

Abstract
The apelin receptor (AR or APJ) is a class A (rhodopsin-like) G-protein coupled receptor (GPCR) with wide distribution throughout the human body. Activation of the AR by its cognate peptide ligand, apelin, induces diverse physiological effects including vasoconstriction and dilation; strengthening of heart muscle contractility; angiogenesis; and, regulation of energy metabolism and fluid homeostasis. Recently, another endogenous peptidic activator of the AR, Toddler/ELABELA, was identified as having a crucial role in zebrafish embryonic development. The AR is also implicated in pathologies including cardiovascular disease, diabetes, obesity and cancer, making it a promising therapeutic target. Despite its established importance, the precise roles of AR signalling remain poorly understood. Moreover, little is known about mechanisms of peptide-AR activation. Additional complexity arises from modulation of the AR by two endogenous peptide ligands, both with multiple bioactive isoforms of variable length and distribution. The various apelin and Toddler/ELABELA isoforms may also produce distinct cellular effects. Further complexity arises through formation of functionally distinct heterodimers between the AR and other GPCRs. This minireview outlines key (patho)physiological actions of the AR, addresses what is known about signal transduction downstream of AR activation, and concludes by discussing unique properties of the endogenous peptidic ligands of the AR.
Keywords: apelinergic system, APJ, APLNR, signal transduction, peptide-activated GPCR
1. Overview of the apelinergic system
The apelin receptor (AR) is a class A (rhodopsin-like) G-protein coupled receptor (GPCR) first identified in 1993 based on its sequence homology with the angiotensin II receptor (AT1R) and dubbed APJ (O’Dowd et al. 1993). With the discovery that it was not activated by angiotensin II, and in the absence of an identified cognate ligand, the receptor was deemed an orphan GPCR. The AR retained this status until 1998, when its endogenous ligand, apelin, was first isolated from bovine stomach extracts (Tatemoto et al. 1998). The 77 amino acid protein preproapelin was identified as the precursor of bioactive apelin. Three shorter isoforms, apelin-36, -17, and -13 (nomenclature based on the number of C-terminal preproapelin amino acids retained), were hypothesized based on the presence of basic residues in the peptide sequence suggestive of recognition sites for proteolytic cleavage (sequences in Table 1). Backing up this hypothesis, synthetic apelin-13, -17 and -36 peptides were all shown to activate the AR. We recently demonstrated the first enzymatic cleavage of proapelin to an active apelin isoform (Shin et al. 2013). Specifically, proprotein convertase subtilisin kexin 3 (PCSK3 or furin) preferentially cleaves proapelin directly into apelin-13. It had been previously suggested that the longest cleavage product, apelin-36, is an intermediate for production of shorter isoforms (Kleinz and Davenport 2005). Our studies suggest that apelin processing may be more complex, with different isoforms being produced by disparate, as yet uncharacterized, mechanisms.
Table 1.
Sequences of human apelin and Toddler/ELABELA, the endogenous peptide ligands of the AR. All isoforms of each ligand retain identical C-termini, and various lengths of peptide are presented based on biological detection or the presence of proposed dibasic proteolytic cleavage sites. Apelin-12 is most likely not produced endogenously, but is the shortest possible synthetic isoform that retains biological activity (Tatemoto et al. 2001).
| Apelin Peptide | Sequence |
|---|---|
| Preproapelin (Apelin-77) | MNLRLCVQALLLLWLSLTAVCGGSLMPLPDGNGLEDGNVRHLVQPRGSRNGPGPWQGGRRKFRRQRPRLSHKGPMPF |
| Proapelin (Apelin-55) | GSLMPLPDGNGLEDGNVRHLVQPRGSRNGPGPWQGGRRKFRRQRPRLSHKGPMPF |
| Apelin-36 | LVQPRGSRNGPGPWQGGRRKFRRQRPRLSHKGPMPF |
| Apelin-17 | KFRRQRPRLSHKGPMPF |
| Pyr-apelin-13 | Pyr–RPRLSHKGPMPF |
| Apelin-13 | QRPRLSHKGPMPF |
| Apelin-12 | RPRLSHKGPMPF |
| Toddler/ELABELA Peptide | Sequence |
| Preprotoddler (Toddler-54) | MRFQQFLFAFFIFIMSLLLISGQEPVNLTMRRKLRKHNCLQRRCMPLHSRVPFP |
| Protoddler (Toddler-32) | QEPVNLTMRRKLRKHNCLQRRCMPLHSRVPFP |
| Toddler-22a | KLRKHNCLQRRCMPLHSRVPFP |
| Toddler-11 | CMPLHSRVPFP |
Toddler-22 is shown by analogy to apelin-17; zebrafish Toddler-21 and the corresponding apelin-16 isoform were shown to be active by Pauli et al. (2014), with divergent sequences in the N-terminal region compared to the human peptides.
Both apelin and the AR are expressed across a wide range of eukaryotes, including humans, and many relevant studies have been conducted in rodent or other animal models, as well as in cell or tissue culture. Consistently high levels of expression have been observed in the central nervous system (CNS), and both receptor and ligand are widely present throughout the peripheral tissue (Azizi et al. 2008; El Messari et al. 2004; Kawamata et al. 2001; Maguire et al. 2009; Reaux et al. 2002). The importance of apelinergic signalling is well established in areas as diverse as cardiovascular regulation, energy metabolism, fluid homeostasis, angiogenesis, human immunodeficiency virus-1 (HIV-1) infection, and the neuroendocrine stress response, among others (Masri et al. 2005; O’Carroll et al. 2013; Pitkin et al. 2010). Very recently, the AR has been shown to be activated by another endogenous peptide ligand, known as both Toddler and ELABELA, with a central role in zebrafish embryonic development (Chng et al. 2013; Pauli et al. 2014).
Thus, various physiological and pathophysiological roles have been attributed to the apelinergic system, but the overarching theme of apelin-AR research to date is that of a complex and important regulator of physiology, the functions of which are only ambiguously characterized. The goal of this minireview is to describe some of the main physiological and pathological roles of the AR, with a focus on the cell signalling pathways downstream of AR activation that may be involved in the varied and context-dependent behaviour of this GPCR. Finally, the minireview concludes with a comparison of the multiple endogenous ligands that regulate the AR, and how their distinct properties may influence the signals transmitted by this important receptor.
2. Physiology and pathophysiology of the AR
Having given a general introduction to GPCR-mediated signal transduction, we now describe a number of the key normal physiological and pathological actions of the AR. Since specific AR-mediated signalling pathways have not always been conclusively linked to particular physiological processes, this is followed by a review of the primary cell signalling events mediated by the AR that have been characterized to date.
2.1 The AR in the cardiovascular system
Some of the most compelling aspects of apelin-AR physiology, from a drug development perspective, are related to its effects in the cardiovascular system. Apelin is one of the most potent known inotropic agents (Szokodi et al. 2002), and doses in mice have been shown to increase myocardial contraction and reduce cardiac load without inducing pathological hypertrophy (Ashley et al. 2005). This GPCR is also involved in an apelin-independent response to mechanical stretch (Scimia et al. 2012). Notably, this stretch-induced response is pathological when the heart is experiencing chronic pressure overload, and AR knockout (KO) mice show reduced risk of cardiac hypertrophy under such conditions.
Apelin-AR signalling is also reported to have cardioprotective effects; for instance, administration of apelin-13 to glucose-deprived cultured cardiomyocytes had a significant inhibitory effect on apoptosis (Zhang et al. 2009b). Likewise, myocardial apelin levels are reduced in patients with ischemic heart failure, and these lower levels are associated with greater mortality rates and less effective cardiac remodelling post-injury (Wang et al. 2013). Conversely, spontaneously hypertensive rats exhibit elevated apelin levels in the rostral ventrolateral medulla (RVLM), the region of the brainstem responsible for regulating blood pressure and other autonomic functions (Zhang et al. 2009a). Microinjection of apelin into the RVLM caused chronic increases in mean arterial blood pressure (MABP) and cardiac hypertrophy. In contrast to these CNS effects, peripherally administered apelin has typically been shown to decrease MABP (Cheng et al. 2003; Dai et al. 2013; Lee et al. 2000; Lee et al. 2005; Reaux et al. 2001; Tatemoto et al. 2001). Interestingly, this effect is fully blocked by the C-terminally modified apelin-13 analogue F13A, which acts as an antagonist at the AR, preventing the typical apelin-induced MABP reduction (Lee et al. 2005).
Moreover, while peripheral administration of apelin has typically yielded a decrease in MABP, this is not without exception: for example, blood vessels denuded of endothelium show a vasoconstrictive response to apelin, directly opposite to the vasodilation observed in endothelium-intact vessels (Maguire et al. 2009). Similarly, studies of peripheral apelin administration in conscious rats have reported both increases and decreases in MABP (Cheng et al. 2003; Kagiyama et al. 2005). These are ambiguous results in comparison to the MABP decreases consistently observed in anesthetized rats (Cheng et al. 2003; Dai et al. 2013; Lee et al. 2000; Lee et al. 2005; Reaux et al. 2001; Tatemoto et al. 2001). Interestingly, and perhaps of relevance to the different responses reported in rats, a study in sheep given a large systemic dose of apelin-13 led to a biphasic response in blood pressure (BP), with an initial transient decrease in BP immediately giving way to an increase in BP. The dose also led to increased plasma concentrations of the vasoactive hormones vasopressin, adrenocorticotrophin, aldosterone, cortisol, atrial and brain natriuretic peptides, cyclic GMP and cyclic AMP (Charles et al. 2006).
The variety of apelin-AR effects summarized above emphasizes the need to consider apelin-AR signalling in the context of whole organism physiology, as the functional readouts chosen may reflect, for example, the organism’s compensatory response to the effects of apelin-AR signalling, rather than the initial signalling itself. Overall, the emerging picture of the AR and apelin in the cardiovascular system is one of complex and important regulators of the heart and vasculature. However, much more research is needed if we are to understand their subtle and often seemingly contradictory modes of action.
2.2 The AR in energy metabolism
Another key aspect of apelin and AR physiology, not least from a therapeutic perspective, is their influence on energy metabolism. Similarly to other systems, apelin-AR signalling appears important in both normal and pathological processes. Both the GPCR (Wei et al. 2005) and its ligand (Boucher et al. 2005; Wei et al. 2005) are expressed in adipose tissue. Apelin is also secreted by adipose tissue (Boucher et al. 2005), leading to its description as an adipokine. Correspondingly, elevated levels of apelin in the blood plasma are correlated to obesity in mice (Boucher et al. 2005) and humans (Boucher et al. 2005; Heinonen et al. 2005) and several additional studies, as recently reviewed in (Castan-Laurell et al. 2012)).
Insulin and apelin have been shown to have co-regulatory affects. Administration of apelin-36 either to live mice or to isolated pancreatic islets inhibits glucose-stimulated insulin secretion (Sorhede Winzell et al. 2005). Thus, it has been proposed that the AR may be present at the membrane of islet β-cells, with its activation by apelin leading to reduced insulin secretion, and to the resulting impairment of glucose elimination. Insulin, on the other hand, binds its receptor on adipocytes, inducing apelin expression and providing a negative feedback mechanism for insulin production (Sorhede Winzell et al. 2005). With respect to feeding behaviour, adipocyte apelin mRNA levels are significantly decreased (6.2-fold) in fasting mice, following the same trend observed for plasma insulin levels (5.4-fold reduction). In turn, levels of both return to normal after food intake (Boucher et al. 2005). More recently, apelin-13 has been reported to increase pancreatic islet cell mass and β-cell insulin content in mice with type I diabetes, improving these two symptoms of the disease (Chen et al. 2011). Likewise, administration of apelin alleviates the impaired insulin sensitivity of both apelin KO mice (Yue et al. 2010) and obese hyperinsulinaemic mice (Attane et al. 2012).
Clearly, apelin and the AR have an important regulatory role in energy metabolism. Both are up-regulated in metabolic disorders, and their actions have until now generally been considered beneficial. However, further research is needed to establish in more depth both the causes and consequences of these altered expression patterns.
2.3 The AR in fluid homeostasis
A number of studies suggest that apelin-AR signalling has an important role in maintaining fluid homeostasis. As mentioned above, both ligand and receptor are present in high levels in the CNS. In particular, apelin-36 is highly present in the rat paraventricular nucleus (PVN) and supraoptic nucleus (SON) of the hypothalamus (Brailoiu et al. 2002), bodies of neurons that project into the pituitary gland, producing secretory hormones to be released into systemic circulation. Production of the antidiuretic hormone vasopressin is a hallmark action of these nuclei, and the co-localization of apelin with vasopressin here suggests a potential shared regulatory role of these peptides on fluid balance. Some studies have observed a clear diuretic effect for apelin, opposing vasopressin’s anti-diuresis and inducing decreased vasopressin expression (De Mota et al. 2004; Reaux et al. 2001). Contrastingly, others have reported anti-diuresis mediated by the AR, with AR knockout mice showing an inability to reduce urine volume in response to water deprivation (Roberts et al. 2010). Regarding drinking behaviour, some studies report increased water intake by water-replete animals in response to central apelin administration (Taheri et al. 2002), whereas others report reduced intake (Clarke et al. 2009), and still others report no effect (Reaux et al. 2001). Thus, apelin and the AR appear to be important physiological regulators of fluid homeostasis, but, as with their actions in other systems, further study is required both to verify their exact involvement and to unravel the complexity of their signalling.
2.4 The AR in angiogenesis
The actions of apelin through the AR have been shown to be angiogenic, both in normal physiology and in the pathology of cancer. The presence of both receptor and ligand is necessary for the normal development of frog and mouse heart vasculature, and their absence causes vessel disruption in a majority of embryos (Cox et al. 2006). On the other hand, apelin and the AR induce premature angiogenesis in Xenopus, and both are up-regulated in microvascular proliferations of malignant gliomas (Kalin et al. 2007). Recently, apelinergic signalling has been implicated in the growth of colon cancer, with apelin expression up-regulated in half of colon adenocarcinomas (Picault et al. 2014). Moreover, both apelin and the AR are expressed in LoVo cells, a colorectal cancer cell line; in these cells, apelin-13 administration prevents apoptosis by inactivating a caspase pathway, while the aforementioned antagonistic apelin-13 mutant F13A significantly reduces cellular proliferation (Picault et al. 2014). The role of this system in tumour angiogenesis has led to identification of apelin and the AR as anticancer therapeutic targets or, at the very least, as potential diagnostic biomarkers.
2.5 Additional physiological actions
The actions discussed above represent some of the most studied roles of apelin and the AR to date. However, a number of additional roles have been described (recently reviewed by (O’Carroll et al. 2013)) including, among others, a role for the AR as co-receptor with CD4 for HIV infection (Choe et al. 1998), which is inhibited by the various apelin isoforms (Zou et al. 2000), and an emerging picture of apelin and the AR as regulators of the stress response via the hypothalamic-pituitary axis (Newson et al. 2013). Thus, while the physiological importance of apelin-AR signalling has been well established, much remains to be determined regarding its harmful and beneficial effects, and the actual mechanisms of signal transduction through the AR remain central and as yet unanswered questions.
3. GPCR signal transduction pathways
To understand how apelin-AR signalling may be involved in the many physiological effects introduced above, we first introduce the general downstream signalling pathways coupled to GPCRs. The classical pathways downstream of GPCRs involve coupling to a heterotrimeric guanine nucleotide binding protein (G-protein), which, upon activation, goes on to regulate various cellular events. For a given GPCR, G-protein-coupling preference is usually defined in terms of the Gα subunit, of which there are four main subfamilies: Gαs, Gαi/o, Gαq/11, and Gα12/13 (Figure 1). Each family, in turn, is associated with a hallmark cellular effect: adenylyl cyclase (AC) activation for Gαs; AC inhibition for Gαi/o; phospholipase C-β (PLCβ) activation and increased intracellular [Ca2+] for Gαq/11; and, regulation of Rho GTPases, which are known for their role in regulating the actin cytoskeleton, for Gα12/13 (Ahmed and Angers 2013).
Figure 1.
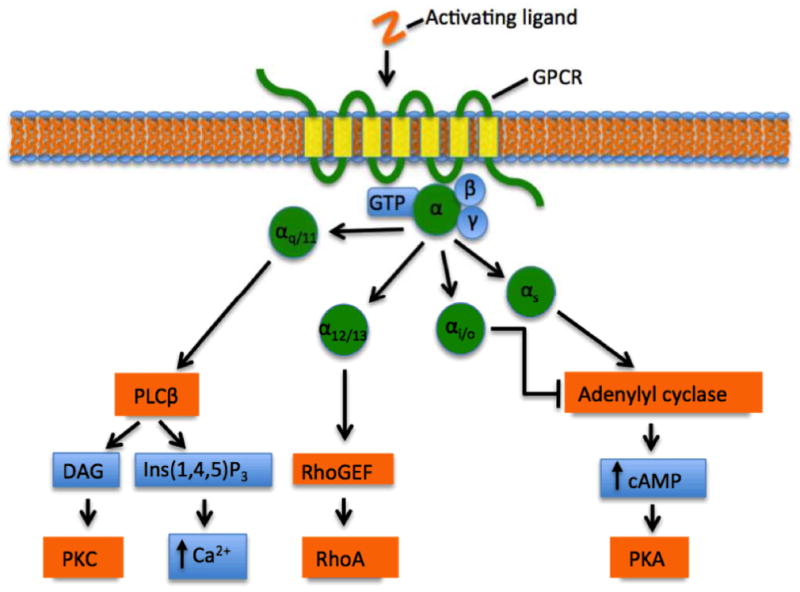
Canonical Gα-mediated signalling at GPCRs typically following ligand binding and activation. The heterotrimeric (i.e., αβγ trimer) G-protein coupling preference and downstream cellular effect of a GPCR is defined by the Gα subunit (Gαs, Gαi/o, Gαq/11, and Gα12/13). Gαs stimulates adenylyl cyclase, producing cAMP and inducing PKA (protein kinase A) activation; Gαi/o inhibits adenylyl cyclase activity; Gαq/11 activates PLCβ, producing DAG (diacyl glycerol) and Ins(1,4,5)P3 (inositol 1,4,5-triphosphate), causing an increase in intracellular [Ca2+] and PKC activation; and, Gα12/13 activates RhoGEF, causing activation of RhoA. Arrowheads indicate activation and blunted arrows indicate inhibition. (Colour available online.)
Although a helpful starting point, this simple picture belies the real situation. These classical pathways account for only a fraction of the many pathways activated downstream of GPCRs. Moreover, while they provide measurable endpoints for functional readouts of GPCR activation (e.g., extracellular signal-regulated kinase (ERK) activation, changes in intracellular [Ca2+] or [cAMP], etc.), these readouts can give very few details about spatiotemporal cellular dynamics, pathway selectivity, degree of signal branching, divergence and re-convergence of various branches, and so on. In other words, with only a rough snapshot of a given downstream event, there is no way to determine which upstream signalling events led to the signal being measured.
Thus, in addition to the canonical pathways mentioned above, there are several others that must be considered. It is now known that in addition to their role in receptor desensitization, β-arrestins are signal transducers in their own right (Figure 2a), and are one of the main mediators of the phenomenon known as “biased agonism” or “functional selectivity” at GPCRs (Kenakin 2011; Luttrell and Lefkowitz 2002). “Biased agonists” are those that preferentially activate one G-protein-coupled pathway over another, leading to distinct cellular effects. Interestingly, for example, a study of biased agonism at the AT1R revealed that G-protein- and β-arrestin-mediated signalling induce distinct spatiotemporal patterns of ERK activation (ERK → pERK) in HEK-293 cells (Ahn et al. 2004). More specifically, Gαq/11 induces a transient spike in pERK localized in the nucleus (where it presumably regulates gene expression); conversely, β-arrestin, in complex with internalized AT1R and other proteins, induces a sustained cytosolic pool of pERK, which presumably has distinct signalling properties. The various Gβγ heterodimers have likewise emerged as important mediators of signalling downstream of GPCRs (Figure 2b). In particular, free Gβγ subunits of the Gαi/o pathways directly interact with a number of effectors, including PLCβ, K+ channels, AC, and phosphoinositol 3-kinase (PI3K); they also indirectly signal to the small GTPase Ras, promoting ERK activation (Neves et al. 2002).
Figure 2.

Non-Gα-mediated signalling at GPCRs. (a) β-arrestin-mediated internalization and signalling at GPCRs. An activated GPCR is phosphorylated (circled P) by GRKs (GPCR kinases, not shown), leading to β-arrestin recruitment at the GPCR intracellular loops and C-terminus and occluding further G-protein binding and subsequent activation. This triggers internalization of the GPCR via endosome formation, in turn mediating activation of cytosolic ERK and other downstream signalling pathways. The internalized GPCR is eventually either recycled to the cell surface, or trafficked to a lysosome for degradation. (b) Gβγ-mediated signalling at GPCRs. In addition to Gαi activation heterotrimeric G-protein activation also leads to activation of the Gβγ dimer. The Gβγ dimer may in turn directly activate or inhibit adenylyl cyclase, inhibit Ca2+ or K+ channels, activate Src, PI3K, and PLCβ, as well as indirectly activate other pathways, such as the ERK cascade, by routes such as RTK transactivation (e.g., panel c). (c) Receptor tyrosine kinase transactivation, or triple-membrane-pass signalling, occurs following activation of a transactivation mediator as a result of G-protein activation. The mediator then activates a membrane-bound matrix metalloproteinase, which in turn cleaves the extracellular N-terminus from a membrane-bound RTK proligand, yielding a soluble activated ligand (e.g., EGF). This ligand is then free to activate its RTK (e.g., EGFR) in an autocrine or paracrine manner. Thus, the initial ligand signal to the GPCR ‘passes’ across the membrane three times before the activated RTK transduces the signal to downstream effectors pathways. Arrowheads indicate activation and blunted arrows indicate inhibition. (Colour available online.)
A third important and incompletely understood event downstream of GPCR activation is the transactivation of receptor tyrosine kinases (RTKs, e.g., epidermal growth factor receptor (EGFR)) and, more recently, receptor serine/threonine kinases (RS/TKs, e.g., transforming growth factor β type I receptor (TβRI);(Burch et al. 2013)). RTK transactivation (Figure 2c), also known as triple-membrane-pass signalling, may occur roughly as follows: a ligand-activated GPCR activates the G-protein, causing activation of an RTK transactivation mediator (e.g., dissociation of Gβγ, followed by its activation of the non-membrane tyrosine kinase Src). The mediator then activates a membrane-bound matrix metalloproteinase, which in turn cleaves the extracellular N-terminus from a membrane-bound RTK proligand, yielding a soluble activated ligand (e.g., EGF). This ligand is then free to activate its RTK (e.g., EGFR) in an autocrine or paracrine manner. Thus, the initial ligand signal to the GPCR ‘passes’ across the membrane three times before the activated RTK transduces the signal to downstream effectors (Liebmann 2011).
RTK transactivation has been proposed to be important for ERK activation and other important proliferative signalling. However, because of the complexity in crosstalk between GPCR and RTK pathways (i.e., the potential for tuning the GPCR signal by GPCR-interacting proteins, the total cell-type dependence of the signalling machinery, etc.), the question of how significant RTK transactivation is to overall GPCR signal transduction remains controversial (Liebmann 2011). Regarding ERK signalling, for example, RTK transactivation with no Gαi/o involvement has been reported to be the exclusive pathway responsible for pERK production in mouse inner medullary collecting duct (mIMCD-3) cells via the bradykinin receptor (B2R) (Mukhin et al. 2003). By contrast, others have reported ERK activation via B2R and m1 and m2 muscarinic receptors relying solely on cooperation between Gαi/o and Gαq/11 in several cell types, whereas Gβγ and RTK transactivation had no influence (Blaukat et al. 2000).
All of the above pathways are important, and the diversity of signalling possibilities must be emphasized. The subunit composition of the G-protein alone provides hundreds of permutations, each presumably with differing cellular abundance and affinity for various interacting partners. Thus, together with the unique infrastructure of a given cell type, the main and preferred routes of signal transduction (which again may vary with the cell cycle and subcellular location), the environmental conditions, etc., GPCR signalling provides cells with an extremely dynamic, tunable, and specific system for maintaining homeostasis.
4. Signal transduction pathways coupled to the AR
As described in Section 3, the signalling cascades downstream of GPCR activation are highly complex. As a result, while physiological effects and signal transduction events may be characterized for a given GPCR, the links between these organism- and molecular-level phenomena are usually less well understood, and current knowledge of the AR is no exception. As described below, AR signalling is heterologous, with the receptor showing preference for multiple G-proteins and mediating different effects in different cell types. In addition, the various isoforms of the two currently known endogenous ligands, apelin and toddler/ELABELA, may induce qualitatively different signals at the AR, as we will detail in section 5. Considering AR signalling in this light, it is less surprising that the AR is able to mediate so many different, sometimes seemingly contradictory, effects.
4.1 AR coupling to Gαi/o
The primary Gα coupling preference of the AR is for Gαi/o (Figure 3) – in particular Gαi1 and Gαi2, but typically not Gαi3 (Masri et al. 2006). However, these interactions have been shown to vary with cell type; for instance, AR coupling to Gαi3 is observed in human umbilical vein endothelial cells (HUVECs) (Kang et al. 2013). Signal transduction via the AR in Chinese hamster ovary (CHO) cells has been linked to classical Gαi/o-mediated downstream effects such as ERK and Akt activation and decreased cAMP production. Of note, this AR-mediated ERK phosphorylation appears not to be Gβγ-dependent, although such dependency is commonly observed for GPCRs (Masri et al. 2002).
Figure 3.

Overview of AR-mediated signalling pathways. Apelin-activated AR couples to Gαi and Gαq. Gαi may inhibit adenylyl cyclase, reducing cAMP production and thereby inhibiting PKA activation; activate PI3K, leading to Akt activation; or, directly activate PKC, causing activation of the mitogenic ERK pathway. Gαq activates PLCβ, inducing DAG and Ins(1,4,5)P3 production. DAG and Ins(1,4,5)P3 activate the PKC cascade and the release of intracellular Ca2+, respectively. Ca2+ release in turn activates calmodulin, which then activates nitrous oxide synthase. Finally, the AR also mediates a mechanosensory response via a β-arrestin-dependent and G-protein-independent pathway. Arrowheads indicate activation and blunted arrows indicate inhibition. (Colour available online.)
4.2 AR coupling to Gαq/11 and/or free Gβγ subunits
In addition to its interaction with pertussis toxin-sensitive Gαi/o proteins, there is also evidence that the AR couples to pertussis toxin-insensitive G proteins, namely Gαq/11 (Szokodi et al. 2002). This is based on the finding that the AR’s contractile effects on cardiomyocytes are only partially attenuated by pertussis toxin, whereas addition of PLCβ and protein kinase C (PKC) inhibitors also significantly attenuated these effects. Similar attenuation was observed when Na+/H+ and Na+/Ca2+ exchangers were inhibited. Thus, it has been proposed that the inotropic effects of apelin via the AR rely, at least in part, on Gαq/11 coupling to the PLCβ-PKC pathway. Of note, this finding in cardiomyocytes differs from the results of Masri et al. (2002), who did not observe AR-Gαq/11 coupling in CHO cells – once again highlighting the influence of cell type on the qualitative outcome of the AR signal.
The vasodilatory effects of apelin-AR signalling appear to be mediated via nitrous oxide, as inhibition of endothelial nitrous oxide synthase (eNOS) prevents the apelin-induced decrease in MABP (Tatemoto et al. 2001). NOS is activated by calmodulin, which itself is activated by rising intracellular [Ca2+], a downstream product of the PLCβ-PKC cascade. Thus, it may be that these effects are mediated by the AR’s activation of the PLCβ-PKC pathway, either through Gαq/11, or through the free Gβγ subunits of the Gαi/o heterotrimer, or through a combination of both. However, since the AR has also been shown to activate the kinase Akt ((Masri et al. 2006), which can in turn activate eNOS (Fleming and Busse 2003), Gαi/o -mediated Akt activation provides another possible mechanism for the vasodilatory effects of the AR.
4.3 β-arrestin-dependent internalization and G-protein-independent signalling
The AR has been shown to internalize following association with β-arrestin (Evans et al. 2001; Lee et al. 2010) in a clathrin-dependent manner (El Messari et al. 2004; Said El Messari 2004). Interestingly, the stretch activated response of the AR mentioned previously is both β-arrestin-dependent and G-protein-independent (Scimia et al. 2012). Administration of apelin-13 attenuates these harmful effects. This directly implies that mechanical force leads to the adoption of an AR conformation capable of β-arrestin-mediated signalling that is distinct from the G-protein ‘active’ state conformation normally induced by apelin. Whether this stretch-induced conformation would be independently arrived at during β-arrestin-mediated internalization of the AR is not clear.
4.4 AR dimerization/oligomerization and its impact on cell signalling
The study of GPCR multimers, in which two or more receptors associate to form a larger signalling complex, is of growing importance to GPCR research (Wertman and Dupre 2013). These complexes may be homomeric (composed of only one type of GPCR) or heteromeric (in which different GPCR interacting partners associate). Importantly, the signalling of these multimers can be distinct from that of their constituent monomeric receptors.
To date, cases of AR heteromer formation with the AT1R (Chun et al. 2008), κ-opioid receptor (KOR) (Li et al. 2012), and bradykinin 1 receptor (B1R) (Bai et al. 2014) have been reported. In the case of the AR-AT1R heteromer, the interaction appears to induce inhibition of AT1R signalling (Chun et al. 2008). More recently, it was shown that this AR-AT1R heteromerization is induced by apelin, but is not affected by the presence of angiotensin II (Siddiquee et al. 2013). For the AR-KOR heteromer, the interaction of these two GPCRs is constitutive and appears to mediate ligand-induced ERK activation at a higher level than either receptor alone. Moreover, the co-transfection of both receptors in cell culture leads to increased PKC activation and decreased activation of PKA (Li et al. 2012). In the case of the AR-B1R heteromer, the interaction is also constitutive and also leads to increased PKC activation. In addition, co-transfection of the AR and B1R in HUVECs results in increased eNOS activation in response to either apelin or the B1R ligand des–Arg9–BK, suggesting that this AR-B1R heteromer may be involved in the vasodilatory effects of apelin (Bai et al. 2014). Beyond these studies showing heteromer formation, Piairo et al. (2011) observed AR dimers and higher order oligomers in the developing lung using western blotting, but these multimers were not conclusively identified as being either homomeric or heteromeric in nature.
In addition to the cases outlined here, the AR may have additional interacting partners, and the impact of multimerization on AR signalling may be significant. Of note, in cases of observed heteromerization, it is difficult to determine with certainty what portion of the observed effects are a direct result of physical interaction between the receptors, as opposed to additional crosstalk between signalling pathways. As such, this aspect of AR research warrants further investigation.
4.5 Summary of AR-mediated cell signalling
The AR-mediated cell signalling events discussed above explain only a portion of the many physiological effects that occur in response to AR activation. To date, only a limited number of the AR’s many cellular effects have been connected unambiguously to particular branches of the signalling cascade downstream of the GPCR. Moreover, it seems likely that the type and magnitude of downstream events may frequently be context-dependent, or vary with cell type, as was observed for the AR’s varied Gαi/o and Gαq/11 coupling preferences. Further complicating matters, as detailed in the next section of this minireview, the exact isoform of activating peptide ligand may also directly influence signalling.
5. Comparison of the properties of endogenous AR ligands
The following section describes some of the unique signalling properties that have been reported to date for the variety of known apelin and postulated Toddler/ELABELA bioactive ligands of the AR (Table 1). It should become apparent that numerous important aspects of the potential ligand-specific variability in signalling remain to be characterized.
5.1 Apelin-induced signalling
All isoforms share the same 12 C-terminal residues (Table 1) required for bioactivity (Tatemoto et al. 2001) and show consistent structuring over this region (Langelaan et al. 2009; Langelaan and Rainey 2009). Compellingly, the conserved structural features of the apelin isoforms have been directly employed in design of peptide-based AR antagonists (Macaluso and Glen 2010; Macaluso et al. 2011), and agonists (Murza et al. 2012). Despite the clear importance of the apelin C-terminal dodecapeptide region, the presence of multiple apelin isoforms in the body suggests an evolutionary purpose.
In the initial report of apelin by Tatemoto et al. (1998), the potency of apelin-13, -17 and -36 peptides was directly compared. Notably, the two shorter isoforms had 8 to 60-fold higher activity than apelin-36 with respect to measurements of the AR-induced cellular acidification rate. AR internalization post-activation in HEK cells, human neurons, and microvascular endothelial cells has also been shown to vary depending on the apelin isoform. Apelin-36 induces internalization – and a sustained β-arrestin association – more potently than apelin-13 (Masri et al. 2006). This disparity is attributed to the different affinity of the receptor for each isoform; namely, apelin-13 dissociates more readily than apelin-36, and this rapid dissociation is accompanied by complete recycling of the AR to the cell surface within one hour. By contrast, apelin-36-associated AR remains sequestered in intracellular compartments, with no recycling even two hours after apelin administration (Lee et al. 2010; Zhou et al. 2003). Additional studies have further highlighted differences in binding affinity (Kd) and potency (EC50) for the different isoforms (Cayabyab et al. 2000; Kawamata et al. 2001; Zou et al. 2000).
In addition, there are differences in the tissue distribution of these peptides. For example, apelin-36 seems to be the predominant isoform in the lung, testis, and uterus; conversely, both apelin-36 and pyr-apelin-13 are present at high levels in the mammary gland (Kawamata et al. 2001). By contrast, Pyr-apelin-13 (the N-terminally pyroglutamate-modified form of apelin-13, a spontaneous modification (e.g. (Shin et al. 2013)) has been the only isoform detected in the hypothalamus, and is the predominant form in rat brain extracts (De Mota et al. 2004; El Messari et al. 2004) and human cardiac tissue (Maguire et al. 2009).
In human blood plasma, apelin-13, Pyr-apelin-13, and apelin-17 have all been detected using antibody-based (Reaux et al. 2002) or HPLC-based assays (Azizi et al. 2008). Recently, mass spectrometry (MS), a more reliable means than immunoassay for unambiguously distinguishing the isoforms, was used to demonstrate that Pyr-apelin-13 is the most prevalent isoform in human plasma (Zhen et al. 2013). It should be noted that this directly contrasts with an earlier HPLC-MS study where none of the expected apelin isoforms (Table 1) were detected in human plasma (Mesmin et al. 2010). This latter method, however, was subsequently employed to successfully identify apelin peptides in the bovine colostrum (Mesmin et al. 2011). Interestingly, in this study, many forms of apelin above and beyond the known bioactive isoforms were identified, including peptides lacking one or more C-terminal residues, to a total of 46 isoforms. Results such as these highlight the incompleteness of current knowledge about the distribution and function of the various apelin peptides.
In summary, both the tissue distribution of the different apelin isoforms, as well as their functional behaviour as activators of the AR, are different. Functionally, the shorter apelin isoforms have exhibited higher potency for certain downstream readouts and fast cell-surface recycling post-internalization, while the longer forms have exhibited higher affinity for the AR and increased intracellular retention (Lee et al. 2010; Masri et al. 2006; Zhou et al. 2003). Though the effects of these distinct internalization patterns have not been studied in detail, it seems likely that they should confer unique, isoform-dependent cellular effects (e.g., sustained cytosolic vs. transient nuclear ERK activation) (Ahn et al. 2004). Moreover, despite, or perhaps because of, its lower affinity for the AR, apelin-13 is the more potent activator of Gαi--mediated ERK phosphorylation (Masri et al. 2006). Interestingly, while the affinities of the apelin-13-bound AR for Gαi1 and Gαi2 are roughly equivalent, apelin-36 induces the AR to couple preferentially to the Gαi2 subunit (Masri et al. 2006). Such differences suggest the existence of different conformations or timescales of ‘activated’ AR, which are differentially induced by the various apelin isoforms, and which may have physiological importance. There have been no studies probing for apelin-induced signalling bias towards specific downstream pathways (e.g., Gαi-mediated, β-arrestin-mediated), but it is a definite possibility that the different apelin isoforms direct the AR signal toward different branches of the downstream cascade.
5.2 Toddler/ELABELA-induced signalling
The recent discovery of a new endogenous peptide ligand for AR, currently known as both Toddler (Pauli et al. 2014), and ELABELA (Chng et al. 2013), followed screens to discover signals regulating early development. Although characterized in zebrafish, a high degree of conservation of the Toddler/ELABELA gene in vertebrate species including humans implies likelihood of similar importance in human development, but this has yet to be shown. Like apelin, this peptide exists in multiple endogenous isoforms (Table 1). Toddler/ELABELA signalling is motogenic, and its absence or overproduction reduces the movement of mesendodermal zebrafish cells during gastrulation, inhibiting proper development (Pauli et al. 2014). Moreover, in toddler/ELABELA knockout zebrafish, the cells of the endoderm have impaired differentiation potential, and embryos exhibit stunted or completely absent heart development. This mirrors the phenotype observed in AR knockout embryos (Chng et al. 2013). Apelin knockout embryos, on the other hand, do not have this phenotype. Systemic administration of Toddler/ELABELA in toddler/ELABELA knockout zebrafish rescues the otherwise aberrant phenotype (Pauli et al. 2014).
Receptor activation studies revealed that the zebrafish Toddler-21 peptide acts by binding the AR and inducing receptor internalization (Pauli et al. 2014). Moreover, the expression profiles of Toddler/ELABELA and apelin differ during zebrafish development (Pauli et al. 2014). In particular, during gastrulation Toddler/ELABELA is highly expressed, whereas apelin expression remains low. Following this period, however, Toddler/ELABELA expression drops sharply and apelin levels begin to rise steadily. All of these findings indicate that Toddler/ELABELA is a developmentally critical AR ligand whose signalling behaviour differs significantly from that of apelin. The exact intracellular signalling mechanism(s) of toddler/ELABELA remain unknown.
5.3 Scope of AR ligand functionality
In summary, major differences are observed with the various bioactive AR ligands. These differences range from changes in expression profiles of Toddler/ELABELA and apelin during embryonic development to the variations in tissue distribution of the various apelin isoforms to differences in affinity, potency, and internalization patterns. Despite a consistent, retained C-terminal region with identifiable structural motifs required for AR activation, the various apelin peptides clearly have different functional behaviour with respect to AR signalling. All of these facts point not only to functionally important differences between apelin and Toddler/ELABELA but also to the ability to fine-tune cellular response through the exact peptide isoform being produced. To rationally design synthetic ligands capable of modulating AR activity for a defined therapeutic benefit, it will therefore be beneficial to better understand the ligand-AR interaction, including consideration of the role of the variable N-terminal region in modifying AR signalling.
6. Summary
It should now be clear that the AR is an important GPCR that mediates a wide array of normal physiological and pathological processes. With involvement in areas ranging from cardiovascular regulation, angiogenesis, and energy metabolism to fluid homeostasis and the neuroendocrine stress response, AR signalling is implicated in several pathologies, including heart disease, diabetes, obesity, and cancer. Yet, relatively little is currently known about the way in which the receptor is activated by its various endogenous peptidic ligands, or about the downstream cellular signalling pathways that lead to the diverse physiological effects that have inspired intense study of the AR. Future AR research should be both functional and structural in its scope, with particular focus on understanding of the unique signalling properties of the different bioactive endogenous ligand isoforms capable of activating the AR. Such studies will help unravel the mechanisms of the complex and context-dependent signalling behaviour of the AR, and will aid in the design of future therapeutics targeting this important receptor.
Acknowledgments
This work was supported by a Canadian Institutes of Health Research (CIHR) Operating Grant (MOP-111138 to J.K.R.); a Nova Scotia Health Research Foundation (NSHRF) Scotia Support Grant (to J.K.R.); and, a Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery grant (RGPIN 355310-2013) (to D.J.D.) J.K.R. is grateful for support from a CIHR New Investigator Award. Thanks to Kyungsoo Shin for his constructive comments on the manuscript.
References
- Ahmed SM, Angers S. Emerging non-canonical functions for heterotrimeric G proteins in cellular signaling. J Recept Signal Transduct Res. 2013;33(3):177–183. doi: 10.3109/10799893.2013.795972. [DOI] [PubMed] [Google Scholar]
- Ahn S, Shenoy SK, Wei H, Lefkowitz RJ. Differential kinetic and spatial patterns of beta-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J Biol Chem. 2004;279(34):35518–35525. doi: 10.1074/jbc.M405878200. [DOI] [PubMed] [Google Scholar]
- Ashley EA, Powers J, Chen M, Kundu R, Finsterbach T, Caffarelli A, Deng A, Eichhorn J, Mahajan R, Agrawal R, Greve J, Robbins R, Patterson AJ, Bernstein D, Quertermous T. The endogenous peptide apelin potently improves cardiac contractility and reduces cardiac loading in vivo. Cardiovasc Res. 2005;65(1):73–82. doi: 10.1016/j.cardiores.2004.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attane C, Foussal C, Le Gonidec S, Benani A, Daviaud D, Wanecq E, Guzman-Ruiz R, Dray C, Bezaire V, Rancoule C, Kuba K, Ruiz-Gayo M, Levade T, Penninger J, Burcelin R, Penicaud L, Valet P, Castan-Laurell I. Apelin treatment increases complete Fatty Acid oxidation, mitochondrial oxidative capacity, and biogenesis in muscle of insulin-resistant mice. Diabetes. 2012;61(2):310–320. doi: 10.2337/db11-0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azizi M, Iturrioz X, Blanchard A, Peyrard S, De Mota N, Chartrel N, Vaudry H, Corvol P, Llorens-Cortes C. Reciprocal regulation of plasma apelin and vasopressin by osmotic stimuli. J Am Soc Nephrol. 2008;19(5):1015–1024. doi: 10.1681/asn.2007070816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai B, Liu L, Zhang N, Wang C, Jiang Y, Chen J. Heterodimerization of human apelin and bradykinin 1 receptors: novel signal transduction characteristics. Cell Signal. 2014;26(7):1549–1559. doi: 10.1016/j.cellsig.2014.03.022. [DOI] [PubMed] [Google Scholar]
- Blaukat A, Barac A, Cross MJ, Offermanns S, Dikic I. G protein-coupled receptor-mediated mitogen-activated protein kinase activation through cooperation of Galpha(q) and Galpha(i) signals. Mol Cell Biol. 2000;20(18):6837–6848. doi: 10.1128/mcb.20.18.6837-6848.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher J, Masri B, Daviaud D, Gesta S, Guigne C, Mazzucotelli A, Castan-Laurell I, Tack I, Knibiehler B, Carpene C, Audigier Y, Saulnier-Blache JS, Valet P. Apelin, a newly identified adipokine up-regulated by insulin and obesity. Endocrinology. 2005;146(4):1764–1771. doi: 10.1210/en.2004-1427. [DOI] [PubMed] [Google Scholar]
- Brailoiu GC, Dun SL, Yang J, Ohsawa M, Chang JK, Dun NJ. Apelin-immunoreactivity in the rat hypothalamus and pituitary. Neurosci Lett. 2002;327(3):193–197. doi: 10.1016/s0304-3940(02)00411-1. [DOI] [PubMed] [Google Scholar]
- Burch ML, Getachew R, Osman N, Febbraio MA, Little PJ. Thrombin-mediated proteoglycan synthesis utilizes both protein-tyrosine kinase and serine/threonine kinase receptor transactivation in vascular smooth muscle cells. J Biol Chem. 2013;288(10):7410–7419. doi: 10.1074/jbc.M112.400259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castan-Laurell I, Dray C, Knauf C, Kunduzova O, Valet P. Apelin, a promising target for type 2 diabetes treatment? Trends Endocrinol Metab. 2012;23(5):234–241. doi: 10.1016/j.tem.2012.02.005. [DOI] [PubMed] [Google Scholar]
- Cayabyab M, Hinuma S, Farzan M, Choe H, Fukusumi S, Kitada C, Nishizawa N, Hosoya M, Nishimura O, Messele T, Pollakis G, Goudsmit J, Fujino M, Sodroski J. Apelin, the natural ligand of the orphan seven-transmembrane receptor APJ, inhibits human immunodeficiency virus type 1 entry. J Virol. 2000;74(24):11972–11976. doi: 10.1128/jvi.74.24.11972-11976.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles CJ, Rademaker MT, Richards AM. Apelin-13 induces a biphasic haemodynamic response and hormonal activation in normal conscious sheep. J Endocrinol. 2006;189(3):701–710. doi: 10.1677/joe.1.06804. [DOI] [PubMed] [Google Scholar]
- Chen H, Zheng C, Zhang X, Li J, Li J, Zheng L, Huang K. Apelin alleviates diabetes-associated endoplasmic reticulum stress in the pancreas of Akita mice. Peptides. 2011;32(8):1634–1639. doi: 10.1016/j.peptides.2011.06.025. [DOI] [PubMed] [Google Scholar]
- Cheng X, Cheng XS, Pang CC. Venous dilator effect of apelin, an endogenous peptide ligand for the orphan APJ receptor, in conscious rats. Eur J Pharmacol. 2003;470(3):171–175. doi: 10.1016/s0014-2999(03)01821-1. [DOI] [PubMed] [Google Scholar]
- Chng SC, Ho L, Tian J, Reversade B. ELABELA: a hormone essential for heart development signals via the apelin receptor. Dev Cell. 2013;27(6):672–680. doi: 10.1016/j.devcel.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Choe H, Farzan M, Konkel M, Martin K, Sun Y, Marcon L, Cayabyab M, Berman M, Dorf ME, Gerard N, Gerard C, Sodroski J. The orphan seven-transmembrane receptor apj supports the entry of primary T-cell-line-tropic and dualtropic human immunodeficiency virus type 1. J Virol. 1998;72(7):6113–6118. doi: 10.1128/jvi.72.7.6113-6118.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun HJ, Ali ZA, Kojima Y, Kundu RK, Sheikh AY, Agrawal R, Zheng L, Leeper NJ, Pearl NE, Patterson AJ, Anderson JP, Tsao PS, Lenardo MJ, Ashley EA, Quertermous T. Apelin signaling antagonizes Ang II effects in mouse models of atherosclerosis. J Clin Invest. 2008;118(10):3343–3354. doi: 10.1172/jci34871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke KJ, Whitaker KW, Reyes TM. Diminished metabolic responses to centrally-administered apelin-13 in diet-induced obese rats fed a high-fat diet. J Neuroendocrinol. 2009;21(2):83–89. doi: 10.1111/j.1365-2826.2008.01815.x. [DOI] [PubMed] [Google Scholar]
- Cox CM, D’Agostino SL, Miller MK, Heimark RL, Krieg PA. Apelin, the ligand for the endothelial G-protein-coupled receptor, APJ, is a potent angiogenic factor required for normal vascular development of the frog embryo. Dev Biol. 2006;296(1):177–189. doi: 10.1016/j.ydbio.2006.04.452. [DOI] [PubMed] [Google Scholar]
- Dai L, Smith PM, Kuksis M, Ferguson AV. Apelin acts in the subfornical organ to influence neuronal excitability and cardiovascular function. J Physiol. 2013;591(Pt 13):3421–3432. doi: 10.1113/jphysiol.2013.254144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Mota N, Reaux-Le Goazigo A, El Messari S, Chartrel N, Roesch D, Dujardin C, Kordon C, Vaudry H, Moos F, Llorens-Cortes C. Apelin, a potent diuretic neuropeptide counteracting vasopressin actions through inhibition of vasopressin neuron activity and vasopressin release. Proc Natl Acad Sci U S A. 2004;101(28):10464–10469. doi: 10.1073/pnas.0403518101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Messari S, Iturrioz X, Fassot C, De Mota N, Roesch D, Llorens-Cortes C. Functional dissociation of apelin receptor signaling and endocytosis: implications for the effects of apelin on arterial blood pressure. J Neurochem. 2004;90(6):1290–1301. doi: 10.1111/j.1471-4159.2004.02591.x. [DOI] [PubMed] [Google Scholar]
- Evans NA, Groarke DA, Warrack J, Greenwood CJ, Dodgson K, Milligan G, Wilson S. Visualizing differences in ligand-induced beta-arrestin-GFP interactions and trafficking between three recently characterized G protein-coupled receptors. J Neurochem. 2001;77(2):476–485. doi: 10.1046/j.1471-4159.2001.00269.x. [DOI] [PubMed] [Google Scholar]
- Fleming I, Busse R. Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am J Physiol Regul Integr Comp Physiol. 2003;284(1):R1–12. doi: 10.1152/ajpregu.00323.2002. [DOI] [PubMed] [Google Scholar]
- Heinonen MV, Purhonen AK, Miettinen P, Paakkonen M, Pirinen E, Alhava E, Akerman K, Herzig KH. Apelin, orexin-A and leptin plasma levels in morbid obesity and effect of gastric banding. Regul Pept. 2005;130(1–2):7–13. doi: 10.1016/j.regpep.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Kagiyama S, Fukuhara M, Matsumura K, Lin Y, Fujii K, Iida M. Central and peripheral cardiovascular actions of apelin in conscious rats. Regul Pept. 2005;125(1–3):55–59. doi: 10.1016/j.regpep.2004.07.033. [DOI] [PubMed] [Google Scholar]
- Kalin RE, Kretz MP, Meyer AM, Kispert A, Heppner FL, Brandli AW. Paracrine and autocrine mechanisms of apelin signaling govern embryonic and tumor angiogenesis. Dev Biol. 2007;305(2):599–614. doi: 10.1016/j.ydbio.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Kang Y, Kim J, Anderson JP, Wu J, Gleim SR, Kundu RK, McLean DL, Kim JD, Park H, Jin SW, Hwa J, Quertermous T, Chun HJ. Apelin-APJ signaling is a critical regulator of endothelial MEF2 activation in cardiovascular development. Circ Res. 2013;113(1):22–31. doi: 10.1161/circresaha.113.301324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamata Y, Habata Y, Fukusumi S, Hosoya M, Fujii R, Hinuma S, Nishizawa N, Kitada C, Onda H, Nishimura O, Fujino M. Molecular properties of apelin: tissue distribution and receptor binding. Biochim Biophys Acta. 2001;1538(2–3):162–171. doi: 10.1016/s0167-4889(00)00143-9. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther. 2011;336(2):296–302. doi: 10.1124/jpet.110.173948. [DOI] [PubMed] [Google Scholar]
- Kleinz MJ, Davenport AP. Emerging roles of apelin in biology and medicine. Pharmacol Ther. 2005;107(2):198–211. doi: 10.1016/j.pharmthera.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Langelaan DN, Bebbington EM, Reddy T, Rainey JK. Structural insight into G-protein coupled receptor binding by apelin. Biochemistry. 2009;48(3):537–548. doi: 10.1021/bi801864b. [DOI] [PubMed] [Google Scholar]
- Langelaan DN, Rainey JK. Headgroup-dependent membrane catalysis of apelin-receptor interactions is likely. J Phys Chem B. 2009;113(30):10465–10471. doi: 10.1021/jp904562q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DK, Cheng R, Nguyen T, Fan T, Kariyawasam AP, Liu Y, Osmond DH, George SR, O’Dowd BF. Characterization of apelin, the ligand for the APJ receptor. J Neurochem. 2000;74(1):34–41. doi: 10.1046/j.1471-4159.2000.0740034.x. [DOI] [PubMed] [Google Scholar]
- Lee DK, Ferguson SS, George SR, O’Dowd BF. The fate of the internalized apelin receptor is determined by different isoforms of apelin mediating differential interaction with beta-arrestin. Biochem Biophys Res Commun. 2010;395(2):185–189. doi: 10.1016/j.bbrc.2010.03.151. [DOI] [PubMed] [Google Scholar]
- Lee DK, Saldivia VR, Nguyen T, Cheng R, George SR, O’Dowd BF. Modification of the terminal residue of apelin-13 antagonizes its hypotensive action. Endocrinology. 2005;146(1):231–236. doi: 10.1210/en.2004-0359. [DOI] [PubMed] [Google Scholar]
- Li Y, Chen J, Bai B, Du H, Liu Y, Liu H. Heterodimerization of human apelin and kappa opioid receptors: roles in signal transduction. Cell Signal. 2012;24(5):991–1001. doi: 10.1016/j.cellsig.2011.12.012. [DOI] [PubMed] [Google Scholar]
- Liebmann C. EGF receptor activation by GPCRs: an universal pathway reveals different versions. Mol Cell Endocrinol. 2011;331(2):222–231. doi: 10.1016/j.mce.2010.04.008. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Lefkowitz RJ. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci. 2002;115(Pt 3):455–465. doi: 10.1242/jcs.115.3.455. [DOI] [PubMed] [Google Scholar]
- Macaluso NJ, Glen RC. Exploring the ‘RPRL’ motif of apelin-13 through molecular simulation and biological evaluation of cyclic peptide analogues. Chem Med Chem. 2010;5(8):1247–1253. doi: 10.1002/cmdc.201000061. [DOI] [PubMed] [Google Scholar]
- Macaluso NJ, Pitkin SL, Maguire JJ, Davenport AP, Glen RC. Discovery of a competitive apelin receptor (APJ) antagonist. Chem Med Chem. 2011;6(6):1017–1023. doi: 10.1002/cmdc.201100069. [DOI] [PubMed] [Google Scholar]
- Maguire JJ, Kleinz MJ, Pitkin SL, Davenport AP. [Pyr1]apelin-13 identified as the predominant apelin isoform in the human heart: vasoactive mechanisms and inotropic action in disease. Hypertension. 2009;54(3):598–604. doi: 10.1161/hypertensionaha.109.134619. [DOI] [PubMed] [Google Scholar]
- Masri B, Knibiehler B, Audigier Y. Apelin signalling: a promising pathway from cloning to pharmacology. Cell Signal. 2005;17(4):415–426. doi: 10.1016/j.cellsig.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Masri B, Lahlou H, Mazarguil H, Knibiehler B, Audigier Y. Apelin (65–77) activates extracellular signal-regulated kinases via a PTX-sensitive G protein. Biochem Biophys Res Commun. 2002;290(1):539–545. doi: 10.1006/bbrc.2001.6230. [DOI] [PubMed] [Google Scholar]
- Masri B, Morin N, Pedebernade L, Knibiehler B, Audigier Y. The apelin receptor is coupled to Gi1 or Gi2 protein and is differentially desensitized by apelin fragments. J Biol Chem. 2006;281(27):18317–18326. doi: 10.1074/jbc.M600606200. [DOI] [PubMed] [Google Scholar]
- Mesmin C, Dubois M, Becher F, Fenaille F, Ezan E. Liquid chromatography/tandem mass spectrometry assay for the absolute quantification of the expected circulating apelin peptides in human plasma. Rapid Commun Mass Spectrom. 2010;24(19):2875–2884. doi: 10.1002/rcm.4718. [DOI] [PubMed] [Google Scholar]
- Mesmin C, Fenaille F, Becher F, Tabet JC, Ezan E. Identification and characterization of apelin peptides in bovine colostrum and milk by liquid chromatography-mass spectrometry. J Proteome Res. 2011;10(11):5222–5231. doi: 10.1021/pr200725x. [DOI] [PubMed] [Google Scholar]
- Mukhin YV, Garnovsky EA, Ullian ME, Garnovskaya MN. Bradykinin B2 receptor activates extracellular signal-regulated protein kinase in mIMCD-3 cells via epidermal growth factor receptor transactivation. J Pharmacol Exp Ther. 2003;304(3):968–977. doi: 10.1124/jpet.102.043943. [DOI] [PubMed] [Google Scholar]
- Murza A, Parent A, Besserer-Offroy E, Tremblay H, Karadereye F, Beaudet N, Leduc R, Sarret P, Marsault E. Elucidation of the structure-activity relationships of apelin: influence of unnatural amino acids on binding, signaling, and plasma stability. Chem Med Chem. 2012;7(2):318–325. doi: 10.1002/cmdc.201100492. [DOI] [PubMed] [Google Scholar]
- Neves SR, Ram PT, Iyengar R. G protein pathways. Science. 2002;296(5573):1636–1639. doi: 10.1126/science.1071550. [DOI] [PubMed] [Google Scholar]
- Newson MJ, Pope GR, Roberts EM, Lolait SJ, O’Carroll AM. Stress-dependent and gender-specific neuroregulatory roles of the apelin receptor in the hypothalamic-pituitary-adrenal axis response to acute stress. J Endocrinol. 2013;216(1):99–109. doi: 10.1530/joe-12-0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Carroll AM, Lolait SJ, Harris LE, Pope GR. The apelin receptor APJ: journey from an orphan to a multifaceted regulator of homeostasis. J Endocrinol. 2013;219(1):R13–35. doi: 10.1530/joe-13-0227. [DOI] [PubMed] [Google Scholar]
- O’Dowd BF, Heiber M, Chan A, Heng HH, Tsui LC, Kennedy JL, Shi X, Petronis A, George SR, Nguyen T. A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene. 1993;136(1–2):355–360. doi: 10.1016/0378-1119(93)90495-o. [DOI] [PubMed] [Google Scholar]
- Pauli A, Norris ML, Valen E, Chew GL, Gagnon JA, Zimmerman S, Mitchell A, Ma J, Dubrulle J, Reyon D, Tsai SQ, Joung JK, Saghatelian A, Schier AF. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. 2014;343(6172):1248636. doi: 10.1126/science.1248636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piairo P, Moura RS, Nogueira-Silva C, Correia-Pinto J. The apelinergic system in the developing lung: expression and signaling. Peptides. 2011;32(12):2474–2483. doi: 10.1016/j.peptides.2011.10.010. [DOI] [PubMed] [Google Scholar]
- Picault FX, Chaves-Almagro C, Projetti F, Prats H, Masri B, Audigier Y. Tumour co-expression of apelin and its receptor is the basis of an autocrine loop involved in the growth of colon adenocarcinomas. Eur J Cancer. 2014;50(3):663–674. doi: 10.1016/j.ejca.2013.11.017. [DOI] [PubMed] [Google Scholar]
- Pitkin SL, Maguire JJ, Bonner TI, Davenport AP. International Union of Basic and Clinical Pharmacology. LXXIV. Apelin receptor nomenclature, distribution, pharmacology, and function. Pharmacol Rev. 2010;62(3):331–342. doi: 10.1124/pr.110.002949. [DOI] [PubMed] [Google Scholar]
- Reaux A, De Mota N, Skultetyova I, Lenkei Z, El Messari S, Gallatz K, Corvol P, Palkovits M, Llorens-Cortes C. Physiological role of a novel neuropeptide, apelin, and its receptor in the rat brain. J Neurochem. 2001;77(4):1085–1096. doi: 10.1046/j.1471-4159.2001.00320.x. [DOI] [PubMed] [Google Scholar]
- Reaux A, Gallatz K, Palkovits M, Llorens-Cortes C. Distribution of apelin-synthesizing neurons in the adult rat brain. Neuroscience. 2002;113(3):653–662. doi: 10.1016/s0306-4522(02)00192-6. [DOI] [PubMed] [Google Scholar]
- Roberts EM, Pope GR, Newson MJ, Landgraf R, Lolait SJ, O’Carroll AM. Stimulus-specific neuroendocrine responses to osmotic challenges in apelin receptor knockout mice. J Neuroendocrinol. 2010;22(4):301–308. doi: 10.1111/j.1365-2826.2010.01968.x. [DOI] [PubMed] [Google Scholar]
- Said El Messari XI, Fassot Celine, De Mota Nadia, Roesch Darren, Llorens-Cortes Catherine. Functional dissociation of apelin receptor signaling and endocytosis: implications for the effects of apelin on arterial blood pressure. Journal of Neurochemistry. 2004;(90):1290–1301. doi: 10.1111/j.1471-4159.2004.02591.x. [DOI] [PubMed] [Google Scholar]
- Scimia MC, Hurtado C, Ray S, Metzler S, Wei K, Wang J, Woods CE, Purcell NH, Catalucci D, Akasaka T, Bueno OF, Vlasuk GP, Kaliman P, Bodmer R, Smith LH, Ashley E, Mercola M, Brown JH, Ruiz-Lozano P. APJ acts as a dual receptor in cardiac hypertrophy. Nature. 2012;488(7411):394–398. doi: 10.1038/nature11263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin K, Pandey A, Liu XQ, Anini Y, Rainey JK. Preferential apelin-13 production by the proprotein convertase PCSK3 is implicated in obesity. FEBS Open Bio. 2013;3:328–333. doi: 10.1016/j.fob.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiquee K, Hampton J, McAnally D, May L, Smith L. The apelin receptor inhibits the angiotensin II type 1 receptor via allosteric trans-inhibition. Br J Pharmacol. 2013;168(5):1104–1117. doi: 10.1111/j.1476-5381.2012.02192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorhede Winzell M, Magnusson C, Ahren B. The apj receptor is expressed in pancreatic islets and its ligand, apelin, inhibits insulin secretion in mice. Regul Pept. 2005;131(1–3):12–17. doi: 10.1016/j.regpep.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Szokodi I, Tavi P, Foldes G, Voutilainen-Myllyla S, Ilves M, Tokola H, Pikkarainen S, Piuhola J, Rysa J, Toth M, Ruskoaho H. Apelin, the novel endogenous ligand of the orphan receptor APJ, regulates cardiac contractility. Circ Res. 2002;91(5):434–440. doi: 10.1161/01.res.0000033522.37861.69. [DOI] [PubMed] [Google Scholar]
- Taheri S, Murphy K, Cohen M, Sujkovic E, Kennedy A, Dhillo W, Dakin C, Sajedi A, Ghatei M, Bloom S. The effects of centrally administered apelin-13 on food intake, water intake and pituitary hormone release in rats. Biochem Biophys Res Commun. 2002;291(5):1208–1212. doi: 10.1006/bbrc.2002.6575. [DOI] [PubMed] [Google Scholar]
- Tatemoto K, Hosoya M, Habata Y, Fujii R, Kakegawa T, Zou MX, Kawamata Y, Fukusumi S, Hinuma S, Kitada C, Kurokawa T, Onda H, Fujino M. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem Biophys Res Commun. 1998;251(2):471–476. doi: 10.1006/bbrc.1998.9489. [DOI] [PubMed] [Google Scholar]
- Tatemoto K, Takayama K, Zou MX, Kumaki I, Zhang W, Kumano K, Fujimiya M. The novel peptide apelin lowers blood pressure via a nitric oxide-dependent mechanism. Regul Pept. 2001;99(2–3):87–92. doi: 10.1016/s0167-0115(01)00236-1. [DOI] [PubMed] [Google Scholar]
- Wang W, McKinnie SM, Patel VB, Haddad G, Wang Z, Zhabyeyev P, Das SK, Basu R, McLean B, Kandalam V, Penninger JM, Kassiri Z, Vederas JC, Murray AG, Oudit GY. Loss of Apelin exacerbates myocardial infarction adverse remodeling and ischemia-reperfusion injury: therapeutic potential of synthetic Apelin analogues. J Am Heart Assoc. 2013;2(4):e000249. doi: 10.1161/jaha.113.000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, Hou X, Tatemoto K. Regulation of apelin mRNA expression by insulin and glucocorticoids in mouse 3T3-L1 adipocytes. Regul Pept. 2005;132(1–3):27–32. doi: 10.1016/j.regpep.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Wertman J, Dupre DJ. G protein-coupled receptor dimers: look like their parents, but act like teenagers! J Recept Signal Transduct Res. 2013;33(3):135–138. doi: 10.3109/10799893.2012.759591. [DOI] [PubMed] [Google Scholar]
- Yue P, Jin H, Aillaud M, Deng AC, Azuma J, Asagami T, Kundu RK, Reaven GM, Quertermous T, Tsao PS. Apelin is necessary for the maintenance of insulin sensitivity. Am J Physiol Endocrinol Metab. 2010;298(1):E59–67. doi: 10.1152/ajpendo.00385.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Yao F, Raizada MK, O’Rourke ST, Sun C. Apelin gene transfer into the rostral ventrolateral medulla induces chronic blood pressure elevation in normotensive rats. Circ Res. 2009a;104(12):1421–1428. doi: 10.1161/circresaha.108.192302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Yu B, Tao GZ. Apelin protects against cardiomyocyte apoptosis induced by glucose deprivation. Chin Med J (Engl) 2009b;122(19):2360–2365. [PubMed] [Google Scholar]
- Zhen EY, Higgs RE, Gutierrez JA. Pyroglutamyl apelin-13 identified as the major apelin isoform in human plasma. Anal Biochem. 2013;442(1):1–9. doi: 10.1016/j.ab.2013.07.006. [DOI] [PubMed] [Google Scholar]
- Zhou N, Fan X, Mukhtar M, Fang J, Patel CA, DuBois GC, Pomerantz RJ. Cell-cell fusion and internalization of the CNS-based, HIV-1 co-receptor, APJ. Virology. 2003;307(1):22–36. doi: 10.1016/s0042-6822(02)00021-1. [DOI] [PubMed] [Google Scholar]
- Zou MX, Liu HY, Haraguchi Y, Soda Y, Tatemoto K, Hoshino H. FEBS Lett. 2000;473(1):15–18. doi: 10.1016/s0014-5793(00)01487-3. [DOI] [PubMed] [Google Scholar]