

Article first published online 1 April 2015.
Supplemental Digital Content is Available in the Text.
Key Words: phagolysosomes, chemotaxis, ciprofloxacin, doxycycline
Abstract
Background:
Crohn's disease (CD) is associated with defective innate immunity, including impaired neutrophil chemotaxis, and mucosal invasion by bacteria, particularly adherent and invasive Escherichia coli that replicate inside macrophage phagolysosomes. We compared CD and healthy control (HC) macrophages for their abilities to kill E. coli and generate neutrophil chemoattractants and also assessed the effects of hydroxychloroquine (HCQ) and vitamin D on killing of phagocytosed E. coli.
Methods:
Peripheral blood monocyte-derived macrophages from CD and HC were compared for bacterial killing and generation of neutrophil chemoattractants in response to CD-derived E. coli. Escherichia coli replication was also assessed in the presence and absence of HCQ, alone and with antibiotics, and vitamin D.
Results:
Monocyte-derived macrophages from patients with CD were similar to HC in allowing replication of phagocytosed CD-derived E. coli: HM605 {CD: N = 10, mean fold replication in 3 hr = 1.08 (95% confidence interval [CI], 0.39–1.78); HC: N = 9, 1.50 (95% CI, 1.02–1.97); P = 0.15} and also in generation of neutrophil chemoattractants in response to E. coli (mean fold chemotaxis relative to control: CD = 2.55 [95% CI, 2.31–2.80]; HC = 2.65 [95% CI, 2.46–2.85], P = 0.42). HCQ and 1,25 OH2-vitamin D3 both caused dose-dependent inhibition of intramacrophage E. coli replication 3-hour postinfection; HCQ: 73.9% inhibition (P < 0.001) at 1 μg/mL, accompanied by raised intraphagosomal pH, and 1,25 OH2-vitamin D3: 80.7% inhibition (P < 0.05) at 80 nM. HCQ had synergistic effects with doxycycline and ciprofloxacin.
Conclusions:
CD and HC macrophages perform similarly in allowing replication of phagocytosed E. coli and generating neutrophil chemoattractants. Replication of phagocytosed E. coli was substantially decreased by HCQ and vitamin D. These warrant further therapeutic trials in CD in combination with relevant antibiotics.
There is growing consensus that Crohn's disease (CD) results from an altered relationship between the immune system and intestinal bacteria.1 Changes in the fecal microbiota, particularly decreased diversity,2 are also found in animal models of gut inflammation,3 but the mucosa-associated microbiota shows more specific changes, even in inactive disease. These include decreased colonization by the probiotic Faecalibacterium prausnitzii and increase in Escherichia coli.4–6 Similar changes have been found in the treatment-naive mucosa-associated microbiome in pediatric CD.7
CD mucosa-associated E. coli commonly adhere to and invade epithelial cell-lines and are therefore termed adherent–invasive E. coli (AIEC). Although originally isolated from CD ileal mucosa, they are also found in the colon.5,7 AIEC taken up by macrophages are initially destroyed within autophagosomes,8 but some escape this process and replicate within the acidic environment of phagolysosomes.9 Phagocytosed AIEC induce granuloma formation in vitro10,11 and in animal models.12 This, and the common presence of E. coli DNA in CD-associated granulomas,11 suggests a possible pathogenic role. AIEC are thought to translocate through microfold cells (M cells) that account for about 5% of the “dome” epithelium overlying Peyer's patches in the distal ileum, and lymphoid follicles in the colon, the sites of the earliest lesions seen in CD.13,14 CD AIEC commonly express long polar fimbriae essential for M-cell translocation.6,15
Patients with CD commonly have polymorphisms in genes (NOD2/CARD15, IRGM, and ATG16L1) linked to pathogen recognition and autophagy relevant to killing of bacteria within macrophages.16 CD is also associated with defective recruitment of neutrophils to sites of dermal17 and mucosal18 wounding and intradermally injected killed E. coli.19 Chemotaxis of CD neutrophils is normal when studied ex vivo,20–22 and it has been suggested that delayed chemotaxis in vivo results from defective macrophage chemokine release.19 The delay in neutrophil chemotaxis could then contribute to increased bacterial uptake by macrophages.
CD peripheral blood monocytes have normal ability to kill Gram-positive bacteria,23 but there has been no published study of their killing of Gram-negative bacteria. This is important not only because of the possible relevance of Gram-negatives such as E. coli in CD pathogenesis but also because macrophages have distinct mechanisms for killing of Gram-negative organisms. This includes interaction with a signaling lymphocyte-activation molecule involved in activation of NADPH oxidase within phagosomes.24
Proof that intramacrophage E. coli replication may have a pathogenic role in CD will depend on resolution of the disease in response to treatments that target these E. coli. Antibiotic therapy has to date been only modestly effective,25 possibly either because single antibiotics have been used, with multidrug resistance seen in 61.5% of AIEC,26 or because some antibiotics, particularly penicillins and gentamicin, are unable to kill bacteria within macrophage vesicles.27
Macrophages are relatively ineffective at killing bacteria in comparison with neutrophils.28 In Q-fever and Whipple's disease, where bacteria, Coxiella burnetii and Tropheryma whipplei, respectively, replicate inside macrophage phagosomes, addition of hydroxychloroquine (HCQ) to antibiotics has greatly improved eradication rates.29–31 Bacteria, including AIEC, that replicate within macrophage vesicles typically require an acid environment.9 HCQ is a mild base that is concentrated within intracellular vesicles thus raising their pH.31
Vitamin D deficiency has been implicated in conditions such as multiple sclerosis and CD that are more common in countries furthest from the equator.32 Vitamin D is important for normal function of the innate immune system including autophagy and a controlled trial of vitamin D supplementation in CD, regardless of their initial vitamin D status, only just failed (P = 0.06) to significantly reduce relapse.33
Here, we have assessed the ability of CD monocyte-derived macrophages (MDM) to kill E. coli and to generate a neutrophil chemotactic response. We then assessed the ability of vitamin D and HCQ, at clinically achievable concentrations, to enhance macrophage killing of phagocytosed E. coli.
METHODS
Ethical Considerations
Ethical approval was obtained from the National Research Ethics Service Committee North-West England (study approval number 09/H1010/64) to take peripheral venous blood after informed consent from patients with CD recruited from the Royal Liverpool University Hospital and healthy controls (HC) recruited from Liverpool hospital and University staff.
Bacterial Strains and Culture
Two representative CD mucosa-associated AIEC isolates were studied; HM605, a colonic isolate,5 and LF82, an ileal isolate.4 Both isolates replicate within phagolysosomes of murine and human MDM.9,27,34 Laboratory strain E. coli K12 (ATCC 29425; Manassas, VA) and Staphylococcus aureus Oxford stain (NCTC 6571; Public Health England; Porton Down, United Kingdom) were also tested. Isolates were grown overnight on Luria-Bertani (LB) agar at 37°C in air, washed 3 times in sterile phosphate-buffered saline pH 7.3 (Life Technologies; Paisley, United Kingdom), and resuspended to an optical density (OD550 nm) equivalent to 109 bacteria per milliliter.
Murine Macrophage Cell-line Culture
J774A.1 murine macrophages (91051511) obtained from the European Collection of Cell Cultures (Porton Down; United Kingdom) were cultured at 37°C in RPMI medium supplemented with 10% vol/vol fetal bovine serum, 2 mM glutamine, 50 U/mL penicillin, and 50 μg/mL streptomycin, in a humidified atmosphere of 5% CO2 per 95% air. Macrophages were passaged by scraping twice weekly, up to passage 19.
Isolation of Human Peripheral Blood MDM and Neutrophils
Peripheral venous blood was taken after informed consent from 10 patients with CD recruited from the Royal Liverpool University Hospital and 10 HC recruited from Liverpool hospital and University staff. Tables, Supplemental Digital Content 1, http://links.lww.com/IBD/A836, for inclusion/exclusion criteria (see Table 1, Supplemental Digital Content 1, http://links.lww.com/IBD/A836), baseline characteristics (equivalent apart from higher hsCRP in CD) (see Table 2, Supplemental Digital Content 1, http://links.lww.com/IBD/A836), and patient characteristics (see Table 3, Supplemental Digital Content 1, http://links.lww.com/IBD/A836). Five patients with CD had active disease (Harvey–Bradshaw Index > 4). None were receiving immunosuppressants, corticosteroids, or anti-tumor necrosis factor (TNF) therapy, and none had features of sepsis (median hsCRP = 5.9 mg/L; range, 1.04–18.3 mg/L) at the time of sampling. Blood (50 mL) was immediately heparinized using unfractionated heparin sodium 5 U/mL (Wockhardt UK Ltd, Wrexham; Wales), mixed 1:1 with phosphate-buffered saline, layered over Lymphoprep (Alere; Stockport, United Kingdom), and centrifuged at 800g for 20 minutes at room temperature. Mononuclear cells were aspirated, washed, resuspended in RPMI supplemented with 20 mM HEPES pH 7.4, 100 U/mL penicillin, and 100 μg/mL streptomycin, adjusted to 5 × 106 cells per milliliter and seeded into 100 mm Nunc culture dishes (VWR; Lutterworth, United Kingdom). After 2-hour incubation at 37°C, nonadherent cells were removed by washing and adherent monocytes differentiated into macrophages over 5 days.19
Neutrophils were isolated from heparinized venous blood using Polymorphprep (Alere) according to manufacturer's instructions and, after removal of red cells by hypotonic lysis, resuspended in RPMI with 20 mM HEPES pH 7.4, 0.5% vol/vol bovine serum albumin, 2 mM CaCl2, and 2 mM MgCl2.
Intramacrophage Bacterial Killing
Ability of human MDM and J774A.1 macrophages to kill phagocytosed bacteria was assessed by a gentamicin protection assay.27 Macrophage monolayers were inoculated with bacteria at a multiplicity of infection of 25 (for MDM) or 10 (J774.A1). After 2-hour infection, macrophages were washed thrice with sterile phosphate-buffered saline and incubated for 1 hour in RPMI containing 10% vol/vol fetal bovine serum, 2 mM glutamine (for J774.A1) or in X-Vivo 15 (for MDMs), and 20 μg/mL gentamicin to kill adherent, noninternalized bacteria. Macrophages were washed and lysed with 1% vol/vol Triton X-100 for 5 minutes to release internalized bacteria. Bacteria colony-forming units were counted after overnight culture on LB agar. Parallel plates were cultured for a further 3 hours after 1-hour incubation with gentamicin (6 hr in total), again followed by lysis and overnight colony-forming units counting. Data were expressed as fold change of recovered intramacrophage bacteria at 6 hours relative to 3 hours.
Effects of HCQ, with and Without Antibiotics, and Vitamin D on Intramacrophage Survival of CD AIEC
Doxycycline and ciprofloxacin were tested at Cmax (peak serum concentration achieved with standard oral dosing) and 10% Cmax, based on published data.35,36 HCQ concentrations were based on published steady state concentrations.37 Concentrations of 1,25 OH2-vitamin D3 were those defined as deficient in human serum (<20 nM)32,38 and that required for optimal immune cell function (>80 nM).32,39
Cytokine Enzyme-linked Immunosorbent Assays
Interleukin 6 (IL-6), IL-8, and TNF-α were quantified by enzyme-linked immunosorbent assay as per manufacturer's instructions (R&D systems; Abingdon, United Kingdom; IL-6, D6050; IL-8, D800C; TNF-α, DTA00C). Murine IL-6 and TNF-α released by J774A.1 macrophages were also determined by enzyme-linked immunosorbent assay (R&D systems; IL-6, M6000B; TNF-α, SMTA00B). OD450 nm was measured using a Sunrise microplate reader (Tecan; Theale, United Kingdom).
Neutrophil Chemotaxis Assay
Neutrophil chemotaxis was quantified using a CytoSelect 96-Well Cell Migration Assay, 3 μm pore diameter (Cambridge Bioscience; Cambridge, United Kingdom) as per manufacturer's instructions. Chemoattractant in the lower chamber was X-Vivo 15 medium from 105 MDM either uninfected or infected for 6 hours with E. coli HM605 or K12 (multiplicity of infection of 25). CyQuantGR fluorescence was measured using an F200 microplate reader (Tecan); excitation λ 485 nm, emission λ 535 nm.
Effect of HCQ on Macrophage Intraphagolysosome pH
J774A.1 macrophages cultured in 35 mm CELLview dishes (Greiner Bio-One; Stonehouse, United Kingdom) were washed and incubated in phenol red-free RPMI containing 10 μg/mL pHrodo-conjugated E. coli bioparticles and HCQ (0–10 μg/mL) for 3 hours at 37°C. Imaging was by a Zeiss LSM-510 meta-laser confocal microscope with atmosphere- and temperature-controlled stage (Carl Zeiss AG, Oberkochen, Germany). Ten random high-powered fields were imaged for each treatment, processed using LSM-5 software (Zeiss AG), and fluorescence intensity analyzed with AQM-6 (Kinetic Imaging; Nottingham, United Kingdom). A pH-calibration curve was constructed by incubation of pHrodo bioparticle-loaded macrophages with buffers (pH 4–7) containing ionophores nigericin (10 μM) and vanilomycin (10 μM) plus the vacuolar H+-ATPase inhibitor bafilomycin (0.1 μM).40 NH4Cl (2 mM) was used as an intraphagolysosome alkalinization control.
Influence of Intracellular Iron Availability on the Action of HCQ
To determine whether the antibacterial actions of HCQ were mediated through pH-mediated restriction of intramacrophage iron, macrophages were pretreated with either 10 μM ferric citrate, which exhibits pH-dependent solubility and release of free ferric iron, or 10 μM ferric nitrilotriacetic acid (FeNTA), which releases free ferric iron irrespective of pH.41
Genomic Analysis for CD-associated Polymorphisms in NOD2/CARD15, ATG16L1, and IRGM
Genomic DNA was extracted using a Nucleon BACC3 kit (Gen-Probe; Livingston, United Kingdom) from EDTA-treated peripheral venous blood taken from patients with CD and HC in whom macrophage function studies were also performed. DNA yield was determined using PicoGreen (Life Technologies), and samples normalized to 10 ng/μL. Single nucleotide polymorphisms were analyzed by TaqMan quantitative polymerase chain reaction for NOD2/CARD15 (rs2066844, rs2066845, and rs2066847), ATG16L1 (rs2241880), and IRGM (rs13361189).42
Cytotoxicity Assay
Macrophage viability was assessed by measuring release of intracellular adenylate kinase into supernatants over 6 hours after drug treatments and/or bacterial infection using the ToxiLight bioassay kit as per manufacturer's instructions (Lonza; Slough, United Kingdom). Luminescence was measured on a Tecan F200 microplate reader.
Statistical Analysis
N indicates number of patients or independent experiments performed with cells seeded in triplicate for each experiment. Sample groups were assessed for normality and equality of variances. Comparison of CD patient data versus HC was by Mann–Whitney U test. Dose–response experiments were analyzed using Cuzick's test for trend, followed, where significant, by Dunnett's test. Other multiple treatment experiments were analyzed using one-way analysis of variance followed by selected pair-wise comparison of treatment means (StatsDirect; Sale, United Kingdom). Differences were considered significant when two-tailed, P < 0.05.
RESULTS
MDM from HC and Patients with CD Have Equivalent Ability to Kill Intracellular Bacteria
Neither CD nor HC peripheral blood MDM were very effective at killing E. coli. CD MDM allowed net replication of phagocytosed CD-derived E. coli HM605 (CD: N = 10 patients, mean fold replication over 3 hours = 1.08 [95% CI, 0.39–1.78]) as did HC MDM (N = 9, 1.50 [95% CI, 1.02–1.97]; P = 0.15; Mann–Whitney U test). Similar results were obtained for replication of phagocytosed E. coli K12 (CD: N = 10, mean fold replication over 3 hours = 0.54 [95% CI, 0.24–0.84]; HC: N = 10, 0.86 [95% CI, 0.47–1.26]; P = 0.14) and also for phagocytosed S. aureus (CD: N = 10, mean fold replication = 0.37 [95% CI, 0.18–0.55]; HC: N = 10, 0.48 [95% CI, 0.39–0.57]; P = 0.09) (Fig. 1). There was a nonsignificant trend toward increased intramacrophage replication of HM605 among patients with CD with active disease (mean fold replication = 1.74 [95% CI, 0.67–2.81]) compared with those with inactive disease (mean = 0.43 [95% CI, 0.0–0.87], P = 0.10), but replication of HM605 in patients with active disease was similar to that seen in HC (Fig. 2). No correlation was seen between the total number of affected NOD2/CARD15 and ATG16L1 alleles and ability to kill intramacrophage HM605, but none of the patients with CD studied were homozygous for affected NOD2/CARD15 or IRGM alleles, and only 1 was homozygous for affected ATG16L1 (Fig. 2).
FIGURE 1.
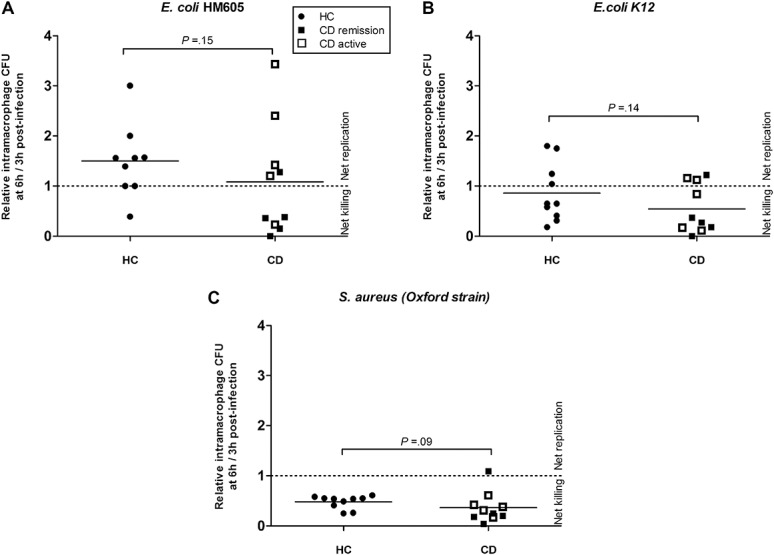
MDM isolated from patients with CD and HC possess similar ability to kill phagocytosed bacteria. No significant differences were seen between CD and HC in the killing of any of the bacteria tested; (A) AIEC HM605, (B) E. coli K12, and (C) S. aureus. Each point represents individual patient mean of triplicate samples, horizontal bar indicates overall mean.
FIGURE 2.
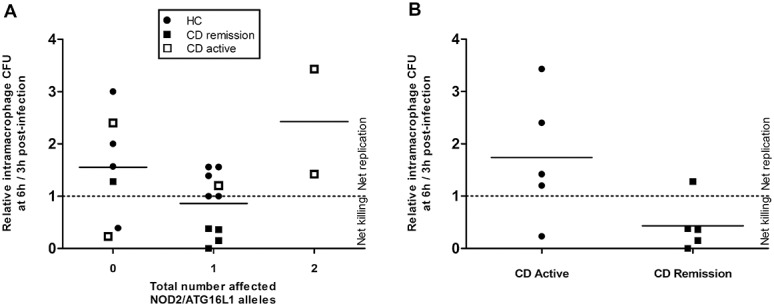
Presence of NOD2/CARD15 and ATG16L1 variants were not associated with altered ability to kill intramacrophage CD-derived E. coli HM605. Patient NOD2/CARD15 and ATG16L1 status reported as number of disease associated alleles. A, Killing of intramacrophage HM605 was unaffected by the total number of variant NOD2 and ATG16L1 alleles (possible range, 0–4: zero affected, N = 7; one affected, N = 10; two affected, N = 2) (zero versus one affected allele, P = 0.18; zero versus two affected alleles, P = 0.65; one versus two affected alleles, P = 0.20, analysis of variance). B, Effect of disease activity on killing of E. coli HM605 (P = 0.10, Mann–Whitney U test).
MDM from HC and Patients with CD Have Equivalent Ability to Generate Neutrophil Chemoattractants in Response to E. coli
HC and CD E. coli–infected MDM were more able to induce neutrophil chemotaxis than uninfected MDM but with no significant difference between CD (N = 10 patients) and HC (N = 7): HM605 (CD: mean fold change in neutrophil chemotaxis = 2.55 [95% CI, 2.31–2.80]; HC: 2.65 [95% CI, 2.46–2.85], P = 0.42 Mann–Whitney U test); K12 (CD: 2.20 [95% CI, 1.78–2.63]; HC: 2.23 [95% CI, 1.86–2.60], P = 0.89) (Fig. 3). No significant difference was seen in the chemotactic response generated by MDM derived from patients with active CD compared with those with inactive disease (P = 0.60) (see Table 3, Supplemental Digital Content 1, http://links.lww.com/IBD/A836).
FIGURE 3.

E. coli-infected MDM from patients with CD and HC have equivalent ability to generate neutrophil chemoattractants. A, MDM-induced neutrophil chemotaxis was equivalent between CD (N = 10) and HC (N = 9) whether uninfected or infected with E. coli (either AIEC HM605 or K12). Infected macrophages induced greater neutrophil chemotaxis than uninfected controls (P < 0.001; analysis of variance). MDM production of (B) IL-8, (C) IL-6, and (D) TNF-α in response to HM605 infection were also equivalent between CD (N = 10) and HC (N = 7). All assays performed in triplicate. P values by Mann–Whitney U test.
Production of cytokines by human MDM in response to HM605 infection was also equivalent between CD (N = 10 patients) and HC (N = 7); TNF-α (CD: mean = 1915.3 pg/mL [95% CI, 1711.6–2119.0]; HC: mean = 1823.2 pg/mL [95% CI, 1590.6–2055.7], P = 0.27); IL-6 (CD: mean = 1720.1 pg/mL [95% CI, 1178.0–2262.3]; HC: mean = 1209.9 pg/mL [95% CI, 710.6–1709.1], P = 0.23); IL-8 (CD: mean = 6643.9 pg/mL [95% CI, 5065.5–8222.4]; HC: mean = 6639.0 pg/mL [95% CI, 4064.6–9213.4], P = 0.89).
HCQ Enhances Intramacrophage Killing of AIEC in Murine and Human Macrophages
HCQ induced a dose-dependent enhancement of intracellular killing of HM605 by J774A.1 macrophages (P < 0.001, Cuzick's test for trend). Net replication of E. coli (mean fold replication over 3 hr: 3.59 ± 0.35) occurred in the untreated control, but, at concentrations of HCQ at ≥2 μg/mL, net killing of E. coli occurred (Fig. 4A). Similar results were obtained with nonhydroxylated chloroquine (see Fig, Supplemental Digital Content 2, http://links.lww.com/IBD/A837). HCQ also induced dose-dependent enhancement of intracellular killing of HM605 in human MDM (P < 0.001, Cuzick's test) (Fig. 4B). It should be noted that HCQ, up to 10 μg/mL, showed no significant direct effect on growth of HM605 in broth over 7 hours monitored at OD600 nm (Fig. 4C) and by colony-forming units on LB agar (see Fig, Supplemental Digital Content 3, http://links.lww.com/IBD/A838).
FIGURE 4.
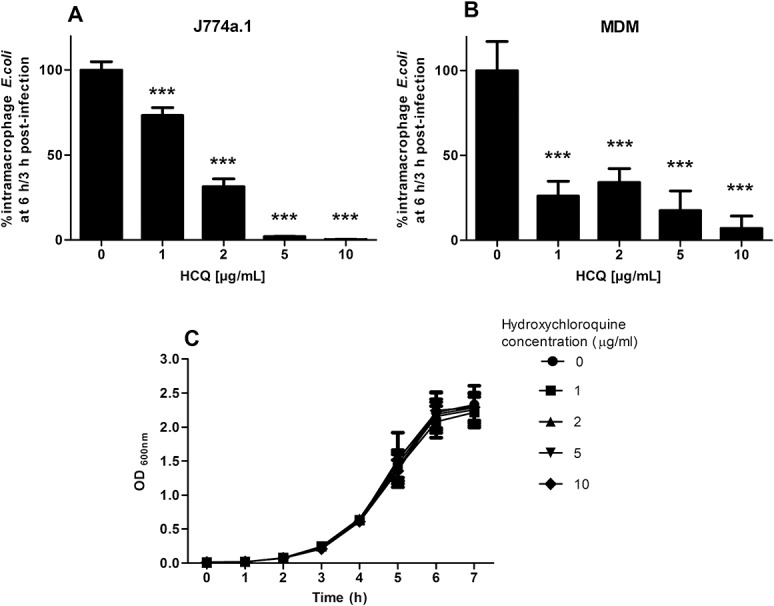
HCQ enhances intramacrophage killing of AIEC. HCQ treatment causes a dose-dependent decrease in survival of intramacrophage AIEC HM605 in (A) J774A.1 murine macrophages (N = 3, Cuzick's test for trend P < 0.001) and (B) healthy human MDM (N = 5, Cuzick's test; P < 0.001). ***P < 0.001, Dunnett's test versus control. C, HCQ did not have a direct bactericidal action as observed by growth in broth, measured at OD600 nm (N = 3).
HCQ Raises Phagolysosomal pH and Alters Intracellular Iron Metabolism
Addition of HCQ to HM605-infected J774A.1 macrophages caused a dose-dependent increase in phagolysosomal pH (N = 6, P < 0.005, Cuzick's test). From a baseline of 5.21 ± 0.31 (mean ± SEM), pH increased stepwise to 5.56 ± 0.36 at HCQ 5 μg/mL and 7.25 ± 0.47 at 10 μg/mL (Fig. 5).
FIGURE 5.

HCQ leads to alkalinization of macrophage phagolysosomes, and its effects on killing of phagocytosed E. coli are only partly reversed by iron supplementation. A, HCQ increased phagolysosomal pH in AIEC HM605-infected J774A.1 macrophages as assessed by confocal microscopy (n = 6, Cuzick's test for trend P < 0.005). **P < 0.01 Dunnett's test versus control. B, Representative standard curve of mean confocal-acquired fluorescence of pHrodo E. coli bioparticle-loaded macrophages with phagolysosomes equalized in calibration buffers at pH 4 to pH 7 (n = 3). C, Confocal images illustrating increased phagolysosomal pH with 10 μg/mL HCQ (HCQ10) as indicated by decreased pHrodo fluorescence compared with untreated control (left panel). Onset of the change in pH occurs within 30 minutes (right panel). D, Iron supplementation only partially antagonizes the effect of HCQ on intracellular survival of AIEC HM605 in J774A.1 macrophages (N = 3; *P < 0.05, **P < 0.01, ***P < 0.001 analysis of variance).
Cotreatment with the iron substrates, ferric citrate (allowing pH-dependent release of ferric iron) and FeNTA (allowing release of ferric iron irrespective of pH), only partially reversed the effect of HCQ, with a significant but partial effect seen only at 5 to 10 μg/mL HCQ (Fig. 5).
A modest trend toward increased cathepsin G (P = 0.03, Cuzick's test) and macrophage elastase MMP12 (P = 0.04) activity was seen in whole cell lysates of HM605-infected macrophages treated with HCQ (see Data, Supplemental Digital Content 4, http://links.lww.com/IBD/A839).
HCQ decreased intracellular respiratory burst as assessed by flow cytometric measurement of dihydrorhodamine fluorescence (P < 0.05, Cuzick's test) but did not affect total respiratory burst as assessed by lucigenin-mediated luminescence (P = 0.73, Cuzick's test); (see Data, Supplemental Digital Content 5, http://links.lww.com/IBD/A840). HCQ neither alter production of TNF-α (P = 0.31, Cuzick's test) nor IL-6 (P = 0.79) from HM605-infected J774A.1 murine macrophages (Fig, Supplemental Digital Content 6, http://links.lww.com/IBD/A841).
HCQ Enhances the Antimicrobial Effect of Doxycycline and Ciprofloxacin Against AIEC Replicating Within Macrophages
Doxycycline at 10% Cmax was ineffective at killing intramacrophage HM605 (89.01% ± 8.57%, mean% ± SEM% survival relative to control, P = 0.08) and only achieved modest decreases in viable bacteria at Cmax (75.5% ± 6.72%, P < 0.01). However, at both 10% Cmax (48.9% ± 5.36%, P < 0.001) and Cmax (34.49 ± 4.71, P < 0.001), cotreatment with HCQ plus doxycycline led to significant enhancement of bacterial killing relative to control and was also significantly more effective than antibiotic monotherapy (P < 0.001 at both 10% Cmax and Cmax; N = 6). (Fig. 6).
FIGURE 6.
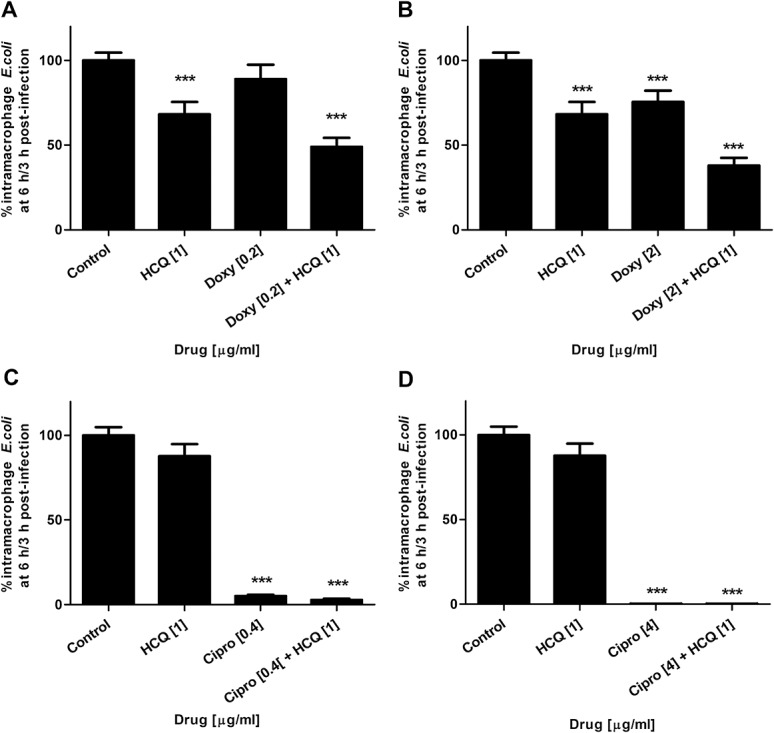
HCQ enhances antibiotic efficacy against intramacrophage AIEC. Combination therapy (antibiotic plus HCQ, 1 μg/mL) was more effective at killing AIEC HM605 within J774.A1 macrophages than antibiotic monotherapy at (A) 10% Cmax doxycycline and (B) Cmax doxycyline [2 μg/mL]; N = 6. Likewise, for (C) 10% Cmax ciprofloxacin. D, No additional benefit was seen with ciprofloxacin at Cmax [4 μg/mL]; N = 3. *P < 0.05, ***P < 0.001, ***P < 0.001; analysis of variance.
Ciprofloxacin effectively killed intramacrophage HM605 at 10% Cmax (4.95% ± 0.92%, mean% ± SEM% survival relative to control, P < 0.001) and led to near complete killing at Cmax (0.2% ± 0.0%, P < 0.001). Combination of HCQ with ciprofloxacin was more effective than antibiotic monotherapy at 10% Cmax (2.8% ± 0.82%, P < 0.05), but at Cmax, no additional effect was seen (0.3% ± 0.1%, P = 0.86) (N = 3). Similar results were obtained for killing of intramacrophage ileal CD AIEC LF82 (see Fig, Supplemental Digital Content 7, http://links.lww.com/IBD/A842).
Vitamin D Enhances Killing of Intramacrophage AIEC
Intramacrophage HM605 survival was decreased by treatment with 1,25 OH2-vitamin D3 in a dose-dependent manner; in human MDM (P = 0.012, Cuzick's test) and in J774A.1 macrophages (P < 0.001); (Fig. 7). Cotreatment with 1,25 OH2-vitamin D3, modestly increased intracellular respiratory burst in J774A.1 macrophages relative to vehicle-treated control (N = 5, P = 0.002, Cuzick's test). No effect was seen on total (intracellular plus extracellular) respiratory burst (see Data, Supplemental Digital Content 5, http://links.lww.com/IBD/A840). No macrophage cytotoxicity was seen as assessed by released adenylate kinase in response to any of the drugs tested or vitamin D.
FIGURE 7.
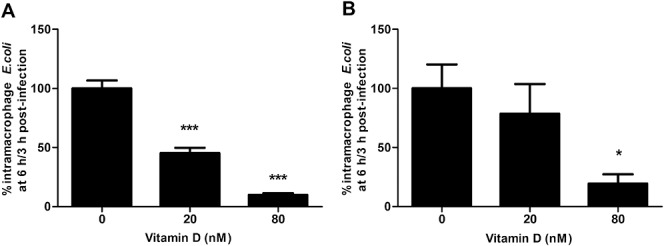
Vitamin D enhances intramacrophage killing of AIEC. 1,25 OH2-vitamin D3 caused dose-dependent decrease in intramacrophage survival of AIEC HM605 in (A) J774A.1 murine macrophages (N = 3, Cuzick's test for trend; P < 0.001) and (B) human MDM (N = 3, Cuzick's test; P = 0.012). *P < 0.05, ***P < 0.001 Dunnett's test versus control.
DISCUSSION
Here, we show that MDM from healthy individuals are ineffective at killing CD mucosally derived AIEC, but that CD MDM are similarly ineffective. Moreover, the CD MDM show no defect in generation of neutrophil chemoattractants in response to E. coli. Since E. coli replicating within macrophage phagolysosomes represent a plausible therapeutic target in CD, and are likely to be relatively resistant to antibiotic therapy, we investigated the effects of HCQ and 1,25 OH2-vitamin D3 on killing of CD E. coli by macrophages. Both HCQ and 1,25 OH2-vitamin D3 are shown to greatly enhance the ability of macrophages to kill phagocytosed E. coli. The effect of HCQ is synergistic with antibiotics, suggesting a plausible new approach to therapy.
Defective neutrophil chemotaxis has been consistently reported in patients with CD. Neutrophils are usually plentiful in CD tissue sections, but it has been suggested that a delayed neutrophil response to an initial bacterial attack may result in bacteria being taken up by macrophages before the neutrophils arrive.18 These macrophages, being much less effective than neutrophils at killing phagocytosed bacteria, may then become chronically infected, resulting in granuloma formation. It has been suggested that the decreased in vivo neutrophil chemotaxis seen in CD may be due to a defective macrophage chemokine response to bacterial triggers.19 Studies have shown defective secretion of IL-8 by CD MDM in response to the NOD2 ligand muramyl dipeptide18 and into CD blister fluid.43 In the latter study, blister fluid concentrations of IL-8, but not those of TNF-α or other cytokines, correlated with decreased chemotaxis. However, subsequent studies of CD MDM have shown decreased secretion of TNF-α and other cytokines but not IL-8.19,44 Here, CD peripheral blood MDM behaved similarly to those from HC in their ability to generate neutrophil chemoattractants in response to E. coli; their E. coli-induced secretion of IL-8, TNF-α, and other cytokines was also similar. A defective macrophage cytokine response has previously been reported in individuals who are homozygous or compound heterozygous for altered NOD2/CARD15 alleles,18,44,45 but this genotype is only present in around 8% of CD and was not present in any patients in our study. The mechanism underlying the defective neutrophil chemotaxis seen in vivo in the majority of patients with CD is therefore still unclear, but the presence of circulating chemotaxis inhibitors remains a possibility.46–48
Peripheral blood MDM from patients with CD have previously been shown to be normal in their ability to kill phagocytosed S. aureus.23 Given the different mechanisms involved in killing of Gram-negative organisms by macrophages, it was important also to assess the ability of CD macrophages in killing AIEC. It is shown here that even healthy macrophages are ineffective at killing phagocytosed E. coli including AIEC but that CD macrophages are no worse. It has recently been shown that MDM from patients with CD, homozygous for the ATG16L1 risk allele show impaired killing of phagocytosed AIEC when studied in the presence of phorbol ester to mimic inflammatory conditions.49 Only 1 of the patients in this study was homozygous for the ATG16L1 risk allele.
The mechanisms by which microbes evade intramacrophage killing include resistance to pH changes, prevention of phagosomal maturation, escape into the cytosol, and resistance to reactive oxygen species.50,51 This is typified by C. burnetii, the agent of Q-fever, which is adapted to survive at the acidic pH within macrophage phagolysosomes.52,53 Although CD AIEC isolates, such as LF82, are rapidly attacked through the autophagocytic pathway,8 some can escape autophagy and then survive and, like C. burnetii, replicate within mature phagolysosomes, which provide them with the acidic pH necessary for their replication.9 The survival of C. burnetii within macrophages is substantially decreased in vitro by HCQ treatment and this translated into clinical response in a randomized trial.54 Similar effects have been seen for HCQ against T. whipplei, the causative organism of Whipple's disease and a combination of doxycycline and HCQ is now recommended as first-line therapy.55 This study shows a marked effect of HCQ on survival of phagocytosed AIEC within macrophages, similar to that previously shown for chloroquine9 and probably mediated largely by increased intravacuolar pH.
We have shown here that HCQ, at concentrations that should be achievable in vivo, has a marked synergistic effect with antibiotics, such as ciprofloxacin and doxycycline that are effective against intracellular E. coli. HCQ has not proved effective when used, mainly on the basis of its anti-inflammatory effects, as a single agent in CD,56 although it has been successfully used in the treatment of chronic granulomatous disease, an inherited condition associated with defective neutrophil function, impaired NADPH oxidase free-radical production and granulomatous colitis that mimics CD.57 Eradication of E. coli replicating within tissue macrophages, in analogy with the causative organisms of Whipple's disease and Q-fever, will almost certainly require that HCQ is combined with appropriate antibiotics. This approach has a proven safety record even with long-term treatment.58
In view of the associations between CD and polymorphisms in genes such as NOD2/CARD15, ATG16L1, and IGRM, all of which impact on autophagy, it has been suggested that stimulation of autophagy would be a plausible therapeutic strategy.8 It might therefore seem counterintuitive to consider a drug such as HCQ that, like other quinolone antimalarials, inhibits the late stages of autophagy, particularly fusion of autophagosomes with lysosomes.59 This effect is dose-dependent, however, and it has been reported that chloroquine may even enhance fusion at lower concentrations.60 In support of this, it has been shown to inhibit intramacrophage replication of Mycobacterium tuberculosis and to enhance the antituberculosis protectiveness of both isoniazid and 25 OH-vitamin D3.61 Moreover CD-derived AIEC can replicate within phagolysosomes, having escaped autophagy,9 whereas rapamycin, which stimulates autophagy, becomes ineffective once intramacrophage replication of AIEC has become established.8
Vitamin D has been shown to be important to autophagy and other aspects of the innate immune response.32,39,62 We have shown here that vitamin D, also at concentrations readily achievable with oral supplementation, substantially enhances the ability of human MDM to kill phagocytosed E. coli. There is clear justification for further studies of vitamin D supplementation in CD.
It can currently still be argued that the increase in mucosally associated E. coli commonly found in CD may be a secondary phenomenon. Proof of the direct involvement of E. coli in CD pathogenesis now depends on showing that a therapy targeted against E. coli replicating within macrophages can induce remission in CD. A controlled trial of combination ciprofloxacin/doxycycline/HCQ therapy in active CD has therefore been commenced (ClinicalTrials.gov NCT01783106).
Supplementary Material
ACKNOWLEDGMENTS
AIEC LF82 was a kind gift from the late Professor Arlette Darfeuille-Michaud (Pathogénie Bactérienne Intestinale, Université d'Auvergne; Clermont-Ferrand, France). The authors are extremely grateful for experimental support from Marco Marcello (Centre for Cell Imaging, University of Liverpool, Liverpool, United Kingdom), Lee Murphy (The Wellcome Trust Clinical Research Facility Edinburgh, United Kingdom), and Eileen Marks (Clinical Biochemistry, Royal Liverpool University Hospital, United Kingdom).
Author contributions: B. J. Campbell, H. L. Wright, J. M. Rhodes, P. K. Flanagan, S. Subramanian, and S. W. Edwards obtained funding/designed study; A. Alswied, D. Chiewchengchol, and P. K. Flanagan acquired data; B. J. Campbell, H. L. Wright, J. M. Rhodes, J. Satsangi, P. K. Flanagan, S. Subramanian, and S. W. Edwards analyzed and interpreted data. B. J. Campbell, P. K. Flanagan, and J. M. Rhodes drafted the article, and all authors revised/approved the manuscript.
B. J. Campbell has received research support from Provexis plc and Bo & Vera Ax:son Johnson Foundation and Arcis:Altos and has received a speaking honorarium from Amgen. J. M. Rhodes has been a member of advisory boards for Atlantic, Procter & Gamble and Falk, has received speaking honoraria from Abbott, Falk, Ferring, GlaxoSmithKline, Procter & Gamble, and Schering-Plough and, with the University of Liverpool and Provexis plc, holds a patent for use of a soluble fiber preparation as maintenance therapy for Crohn's disease. J. Satsangi has received honoraria for lectures or consultancy work from AbbVie, MSD, Takeda, Ferring, and travel support from Shire. P. K. Flanagan has received travel support from AbbVie. S. Subramanian has received speaker honoraria from Falk, Shire, AbbVie, MSD, and Warner Chilcott, served as an advisory board member for AbbVie, MSD, and Vifor and received educational grants from AbbVie, MSD, and Warner Chilcott. S. W. Edwards has received a speaking honorarium from Pfizer. The remaining authors have no conflicts of interest to disclose.
Footnotes
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.ibdjournal.org).
P. K. Flanagan was supported by the National Institute for Health Research (NIHR), Biomedical Research Center for Microbial Diseases, Liverpool (01CD1), and an NIHR Biomedical Research fellowship (BRF-2011-025). P. K. Flanagan and B. J. Campbell acknowledge support of a Shire Innovation Fund award. D. Chiewchengchol was supported by a Thai Government Scholarship and Chulalongkorn University, Bangkok. H. L. Wright was supported by Arthritis Research UK (Grant 19437).
Author disclosures are available in the Acknowledgments.
REFERENCES
- 1.Cader MZ, Kaser A. Recent advances in inflammatory bowel disease: mucosal immune cells in intestinal inflammation. Gut. 2013;62:1653–1664. [DOI] [PubMed] [Google Scholar]
- 2.Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology. 2014;146:1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwab C, Berry D, Rauch I, et al. Longitudinal study of murine microbiota activity and interactions with the host during acute inflammation and recovery. ISME J. 2014;8:1101–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Darfeuille-Michaud A, Neut C, Barnich N, et al. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn's disease. Gastroenterology. 1998;115:1405–1413. [DOI] [PubMed] [Google Scholar]
- 5.Martin HM, Campbell BJ, Hart CA, et al. Enhanced Escherichia coli adherence and invasion in Crohn's disease and colon cancer. Gastroenterology. 2004;127:80–93. [DOI] [PubMed] [Google Scholar]
- 6.Prorok-Hamon M, Friswell MK, Alswied A, et al. Colonic mucosa-associated diffusely adherent afaC+ Escherichia coli expressing lpfA and pks are increased in inflammatory bowel disease and colon cancer. Gut. 2014;63:761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gevers D, Kugathasan S, Denson LA, et al. The treatment-naive microbiome in new-onset Crohn's disease. Cell Host Microbe. 2014;15:382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lapaquette P, Bringer MA, Darfeuille-Michaud A. Defects in autophagy favour adherent-invasive Escherichia coli persistence within macrophages leading to increased pro-inflammatory response. Cell Microbiol. 2012;14:791–807. [DOI] [PubMed] [Google Scholar]
- 9.Bringer MA, Glasser AL, Tung CH, et al. The Crohn's disease-associated adherent-invasive Escherichia coli strain LF82 replicates in mature phagolysosomes within J774 macrophages. Cell Microbiol. 2006;8:471–484. [DOI] [PubMed] [Google Scholar]
- 10.Meconi S, Vercellone A, Levillain F, et al. Adherent-invasive Escherichia coli isolated from Crohn's disease patients induce granulomas in vitro. Cell Microbiol. 2007;9:1252–1261. [DOI] [PubMed] [Google Scholar]
- 11.Ryan P, Kelly RG, Lee G, et al. Bacterial DNA within granulomas of patients with Crohn's disease—detection by laser capture microdissection and PCR. Am J Gastroenterol. 2004;99:1539–1543. [DOI] [PubMed] [Google Scholar]
- 12.Simpson KW, Dogan B, Rishniw M, et al. Adherent and invasive Escherichia coli is associated with granulomatous colitis in boxer dogs. Infect Immun. 2006;74:4778–4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krauss E, Agaimy A, Neumann H, et al. Characterization of lymphoid follicles with red ring signs as first manifestation of early Crohn's disease by conventional histopathology and confocal laser endomicroscopy. Int J Clin Exp Pathol. 2012;5:411–421. [PMC free article] [PubMed] [Google Scholar]
- 14.Roberts CL, Keita AV, Duncan SH, et al. Translocation of Crohn's disease Escherichia coli across M-cells: contrasting effects of soluble plant fibres and emulsifiers. Gut. 2010;59:1331–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chassaing B, Rolhion N, de Vallee A, et al. Crohn disease–associated adherent-invasive E. coli bacteria target mouse and human Peyer's patches via long polar fimbriae. J Clin Invest. 2011;121:966–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Segal AW, Loewi G. Neutrophil dysfunction in Crohn's disease. Lancet. 1976;2:219–221. [DOI] [PubMed] [Google Scholar]
- 18.Marks DJ, Harbord MW, MacAllister R, et al. Defective acute inflammation in Crohn's disease: a clinical investigation. Lancet. 2006;367:668–678. [DOI] [PubMed] [Google Scholar]
- 19.Smith AM, Rahman FZ, Hayee B, et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn's disease. J Exp Med. 2009;206:1883–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O'Morain CO, Segal AA, Walker D, et al. Abnormalities of neutrophil function do not cause the migration defect in Crohn's disease. Gut. 1981;22:817–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rhodes JM, Jewell DP. Motility of neutrophils and monocytes in Crohn's disease and ulcerative colitis. Gut. 1983;24:73–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wandall JH, Binder V. Leucocyte function in Crohn's disease. Studies on mobilisation using a quantitative skin window technique and on the function of circulating polymorphonuclear leucocytes in vitro. Gut. 1982;23:173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mee AS, Szawatakowski M, Jewell DP. Monocytes in inflammatory bowel disease: phagocytosis and intracellular killing. J Clin Pathol. 1980;33:921–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berger SB, Romero X, Ma C, et al. SLAM is a microbial sensor that regulates bacterial phagosome functions in macrophages. Nat Immunol. 2010;11:920–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khan KJ, Ullman TA, Ford AC, et al. Antibiotic therapy in inflammatory bowel disease: a systematic review and meta-analysis. Am J Gastroenterol. 2011;106:661–673. [DOI] [PubMed] [Google Scholar]
- 26.Dogan B, Scherl E, Bosworth B, et al. Multidrug resistance is common in Escherichia coli associated with ileal Crohn's disease. Inflamm Bowel Dis. 2013;19:141–150. [DOI] [PubMed] [Google Scholar]
- 27.Subramanian S, Roberts CL, Hart CA, et al. Replication of colonic Crohn's disease mucosal Escherichia coli isolates within macrophages and their susceptibility to antibiotics. Antimicrob Agents Chemother. 2008;52:427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoidal JR, Schmeling D, Peterson PK. Phagocytosis, bacterial killing, and metabolism by purified human lung phagocytes. J Infect Dis. 1981;144:61–71. [DOI] [PubMed] [Google Scholar]
- 29.Boulos A, Rolain JM, Raoult D. Antibiotic susceptibility of Tropheryma whipplei in MRC5 cells. Antimicrob Agents Chemother. 2004;48:747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kersh GJ. Antimicrobial therapies for Q fever. Expert Rev Anti Infect Ther. 2013;11:1207–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lagier JC, Fenollar F, Lepidi H, et al. Treatment of classic Whipple's disease: from in vitro results to clinical outcome. J Antimicrob Chemother. 2014;69:219–227. [DOI] [PubMed] [Google Scholar]
- 32.Hewison M. Vitamin D and immune function: an overview. Proc Nutr Soc. 2012;71:50–61. [DOI] [PubMed] [Google Scholar]
- 33.Jorgensen SP, Agnholt J, Glerup H, et al. Clinical trial: vitamin D3 treatment in Crohn's disease—a randomized double-blind placebo-controlled study. Aliment Pharmacol Ther. 2010;32:377–383. [DOI] [PubMed] [Google Scholar]
- 34.Mpofu CM, Campbell BJ, Subramanian S, et al. Microbial mannan inhibits bacterial killing by macrophages: a possible pathogenic mechanism for Crohn's disease. Gastroenterology. 2007;133:1487–1498. [DOI] [PubMed] [Google Scholar]
- 35.Agwuh KN, MacGowan A. Pharmacokinetics and pharmacodynamics of the tetracyclines including glycylcyclines. J Antimicrob Chemother. 2006;58:256–265. [DOI] [PubMed] [Google Scholar]
- 36.Saravolatz L, Manzor O, Pawlak J, et al. Antimicrobial activity and a comparison of published pharmacodynamics of gemifloxacin and eight fluoroquinolones against Streptococcus pneumoniae. Int J Antimicrob Agents. 2005;26:81–84. [DOI] [PubMed] [Google Scholar]
- 37.Carmichael SJ, Charles B, Tett SE. Population pharmacokinetics of hydroxychloroquine in patients with rheumatoid arthritis. Ther Drug Monit. 2003;25:671–681. [DOI] [PubMed] [Google Scholar]
- 38.Rosen CJ. Clinical practice. Vitamin D insufficiency. N Engl J Med. 2011;364:248–254. [DOI] [PubMed] [Google Scholar]
- 39.Fabri M, Stenger S, Shin DM, et al. Vitamin D is required for IFN-gamma-mediated antimicrobial activity of human macrophages. Sci Transl Med. 2011;3:104ra102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Di A, Brown ME, Deriy LV, et al. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat Cell Biol. 2006;8:933–944. [DOI] [PubMed] [Google Scholar]
- 41.Byrd TF, Horwitz MA. Chloroquine inhibits the intracellular multiplication of Legionella pneumophila by limiting the availability of iron. A potential new mechanism for the therapeutic effect of chloroquine against intracellular pathogens. J Clin Invest. 1991;88:351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Limbergen J, Russell RK, Nimmo ER, et al. Autophagy gene ATG16L1 influences susceptibility and disease location but not childhood-onset in Crohn's disease in Northern Europe. Inflamm Bowel Dis. 2008;14:338–346. [DOI] [PubMed] [Google Scholar]
- 43.Harbord MW, Marks DJ, Forbes A, et al. Impaired neutrophil chemotaxis in Crohn's disease relates to reduced production of chemokines and can be augmented by granulocyte-colony stimulating factor. Aliment Pharmacol Ther. 2006;24:651–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sewell GW, Rahman FZ, Levine AP, et al. Defective tumor necrosis factor release from Crohn's disease macrophages in response to toll-like receptor activation: relationship to phenotype and genome-wide association susceptibility loci. Inflamm Bowel Dis. 2012;18:2120–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Heel DA, Hunt KA, King K, et al. Detection of muramyl dipeptide-sensing pathway defects in patients with Crohn's disease. Inflamm Bowel Dis. 2006;12:598–605. [DOI] [PubMed] [Google Scholar]
- 46.Van Dyke TE, Dowell VR, Jr, Offenbacher S, et al. Potential role of microorganisms isolated from periodontal lesions in the pathogenesis of inflammatory bowel disease. Infect Immun. 1986;53:671–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.D'Amelio R, Pallone F, Le Moli S, et al. Humoral inhibition of neutrophil chemotaxis in Crohn's disease. Scand J Immunol. 1985;22:597–602. [DOI] [PubMed] [Google Scholar]
- 48.Rhodes JM, Potter BJ, Brown DJ, et al. Serum inhibitors of leukocyte chemotaxis in Crohn's disease and ulcerative colitis. Gastroenterology. 1982;82:1327–1334. [PubMed] [Google Scholar]
- 49.Sadaghian Sadabad M, Regeling A, de Goffau MC, et al. The ATG16L1-T300A allele impairs clearance of pathosymbionts in the inflamed ileal mucosa of Crohn's disease patients. Gut. [published online ahead of print September 24, 2014]. doi: 10.1136/gutjnl-2014-307289. [DOI] [PubMed] [Google Scholar]
- 50.Diacovich L, Gorvel JP. Bacterial manipulation of innate immunity to promote infection. Nat Rev Microbiol. 2010;8:117–128. [DOI] [PubMed] [Google Scholar]
- 51.Flannagan RS, Cosio G, Grinstein S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol. 2009;7:355–366. [DOI] [PubMed] [Google Scholar]
- 52.Maurin M, Benoliel AM, Bongrand P, et al. Phagolysosomes of Coxiella burnetii-infected cell lines maintain an acidic pH during persistent infection. Infect Immun. 1992;60:5013–5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Winchell CG, Graham JG, Kurten RC, et al. Coxiella burnetii type IV secretion-dependent recruitment of macrophage autophagosomes. Infect Immun. 2014;82:2229–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maurin M, Benoliel AM, Bongrand P, et al. Phagolysosomal alkalinization and the bactericidal effect of antibiotics: the Coxiella burnetii paradigm. J Infect Dis. 1992;166:1097–1102. [DOI] [PubMed] [Google Scholar]
- 55.Fenollar F, Lagier JC, Raoult D. Tropheryma whipplei and Whipple's disease. J Infect. 2014;69:103–112. [DOI] [PubMed] [Google Scholar]
- 56.Louis E, Belaiche J. Hydroxychloroquine (Plaquenil) for recurrence prevention of Crohn's disease after curative surgery. Gastroenterol Clin Biol. 1995;19:233–234. [PubMed] [Google Scholar]
- 57.Arlet JB, Aouba A, Suarez F, et al. Efficiency of hydroxychloroquine in the treatment of granulomatous complications in chronic granulomatous disease. Eur J Gastroenterol Hepatol. 2008;20:142–144. [DOI] [PubMed] [Google Scholar]
- 58.Mavrikakis I, Sfikakis PP, Mavrikakis E, et al. The incidence of irreversible retinal toxicity in patients treated with hydroxychloroquine: a reappraisal. Ophthalmology. 2003;110:1321–1326. [DOI] [PubMed] [Google Scholar]
- 59.Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11:709–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bhat M, Hickey AJ. Effect of chloroquine on phagolysosomal fusion in cultured guinea pig alveolar macrophages: implications in drug delivery. AAPS PharmSci. 2000;2:E34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Crowle AJ, May MH. Inhibition of tubercle bacilli in cultured human macrophages by chloroquine used alone and in combination with streptomycin, isoniazid, pyrazinamide, and two metabolites of vitamin D3. Antimicrob Agents Chemother. 1990;34:2217–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chun RF, Liu PT, Modlin RL, et al. Impact of vitamin D on immune function: lessons learned from genome-wide analysis. Front Physiol. 2014;5:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.