Finally, we sought to determine if the CPMV-PF429242 complexes are able to protect cells from a viral infection. To test specificity, recombinant LCMV virions able to complete their viral lifecycle using an alternative furin serine protease rather than S1P were produced using a previously described protocol (Figure 3A).22 Either LCMVWT virus bearing the S1P recognition site RRLA or mutant LCMVFurin virus encoding a substitution at the S1P recognition site for RRRR, which is recognized by the furin protease and not by S1P, were used to infect BHK-21 baby hamster kidney fibroblast cells. The cells were infected at a multiplicity of infection (MOI) of 0.01. After infection, the cells were then incubated with PF-429242, CPMV, or CPMV-PF429242. The effectiveness of restraining LCMV infection was determined by measuring the titer of the supernatant over the course of 2 days. It was found that 20 μM of PF-429242 packaged within CPMV-PF429242 was as effective as similar doses of unpackaged PF-429242. Due to the slow release profile of PF-429242 from CPMV in solution (see Figure 2C), the similarity in effectiveness of packaged to free drug after both 24 and 48 h can be attributed to cell uptake of CPMV-PF429242. Endocytosis of CPMV and translocation to the endolysosomes is well-documented.29–31 We hypothesize that release of the drug within the cells is triggered by a combination of the lower pH of the endolysosomal compartment and degradation of the CPMV particles. Moreover, with the genetic complementation strategy, we determined that the effect of viral protection was specific to the natural form of LCMV glycoprotein specifically requiring cleavage by S1P and did not occur with S1P-independent LCMVFurin (Figure 3B). We therefore conclude that CPMV-PF429242 can be used to efficiently limit LCMV viral replication in an S1P-dependent fashion.
Figure 3.
CPMV-PF429242 protection against viral infection. (A) Genetic complementation approach to determine specificity of PF-429242 for S1P cleavage of arenavirus glycoprotein. A mutant LCMVFurin virus was reverse engineered to bear the ubiquitously expressed serine protease furin recognition site, RRRR, rather than the S1P recognition site, RRLA, to enable processing of the viral glycoprotein by furin. (B) BHK-21 cells were infected with either LCMVWT (left) or LCMVFurin (right), and the efficacy of PF-429242, free or loaded in CPMV, was tested. The indicated amounts of the S1P inhibitor PF-429242 or CPMV (empty or loaded with PF-429242 to produce 20 μM final concentration of the S1P inhibitor) were then added to the cultures after infection. The supernatant was collected and titered at the indicated time points. PF-429242 packaged within CPMV inhibits S1P cleavage-dependent viral replication (left), whereas LCMVFurin can complete its lifecycle even if S1P activity is inhibited by PF-429242 (right). Average of 2 similar experiments is shown.
In summary, this report addresses the clinically significant use of CPMV as a feasible platform for delivery of therapeutics specifically to APCs. We determined the tropism of CPMV to APCs both in vivo and in freshly isolated human cells. As an example, we demonstrated that this technique could be used to deliver an antiviral protease inhibitor in a mechanistically defined manner. Although we showed here that PF-429242 protects the host from a prototypic LCMV infection, CPMV-PF429242 has the potential to control or eliminate several virus families that require S1P for their life cycle. Conceptually, a much broader group of viral infections could be treated with more potency and less toxicity using this approach. Therefore, advancement of our platform will provide a generally useful technique that will be broadly applicable across fields.
Supplementary Material
Acknowledgments
This study was supported in part by the Skin Diseases Research Center P30AR039750 (DLP), startup institutional funds (DLP), Doris Duke Charitable Foundation Award (DLP), STERIS Corporation grant (DLP, NFS), CNIHR subaward from UAB CFAR 2P30AI027767-26 RA (DLP), Cleveland Foundation (DLP), National Science Foundation grant CMMI 1333651 (NFS), Mt. Sinai Foundation (NFS), and an AHA predoctoral fellowship (AMW).
ABBREVIATIONS
- LCMV
lymphocytic choriomeningitis virus
- APC
antigen presenting cells
- MHC
major histocompatibility complex
- S1P
site-1 protease
- HCV
hepatitis C virus
- CPMV
cowpea mosaic virus
- AF647
Alexa Fluor 647
- PBMC
peripheral blood mononuclear cells
- TEM
transmission electron microscopy
- HPLC
high-performance liquid chromatography
- PFU
plaque-forming units
- FFU
focus-forming units
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsbiomaterials.5b00344.
Experimental details and supporting figures (PDF)
Author Contributions
N.F.S. and D.L.P. conceived and designed the research. A.M.W. loaded and characterized the particle. N.L. and X.Z. performed cell and animal experiments. The manuscript was written through contributions of all authors.
Notes
The authors declare no competing financial interest.
References
- 1.Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell. 2009;138:30–50. doi: 10.1016/j.cell.2009.06.036. [DOI] [PubMed] [Google Scholar]
- 2.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bowen DG, Walker CM. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature. 2005;436:946–952. doi: 10.1038/nature04079. [DOI] [PubMed] [Google Scholar]
- 4.Hogue IB, Bajaria SH, Fallert BA, Qin S, Reinhart TA, Kirschner DE. The dual role of dendritic cells in the immune response to human immunodeficiency virus type 1 infection. J Gen Virol. 2008;89:2228–2239. doi: 10.1099/vir.0.83600-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol. 2012;30:1–22. doi: 10.1146/annurev-immunol-100311-102839. [DOI] [PubMed] [Google Scholar]
- 6.Rahman S, Khan ZK, Jain P. The tug-of-war between dendritic cells and human chronic viruses. Int Rev Immunol. 2011;30:341–365. doi: 10.3109/08830185.2011.561506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oldstone M. Viral persistence: parameters, mechanisms and future predictions. Virology. 2006;344:111–118. doi: 10.1016/j.virol.2005.09.028. [DOI] [PubMed] [Google Scholar]
- 8.Liu B, Woltman A, Janssen H, Boonstra A. Modulation of dendritic cell function by persistent viruses. J Leukocyte Biol. 2009;85:205–214. doi: 10.1189/jlb.0408241. [DOI] [PubMed] [Google Scholar]
- 9.Chitale R. Merck hopes to extend gardasil vaccine to men. J Natl Cancer Inst. 2009;101:222–223. doi: 10.1093/jnci/djp014. [DOI] [PubMed] [Google Scholar]
- 10.Liu TC, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nat Clin Pract Oncol. 2007;4:101–117. doi: 10.1038/ncponc0736. [DOI] [PubMed] [Google Scholar]
- 11.Shirakawa T. Clinical trial design for adenoviral gene therapy products. Drug News Perspect. 2009;22:140–145. doi: 10.1358/dnp.2009.22.3.1354090. [DOI] [PubMed] [Google Scholar]
- 12.Manchester M, Singh P. Virus-based nanoparticles (VNPs): platform technologies for diagnostic imaging. Adv Drug Delivery Rev. 2006;58:1505–1522. doi: 10.1016/j.addr.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 13.Gonzalez MJ, Plummer EM, Rae CS, Manchester M. Interaction of Cowpea mosaic virus (CPMV) nanoparticles with antigen presenting cells in vitro and in vivo. PLoS One. 2009;4:e7981. doi: 10.1371/journal.pone.0007981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rae CS, Khor IW, Wang Q, Destito G, Gonzalez MJ, Singh P, Thomas DM, Estrada MN, Powell E, Finn MG, Manchester M. Systemic trafficking of plant virus nanoparticles in mice via the oral route. Virology. 2005;343:224–235. doi: 10.1016/j.virol.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 15.Singh P, Prasuhn D, Yeh RM, Destito G, Rae CS, Osborn K, Finn MG, Manchester M. Bio-distribution, toxicity and pathology of cowpea mosaic virus nanoparticles in vivo. J Controlled Release. 2007;120:41–50. doi: 10.1016/j.jconrel.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin T, Chen Z, Usha R, Stauffacher CV, Dai JB, Schmidt T, Johnson JE. The refined crystal structure of cowpea mosaic virus at 2.8 Å resolution. Virology. 1999;265:20–34. doi: 10.1006/viro.1999.0038. [DOI] [PubMed] [Google Scholar]
- 17.Wen AM, Shukla S, Saxena P, Aljabali AA, Yildiz I, Dey S, Mealy JE, Yang AC, Evans DJ, Lomonossoff GP, Steinmetz NF. Interior engineering of a viral nanoparticle and its tumor homing properties. Biomacromolecules. 2012;13:3990–4001. doi: 10.1021/bm301278f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Q, Kaltgrad E, Lin T, Johnson JE, Finn MG. Natural supramolecular building blocks. Wild-type cowpea mosaic virus. Chem Biol. 2002;9:805–811. doi: 10.1016/s1074-5521(02)00165-5. [DOI] [PubMed] [Google Scholar]
- 19.Roche PA, Furuta K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol. 2015;15:203–216. doi: 10.1038/nri3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Popkin DL, Teijaro JR, Sullivan BM, Urata S, Rutschmann S, de la Torre JC, Kunz S, Beutler B, Oldstone M. Hypomorphic mutation in the site-1 protease Mbtps1 endows resistance to persistent viral infection in a cell-specific manner. Cell Host Microbe. 2011;9:212–222. doi: 10.1016/j.chom.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sevilla N, McGavern DB, Teng C, Kunz S, Oldstone MB. Viral targeting of hematopoietic progenitors and inhibition of DC maturation as a dual strategy for immune subversion. J Clin Invest. 2004;113:737–745. doi: 10.1172/JCI20243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rojek JM, Pasqual G, Sanchez AB, Nguyen NT, de la Torre JC, Kunz S. Targeting the proteolytic processing of the viral glycoprotein precursor is a promising novel antiviral strategy against arenaviruses. J Virol. 2010;84:573–584. doi: 10.1128/JVI.01697-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brandl K, Rutschmann S, Crozat K, Berger M, Du X, Moresco EMY, Murray A, Beutler B. Record for woodrat. updated 12/12/2013, last accessed 10/17/2015 http://mutagenetix.utsouthwestern.edu.
- 24.Olmstead AD, Knecht W, Lazarov I, Dixit SB, Jean F. Human subtilase SKI-1/S1P is a master regulator of the HCV Lifecycle and a potential host cell target for developing indirect-acting antiviral agents. PLoS Pathog. 2012;8:e1002468. doi: 10.1371/journal.ppat.1002468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blanchet M, Seidah NG, Labonte P. SKI-1/S1P inhibition: A promising surrogate to statins to block Hepatitis C virus replication. Antiviral Res. 2012;95:159–166. doi: 10.1016/j.antiviral.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 26.Urata S, Yun N, Pasquato A, Paessler S, Kunz S, de la Torre JC. Antiviral activity of a small-molecule inhibitor of arenavirus glycoprotein processing by the cellular site 1 protease. J Virol. 2011;85:795–803. doi: 10.1128/JVI.02019-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hawkins J, Robbins M, Warren L, Xia D, Petras S, Valentine J, Varghese A, Wang I, Subashi T, Shelly L, Hay B, Landschulz K, Geoghegan K, Harwood HJ. Pharmacologic inhibition of site 1 protease activity inhibits sterol regulatory element-binding protein processing and reduces lipogenic enzyme gene expression and lipid synthesis in cultured cells and experimental animals. J Pharmacol Exp Ther. 2008;326:801–808. doi: 10.1124/jpet.108.139626. [DOI] [PubMed] [Google Scholar]
- 28.Hay B, Abrams B, Zumbrunn A, Valentine J, Warren L, Petras S, Shelly L, Xia A, Varghese A, Hawkins J, Van Camp J, Robbins M, Landschulz K, Harwood HJ. Aminopyrrolidineamide inhibitors of site-1 protease. Bioorg Med Chem Lett. 2007;17:4411–4414. doi: 10.1016/j.bmcl.2007.06.031. [DOI] [PubMed] [Google Scholar]
- 29.Yildiz I, Lee KL, Chen K, Shukla S, Steinmetz NF. Infusion of imaging and therapeutic molecules into the plant virus-based carrier cowpea mosaic virus: cargo-loading and delivery. J Controlled Release. 2013;172:568–578. doi: 10.1016/j.jconrel.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aljabali AA, Shukla S, Lomonossoff GP, Steinmetz NF, Evans DJ. CPMV-DOX delivers. Mol Pharmaceutics. 2013;10:3–10. doi: 10.1021/mp3002057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wen AM, Infusino M, De Luca A, Kernan DL, Czapar AE, Strangi G, Steinmetz NF. Interface of physics and biology: engineering virus-based nanoparticles for biophotonics. Bioconjugate Chem. 2015;26:51–62. doi: 10.1021/bc500524f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.