

Abstract
Objective
Inflammation is an important component of the response to traumatic brain injury (TBI). Progesterone has been shown to inhibit neuroinflammation following (TBI), and may do so through Toll-like receptor (TLR) -mediated pathways. In vitro studies indicate that 1,25-dihydroxyvitamin D(3) (VDH) may also modulate the inflammatory response through the TLR4 pathway. We tested the hypothesis PROG and VDH would exert additive and synergistic neuroprotective effects compared with individual treatment by modulating TLR4/NF-κB-mediated inflammation pathways after TBI in rats.
Research Design and Methods
Bilateral medial frontal cortical impact injury was induced in young adult Sprague-Dawley rats. Progesterone (i.p., 16 mg/kg body weight) and VDH (1 ug/kg body weight) were injected separately or combined at 1 and 6 h after surgery. Rats were killed 24 h post-surgery and peri-contusional brain tissue harvested for immunostaining and protein measurement.
Results
TLR4, phosphorylation of NF-κB,. neuronal loss, and astrocyte activation were significantly reduced with combination treatment after TBI compared to each agent iven individually.
Conclusions
At 24h after TBI, combination therapy shows greater efficacy in reducing neuroinflammation compared to progesterone and VDH given separately, and does so by modulating the TLR4/NF-κB signaling pathway.
Keywords: Frontal cortex, Toll-like receptors, neurosteroids, cytokines, neuroprotection
Introduction
Traumatic brain injury (TBI) is a complex disease comprised of a primary insult and a progressive secondary cascade of inflammatory and immune responses [1, 2] resulting in neuronal apoptosis, necrosis, neuronal degeneration [3], neurological deficits [4], and chronic behavioural impairments [5]. The mechanisms involved in this injury cascade are still not completely understood, but recent studies have shown that Toll-like receptors (TLR) play an important role in the neuroinflammation activated by TBI [6], cerebral stroke [7], Alzheimer's Disease [8], and subarachnoid hemorrhage (SAH) [9]. Modulating TLR activity may be a potential target for therapeutic innovations for brain injury.
The TLR is an important player in controlling the extent of brain damage because it triggers a cascade of inflammatory signaling events resulting in the activation of NF-κB, and subsequent production of inflammatory mediators [10] that precipitate more damage to vulnerable CNS tissue and reduce the chances for functional recovery. For example, activation of TLR4 stimulates IκB phosphorylation and degradation, which results in the phosphorylation and translocation of NF-κB. Phosphorylated NF-κB then triggers the transcription of downstream genes [11, 12]. NF-κB controls the transcription of a variety of genes involved in inflammation, including TNFα, IL-1β, COX2 and iNOS, and these in turn initiate an acute inflammatory response post-TBI [13-16]. Since most inflammatory cytokines increase within 24 h after the original injury and the secondary injury develops quickly [13, 17], we chose to investigate TLR4/NF-kB-mediated neuroinflammation at 24 h.
The story about the role of inflammation in brain injury is complex because moderate inflammatory responses induced by TLRs are protective and needed for immune defense against exogenous pathogens and tissue repair, while excessive or chronic inflammation mediated by these same receptors will lead to irreversible tissue damage and its consequent pathologies [18]. In a mouse model of transient cerebral ischemia, TLR4 knock-out (KO) mice had lower expression of mediators indicating brain inflammation and damage, including interferon regulatory factor-1(IRF1), matrix metalloproteinase-9 (MMP9), inducible nitric oxide synthase (iNOS) and COX2, significantly smaller infarct volume, and improved neurological behaviour compared to wild-type mice within 24 h after ischemic insult [19, 20]. In general, much of the recent literature on the role of TLRs in neuroinflammation-related disorders suggests that TLR signaling has potential as a therapeutic target against brain injury.
Despite decades of effort, there are still no clinically available neuroprotective agents to combat the injury cascade, but studies from our and other laboratories have consistently demonstrated that progesterone, a potent, pleiotropic developmental neurosteroid, modulates diverse secondary injury mechanisms in TBI, including apoptotic pathways, diffuse axonal injury, lipid peroxidation [21-23], the expression of inflammatory cytokines, and immune cell activation and migration [6, 24-27],
Many studies have now concluded that progesterone is a promising neuroprotective drug in experimental models of stroke and TBI [28, 29], but initial analyses of Phase III clinical trials with progesterone have not been successful on their quality-of-life measures of functional outcome [30, 31]. This failure may be due in part to the exclusive use of monotherapies to treat complex, heterogeneous disorders like brain injury. A recent workshop sponsored by several national public health communities including the National Institute of Neurological Disorders and Stroke, National Institutes of Health Heart Lung and Blood Institute, National Institute of Child Health and Human Development, and the Department of Veterans Affairs recommended that researchers turn their attention to potential combination therapies in TBI treatment, especially during the first 72 h after TBI [32]. The workshop specifically recommended that progesterone be considered as a promising agent whose neuroprotective effects might be enhanced by combination with other agents.
A recent study found that progesterone activates a number of genes that modulate TLR-mediated signaling pathways on mice after TBI [6]. A series of studies of SAH in male rats have shown that progesterone can regulate TLR4 and nuclear factor-kappa B (NF-κB) signaling pathways in early brain injury after SAH, and that post-SAH progesterone administration may attenuate TLR4/NF-κB signaling following SAH [26]. More recently we showed that blocking of TLR4 signaling mediates the expression of inflammatory cytokines and protects the brain from acute damage induced by ischemia/reperfusion injury [33].
Vitamin D hormone (VDH), like progesterone, is a potent secosteroid hormone and has been shown to exert its own anti-inflammatory effects by promoting negative feedback regulation of TLR4 signaling in macrophages [34] and human myometrial cells [35].
We have previously shown that combined treatment with progesterone and VDH has better efficacy than either agent alone in reducing spatial and reference memory impairment after TBI in both adult and middle-aged rats [36-38]. To more fully elucidate the mechanisms underlying these effects, we tested the idea that combining progesterone with VDH would act on inflammatory responses induced by TBI, and that this regulation of inflammation would occur specifically through TLR4/NF-κB-mediated pathways in rats with TBI. Here we used a bilateral medial frontal cortex contusion model to mimic TBI and tested the hypothesis that (1) the combination therapy would inhibit inflammation post-TBI more effectively than each agent given individually; (2) the combination therapy would exert its effects by regulating the TLR/NF-kB-mediated signaling pathway, and would do so more effectively than the individual treatments; (3) the combination therapy would reduce astrocyte activation and neuron loss post-TBI more effectively than individual treatments.
Methods
Animals
Forty male Sprague-Dawley rats with a body weight of 285±20 g were obtained (Charles River Laboratories, Wilmington, MA, USA) and housed in the Division of Animal Resources of Emory University. All animal experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 85-23, revised 1996). Animal care and experimental protocols (#2001032) were approved by the Emory University Institutional Animal Care and Use Committee. Rats were assigned by chance to one of five groups: sham injury control plus vehicle treatment (Sham, n=8), TBI plus vehicle treatment (TBI, n=8), TBI plus progesterone treatment (TBI+P, n=8), TBI plus VDH treatment (TBI+D, n=8), and TBI plus VDH and progesterone treatment (TBI+P+D, n=8).
Surgery and treatment
The animals were anesthetised with isoflurane (5.0% induction, 2.0–2.5% maintenance) and surgery was performed using aseptic techniques. A SurgiVetTM pulse oximeter (model V3304) was placed on the rear paw to monitor and maintain blood SpO2 at or above 90%. A heart rate monitor with its sensor attached to the other hind paw was used to observe and maintain a rate of 300 beats per minute. To prevent hypothermia during surgery, a homeothermic blanket control unit (Harvard Apparatus, Holliston, MA) was used to monitor and maintain core body temperature (37.5° C). Using aseptic techniques, a midline incision was made along the scalp into the skin and fascia covering the skull, exposing the cranium. A 6-mm diameter mid-sagittal bilateral craniotomy was performed 3 mm anterior to the bregma and a controlled cortical impact was produced in the medial frontal cortex by an electromagnetic cortical contusion device (5-mm diameter impactor tip) with an impact velocity of 2.25 m/s and dwelling time of 500 ms, to a depth of 3.5 mm ventral to bregma (Figure 1) as previously described [39]. The incision was sutured closed after all bleeding had stopped. In the sham group, there was no impact and the incisions were sutured closed after comparable time under anesthesia.
Figure 1.
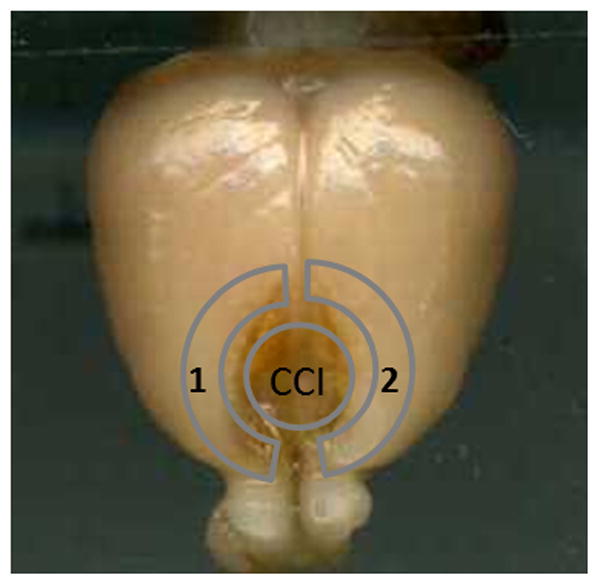
Schematic indication of the cortical contusion injury area (the circle indicates the site of controlled cortical impact (CCI)) induced by an electromagnetic cortical contusion device and the detected area around the injured brain. Label 1 indicates the cortex collected for western blots. Label 2 indicates the cortex collected for ELISA.
In a previous study [37], we found that 16 mg/kg of progesterone combined with 1 μg/kg of VDH had optimal efficacy in sparing spatial learning and memory in adult rats after TBI. We therefore used the same dosing in the present study. A recent dose-response study reported that 1 ug/kg VDH combined with 16 mg/kg progesterone led to greater improvement of behavior outcomes than high-dose VDH (5 μg/kg), which failed to improve spatial learning performance after TBI [37].
The preparation and administration of progesterone and VDH are described in our previous papers [37, 38]. In brief, progesterone was administered in 2-hydropropyl-β-cyclodextrin (HBC; 22.5% w/v solution with 2% ethanol in dH2O) as the solvent. VDH (1,25-dihydroxyvitamin D(3)) was administered in 22.5% HBC with 2% ethanol. All sham and vehicle groups received a volume of 22.5% HBC with 2% ethanol vehicle equal to the volume of the progesterone + VDH dose. Progesterone and VDH separately or combined were injected intraperitoneally 1 h post-injury and a second dose was given at 6 h post-injury.
Tissue preparation
Twenty-four hours after surgery, rats were anesthetised with 5% isoflurane for 5 min. Five animals from each group were perfused with ice-cold 0.05 M phosphate-buffered saline (PBS; 200 ml) via the ascending aorta. The cortex around the contusion was harvested for western blots (Figure 1, label 1) and ELISA (Figure 1, label 2). The other 3 animals from each group were perfused with 0.05 M PBS and fixed with 10% formalin buffer (pH = 7.4, 250 ml). Brains were then extracted from the skull, post-fixed for 24 h at 4°C in the same fixative, and placed in increasing amounts of 0.1 M phosphate-buffered sucrose (10%, 20%, 30%) each day for 3 days. Finally, brains were covered with cryoprotectant, frozen using 2-methyl-butane chilled on dry ice, placed in a cryostat, and cut into 20-μm coronal sections. Sections were stored in cryoprotectant at −80°C [38].
Western blotting
Protein was extracted from the 5 samples from each group, electrophoresed with 4-20% sodium dodecyl sulfate (SDS) -polyacrylamide gradient gel, and transferred onto polyvinylidene fluoride transfer membranes (Immobilon-P, 0.45 μm, Millipore, Billerica, MA, USA). The membranes were incubated with primary antibodies: rabbit anti-COX2 (1:200, ab15191, Abcam, Cambridge, MA, USA), mouse anti-GFAP (1:1000, MAB360, Millipore), rabbit anti-iNOS (1:200, ab15353, Abcam), mouse anti-MAP-2 (1:1000, MAB3418, Millipore), rabbit anti-NF-κB p65(1:1000, #8242, Cell Signaling, Danvers, MA, USA), mouse anti-Phospho-NF-κB p65 (1:1000, #3036, Cell Signaling), mouse anti-IκB (1:1000, #9242, Cell Signaling), mouse anti-Phospho-IκB (1:1000, #9246, Cell Signaling), and rabbit anti-TLR-4 (1:200, sc16240, Santa Cruz Biotechnology, Santa Cruz, CA, USA), followed by incubation with peroxidase-conjugated secondary antibodies. The signals were detected by an enhanced chemiluminescence system (E2400, Denville Scientific, South Plainfield, NJ, USA). The same membranes were incubated with antibody for β-actin as a control after washing with stripping buffer (21059, Thermo Scientific, Waltham, MA, USA). The blotting signals were quantified using ImageJ software (NIH) and presented as adjusted density normalised to density of actin in the same gel.
ELISA
ELISA kits for TNF-α and IL-1β (eBioscience Inc., San Diego, CA, USA) were used to examine these two factors' concentration in brain tissue according to the manufacturer's instructions. The absorbance signals were read on a Synergy HT multi-mode microplate reader (BioTek, Winooski, VT, USA) with a wavelength of 450 nm and a reference of 570. The concentrations of TNF-α and IL-1β were calculated according to the standard curve and presented as pg/mg tissue.
IHC and IF
Three slides from each group at 3 mm to 2 mm anterior to bregma were labeled for TLR-4 using immunohistochemistry (IHC) and the slides from the TBI group were double-labeled for TLR4 with GFAP, CD31, CD68 or MAP-2 using immunofluorescence (IF). For IF double staining, the slides were air-dried for 2 h, washed 3 times in PBS, incubated with blocking buffer (1% BSA in PBS) at RT for 30 min, and then co-incubated with rabbit anti-TLR-4 antibody (1:200, sc16240, Santa Cruz) and mouse anti-rat GFAP primary antibody (1:200, MAB360, Millipore), mouse anti-rat MAP-2 primary antibody (SC32791, Santa Cruz), mouse anti-CD31 primary antibody (1:200, 550300, BD Pharmingen), or mouse anti-CD68 primary antibody (1:200, ab31630, Abcam) for 1h at RT. Slides were washed 3 times in PBS and incubated with goat anti-rabbit IgG (H + L) labeled with Alexa Fluor 594 F(ab′) fragment (A-11071, Molecular Probes, Carlsbad, CA, USA) and goat anti-mouse IgG (H + L) labeled with Alexa Fluor 488 F(ab′) fragment (A-11001, Molecular Probes) for 1 h at RT, then rinsed with PBS for 3 × 5 min and covered with the mounting medium DAPI. For TLR-4 IHC, the slides were air-dried for 2 h, washed 3 times in PBS, incubated with 0.3% H2O2 for 15 min followed by washing 3 times in PBS. The slides were then blocked by protein block (X0909 Dako, Carpinteria, CA, USA) for 1 h and incubated in diluted primary antibodies at 4° C overnight. A peroxidase-based visualisation kit (Universal LSABTM+Kit/HRP, K0690, Dako) and DAB was used to detect the positive signal. The tissue was examined and photographed with a Zeiss Axioskop microscope with an Axiocam camera (Zeiss, Jena, Germany).
Statistical analysis
The results are presented as mean ± SE. One-way ANOVA with Tukey post-hoc comparisons were used to analyse all the data. Sigmaplot 11.0 was used for data analyses and statistical significance was set at p < 0.05.
Results
The effect of combination therapy on the expression of TLR4 after TBI
To detect the activation of TLR4 activity, its expression was evaluated by western blot and immunostaining. Immunostaining for TLR4 in the cytoplasm showed that the sham-operated animals had less positive TLR4 staining than animals in the injury groups (Figure 2C).
Figure 2.
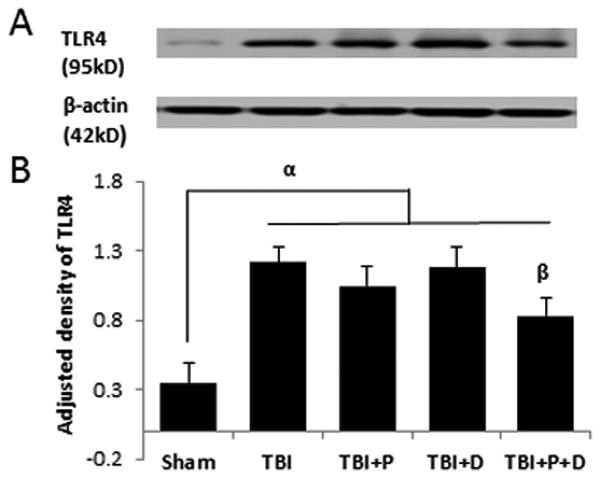
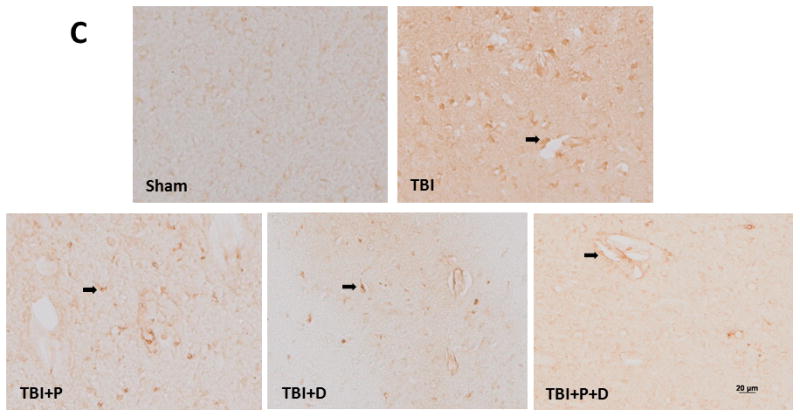
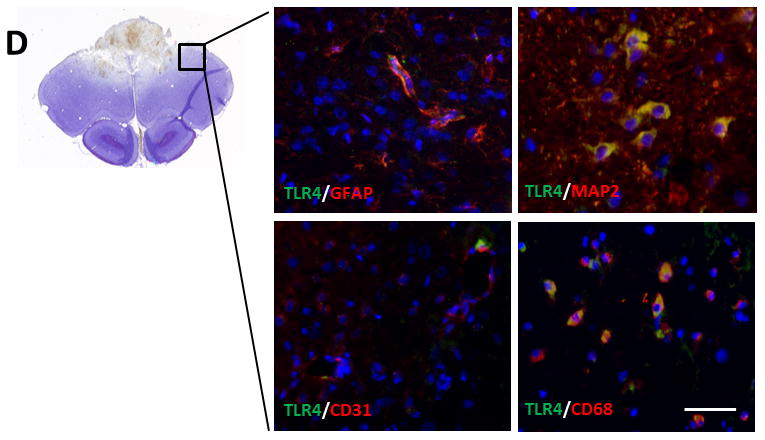
TLR4 expression at 24 h after traumatic brain injury (TBI) and the effect of combination of progesterone and vitamin D (VDH) on TLR4 expression. A. Protein expression of TLR4 assessed by western blot. B. Adjusted density of TLR4. n=5, α: p < 0.05 vs. Sham; β: p < 0.05 vs. TBI. C. Immunostaining of TLR4 (indicated by arrows). D. Double-labeling of TLR4/GFAP, TLR4/MAP2, TLR4/CD31, and TLR4/CD68. Scale bar is 20 μm. Sham: sham with vehicle treatment; TBI: TBI with vehicle treatment; TBI+P: TBI with progesterone treatment; TBI+D: TBI with vitamin D treatment; TBI+P+D: TBI with progesterone and VDH combination treatment, n=5.
Densitometric analysis of western blots revealed a significant difference in the expression of TLR4 among groups (F (4, 20) = 6.419, p<0.01). TBI significantly increased the expression of TLR4 compared to sham-operated animals p<0.01, Figure 2A and B). Neither progesterone nor VDH alone affected TLR4 expression significantly (Figure 2A and B, p>0.05) but combination therapy significantly reduced TLR4 expression compared with vehicle and VDH as detected by western blot (Figure 2A and B, p<0.05).
As revealed by IF double staining, TLR4 was expressed in GFAP-, MAP-2-, and CD68-positive cells proximal to the lesion site (Figure 2D).
The modulation of nuclear transcription factor activation by combination therapy
Western blotting showed that neither NF-κB nor IκBα total protein differed significantly between any treatment groups (Figure 3A, B, D, and E), but the expression of pNF-κB and pIκBα was significantly different among the groups (F (4, 20)=5.629, p<0.05; F (4, 20) =5.701, p<0.05, respectively). TBI induced significantly higher expression of pNF-κB compared with Sham treatment (Figure 3A and C, p < 0.01). Neither progesterone nor VDH alone affected the expression of pNF-κB compared with vehicle treatment. However, both progesterone and VDH reduced pIκBα expression significantly compared to vehicle (p < 0.05). The combination therapy inhibited the phosphorylation of both NF-κB and IκBα significantly compared to vehicle, progesterone alone, or VDH treatment alone (p < 0.05) (Figure 3F).
Figure 3.
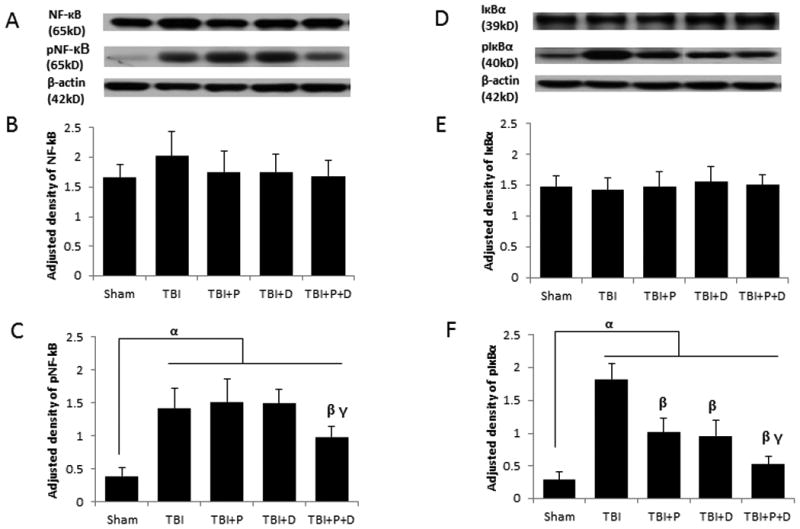
The effect of treatments on NF-κB and IκBα activation at 24 h after TBI. A. Representative bands of western blots of NF-κB and pNF-κB. B. Adjusted density of NF-κB. C. Adjusted density of pNF-κB. D. Representative bands of western blots of IκBα and pIκBα. E. Adjusted density of IκB. F. Adjusted density of pIκBα. n=5, α: p < 0.05 vs. Sham; β: p < 0.05 vs. TBI; γ: p < 0.05 vs.compared with TBI+P and TBI+D. Sham: sham with vehicle treatment group; TBI: TBI with vehicle treatment group; TBI+P: TBI with progesterone treatment group; TBI+D: TBI with vitamin D treatment group; TBI+P+D: TBI with progesterone and vitamin D combination treatment group, n=5.
The effect of combination therapy on the expression of inflammatory cytokines
After NF-κB phosphorylation and translocation to the cell nucleus, the expression of inflammatory cytokines is stimulated to initiate the inflammatory cascade [12, 16]. As shown in Figure 4, there were significant differences in TNF-α and IL-1β expression among the groups revealed by ELISA (F (4, 20) =3.313, p<0.05; F (4, 20) =4.60, p<0.05, respectively). We observed that pro-inflammatory factors TNF-α and IL-1β were significantly increased after TBI (Figure 4A and D, p < 0.01). The combination therapy, but neither progesterone nor VDH alone, reduced the expression of TNF-α compared with vehicle (p < 0.05). Progesterone, VDH, and combination therapy all down-regulated the expression of IL-1β significantly compared with vehicle alone (p < 0.05). There were no significant differences among the progesterone, VDH, and combination groups in the expression of IL-1 β.
Figure 4.
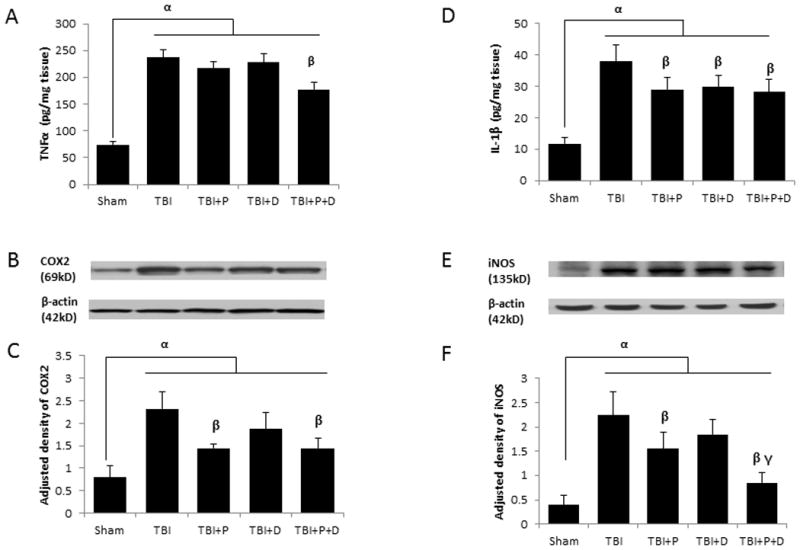
The effect of treatments on the expression of inflammatory factors at 24 h after TBI. A. TNFα expression shown by ELISA. B. Representative bands of COX2 shown by western blot. C. Adjusted density of COX2. D. IL-1β expression assessed by ELISA. E. Representative bands of iNOS shown by western blot. F. Adjusted density of iNOS. n=5, α: p < 0.05 vs. Sham; β: p < 0.05 vs. TBI; γ: p < 0.05 vs. TBI+P and TBI+D. Sham: sham with vehicle treatment group; TBI: TBI with vehicle treatment group; TBI+P: TBI with progesterone treatment group; TBI+D: TBI with vitamin D treatment group; TBI+P+D: TBI with progesterone and vitamin D combination treatment group, n=5.
Activation of NF-κB also induced the expression of COX-2 and iNOS. The expression of COX-2 and iNOS were significantly different among the treatment groups as assessed by ELISA (F (4, 20) =9.339, p<0.05; F (4, 20) =2.776, p<0.05, respectively) and western blotting (F (4, 20)= 6.575, p<0.05; F (4, 20)= 5.867, p<0.05, respectively). Western blotting revealed a significant upregulation of COX-2 and iNOS after TBI-plus-vehicle treatment compared to Shams (Figure 4B, C, E, and F, p < 0.01). Both progesterone alone and the combination therapy led to significant inhibition in the expression of both enzymes compared with TBI-plus-vehicle treatment (p < 0.05), whereas VDH showed no significant effect. The combination therapy also showed a significantly stronger inhibition of iNOS compared to progesterone alone (p < 0.05).
The effect of combination therapy on neuron loss and astrocyte activity
Western blot analysis showed significant differences in MAP-2 and GFAP expression among all treatment groups (F (4, 20) =9.346, p<0.05; F (4, 20) =6.738, p<0.05, respectively). TBI induced significant neuronal loss and astrocyte activation compared with sham treatment (Figure 5A-D, p < 0.01). Only the combination treatment reduced the expression of GFAP compared with TBI, indicating a lower activation of astrocytes (p < 0.05). Progesterone, VDH, and the combination treatment all alleviated the loss of MAP-2 significantly compared with TBI (TBI + P vs. TBI, TBI + D vs. TBI, p < 0.05, and TBI + P + D vs. TBI, p < 0.01). The combination therapy also showed a stronger effect on the sparing of MAP-2 expression compared with either progesterone or VDH treatment alone (p < 0.01).
Figure 5.
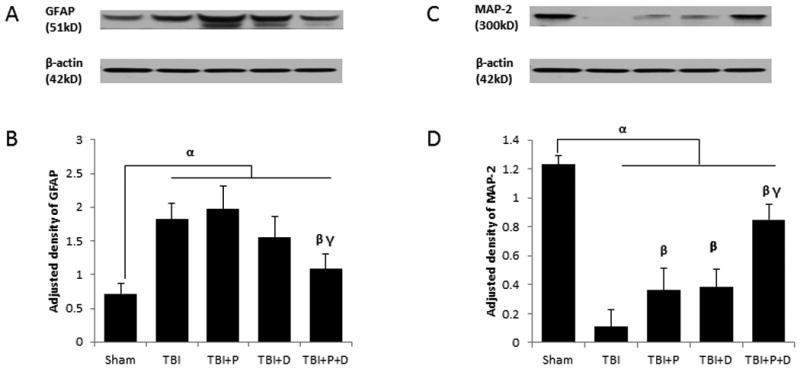
The effect of a combination of progesterone and VDH on the expression of GFAP and MAP-2 at 24 h after TBI. A. Representative bands of GFAP shown by western blot. B. Adjusted density of GFAP. C. Representative bands of MAP-2 shown by western blot. D. Adjusted density of MAP-2. n=5, α: p < 0.05 vs.Sham; β: p < 0.05 vs. with TBI; γ: p < 0.05 vs. with TBI+P and TBI+D. Sham: sham with vehicle treatment group; TBI: TBI with vehicle treatment group; TBI+P: TBI with progesterone treatment group; TBI+D: TBI with vitamin D treatment group; TBI+P+D: TBI with progesterone and vitamin D combination treatment group, n=5.
Discussion
Combination therapy and acute neuroinflammation after TBI
In this report we investigated the activity of the TLR4/NF-κB-mediated pathways at 24 h after TBI. We found that at this acute stage of the injury, progesterone alone did not significantly reduce TLR4 or pNF-κB expression compared with vehicle treatment. This finding is consistent with an earlier study from our lab showing that progesterone does not down-regulate increased TLR mRNA expression or TLR adaptor proteins in traumatically injured brain at 24 h in mice [6]. In contrast, Chen et al. [40] have demonstrated that progesterone reduces TLR2/4 mRNA expression as well as NF-κB activation at 5 days after TBI in rats—an effect different from that observed in the acute stage of the injury.
Interestingly, at 24 h post-TBI, we found that the combination of progesterone and VDH treatment reduced the expression of TLR4 compared with vehicle. The combination therapy more effectively inhibited phosphorylation of NF-κB by reducing phosphorylation and degradation of IκB. Here we propose that in the context of TBI, progesterone alone can exert effects on the TLR4/NF-κB pathway at later time points [40], and when given together with VDH, its effect may be strengthened or advanced to an earlier time point where it is most critical to stem the inflammatory cascade.
TLR4 was expressed in GFAP-positive astrocytes, CD31-positive endothelial cells, CD68-positive microglia and MAP2-positive neurons adjacent to the injury site, and increased significantly together with the phosphorylation and activation of NF-κB and the up-regulation of TNF-α, IL-1β, COX2 and iNOS. All these factors play a critical role in initiating and sustaining the inflammatory cascade that frequently follows significant brain injury. Here we demonstrate that a cocktail of agents like progesterone and vitamin D can reduce this evolving cytotoxicity and that this combination is necessary to prevent injured neurons and their support cells from dying off over time. In a clinical setting, these neuroprotective effects should result in better prognoses and functional recovery.
Earlier studies have reported that progesterone inhibits the neuroinflammatory response after TBI by regulating NF-κB signaling and reducing IL-1β, IL-6 and TNFα expression [41, 42], all of which are implicated in the modulation of TLR4-mediated immune and inflammatory responses [25, 40]. However, given alone, progesterone was only partially successful in attenuating the inflammatory cascade after TBI [40].
In contrast, Chen et al. demonstrated that progesterone reduces TLR2/4 mRNA expression as well as NF-κB activation at 5 days after TBI in rats. Si et al. [43] found that progesterone treatment significantly reduced post-injury inflammatory response, brain edema and Evans blue dye extravasation, and improved neurological scores compared with TBI at 24 h following injury. However, Gilmer et al. [44] found that, at 7 days post-injury, progesterone alone failed to alter cortical edema or tissue sparing at either 8 or 16 mg/kg. These inconsistencies perhaps further strengthen the argument for developing combination therapies with synergistic neuroprotective actions for treatment of CNS injuries.
We take our current data to support our working hypothesis that in the context of TBI, progesterone alone can exert effects on the TLR4/NF-κB pathway at later time points [40], and when given together with VDH, its effect may be strengthened or advanced to an earlier time point where it is most critical to stem the inflammatory cascade. Like progesterone, VDH can also exert some of its anti-inflammatory effects by promoting negative feedback regulation of TLR signaling in macrophages [34] and human myometrial cells [35].
It is generally agreed that TNFα and IL-1β are important inflammatory cytokines modulated by NF-κB and both unleash inflammatory responses to TBI and aggravate tissue damage and neurological deficits [17, 45]. At 24 h post-TBI, the combination therapy reduced the expression of both TNFα and IL-1β significantly compared to vehicle. Although both progesterone and VDH can significantly inhibit the elevation of IL-1β, when given individually in the doses used here, they failed to suppress TNFα compared to vehicle at 24 h.
In the present study, both progesterone and the combination of progesterone and VDH reduced the expression of both COX2 and iNOS compared with vehicle-alone treatment. Furthermore, the combination showed a more robust inhibition of iNOS compared to progesterone alone, indicating that combination with VDH can enhance progesterone's effect. Why is this finding important? COX2 plays an important role in inflammatory responses to TBI by converting arachidonic acid from injured membranes into prostaglandins and releasing ROS [46]. The products of COX2 following TBI may result in damage to the blood-brain barrier and neural membranes [46], triggering more extensive injury. In rat and mouse TBI models, iNOS mRNA can increase as early as several hours after injury and lead to production of NO, which then reinforces the injury cascade and leads to further cell loss and functional disruption [47, 48]. Several studies report that application of iNOS antisense oligonucleotides prior to TBI reduced the release of NO and lesion volume [47, 49].
Combination therapy, neuron loss and glial activation in the acute phase of TBI
Another important component of injury repair is to stabilise the metabolic machinery of the cell. MAP-2 is required for microtubule stability, dendritic elongation, and the interaction of microtubules with other organelles because it facilitates the polymerisation of pure tubulin into microtubules [50]. Therefore, injury-induced MAP2 degradation indicates neuron loss [51, 52]. We observed a rapid degradation of MAP2 proximal to injured cortex at 24 h post-TBI. . Both progesterone and VDH alleviated the loss of MAP2 compared to vehicle, and this protective effect was enhanced with the combined treatment.
At the anatomical level, following TBI, neuroinflammation contributes to ongoing neurodegeneration and neurological impairments by activation of glial cells, leukocyte recruitment and the upregulation of inflammatory mediators (Morganti-Kossmann et al., 2007). Reactive astrocytes migrate to the lesion site, where they can create a dense barrier that inhibits axonal regeneration and prevents restoration of functional neuronal connections required for axonal growth and repair [53]. The over-activation of astrocytes after TBI increases neuronal cell death and can worsen functional outcome after TBI [54]. In our hands, neither progesterone nor VDH alone inhibited the increase in GFAP, a measure of reactive astrocyte activity, compared with vehicle, but combination therapy reduced GFAP expression compared to both vehicle and individual drug treatment.
This better neuroprotection afforded by the effects of combination therapy on astrocyte activity and neuronal loss could be the result of the robust inhibition of TLR4/NF-κB-mediated neuroinflammatory pathways which, if left untended, over time would contribute to further neurodegeneration and impaired recovery instigated by the initial injury.
In conclusion, our current research is but one step in identifying the possible mechanisms of progesterone combined with VDH on acute inflammatory response after TBI. It is becoming evident that progesterone and VDH share some pathways in common but they also mediate different genes in both normal and pathological processes. The combination therapy is likely to modulate other important factors and signaling pathways in the inflammatory cascade after TBI that we have yet to study. The interaction between progesterone and VDH in the context of brain injury is a promising direction for future experiments and for finding a safe and effective therapy for the multiple brain and systemic effects caused by CNS trauma or stroke. Overall, it is becoming increasingly clear that to treat the highly complex and systemic nature of brain injuries, monotherapies or “magic bullet” approaches may not be the answer to the development of a safe and effective treatment for TBI and its related disorders.
Acknowledgments
The authors would like to thank Leslie McCann for the invaluable editorial assistance and Jason S. Lee for his assistance with immunostaining for this project.
Grant support: The research reported in the paper was supported by NIH 5 R01 HD61971 and by gifts from BHR Pharma, The Marcus Foundation, and Allen and Company.
Footnotes
Declaration of Interest: The authors report no conflict of interest.
Roles of co-authors: HT and FH, experimental design, surgery and western blots; JW, surgery assistant and immunology experiment; SY and FA, sample collection and ELISA; IS and DGS, experimental design; HT, IS and DGS, analysis and manuscript preparation.
References
- 1.Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T. Inflammatory response in acute traumatic brain injury: A double-edged sword. Curr Opin Crit Care. 2002;8:101–105. doi: 10.1097/00075198-200204000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Loane DJ, Byrnes KR. Role of microglia in neurotrauma. Neurotherapeutics. 2010;7:366–377. doi: 10.1016/j.nurt.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amor S, Peferoen LA, Vogel DY, Breur M, van der Valk P, Baker D, van Noort JM. Inflammation in neurodegenerative diseases - an update. Immunology. 2013 doi: 10.1111/imm.12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: Translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci. 2010;31:596–604. doi: 10.1016/j.tips.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McIntosh TK. Neuropathological sequelae of traumatic brain injury: Relationship to neurochemical and biomechanical mechanisms. Lab Invest. 1996;74:315–342. [PubMed] [Google Scholar]
- 6.Hua F, Wang J, Ishrat T, Wei W, Atif F, Sayeed I, Stein DG. Genomic profile of toll-like receptor pathways in traumatically brain-injured mice: Effect of exogenous progesterone. J Neuroinflammation. 2011;8:42. doi: 10.1186/1742-2094-8-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hua F, Ma J, Ha T, Xia Y, Kelley J, Williams DL, Kao RL, Browder IW, Schweitzer JB, Kalbfleisch JH, et al. Activation of toll-like receptor 4 signaling contributes to hippocampal neuronal death following global cerebral ischemia/reperfusion. J Neuroimmunol. 2007;190:101–111. doi: 10.1016/j.jneuroim.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Michaud JP, Halle M, Lampron A, Theriault P, Prefontaine P, Filali M, Tribout-Jover P, Lanteigne AM, Jodoin R, Cluff C, et al. Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid a improves alzheimer's disease-related pathology. Proc Natl Acad Sci U S A. 2013;110:1941–1946. doi: 10.1073/pnas.1215165110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanafy KA. The role of microglia and the tlr4 pathway in neuronal apoptosis and vasospasm after subarachnoid hemorrhage. J Neuroinflammation. 2013;10:83. doi: 10.1186/1742-2094-10-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kenny EF, O'Neill LA. Signalling adaptors used by toll-like receptors: An update. Cytokine. 2008;43:342–349. doi: 10.1016/j.cyto.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto Y, Gaynor RB. Ikappab kinases: Key regulators of the nf-kappab pathway. Trends Biochem Sci. 2004;29:72–79. doi: 10.1016/j.tibs.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 12.Karin M, Yamamoto Y, Wang QM. The ikk nf-kappa b system: A treasure trove for drug development. Nat Rev Drug Discov. 2004;3:17–26. doi: 10.1038/nrd1279. [DOI] [PubMed] [Google Scholar]
- 13.Cederberg D, Siesjo P. What has inflammation to do with traumatic brain injury? Childs Nerv Syst. 2010;26:221–226. doi: 10.1007/s00381-009-1029-x. [DOI] [PubMed] [Google Scholar]
- 14.Surh YJ, Chun KS, Cha HH, Han SS, Keum YS, Park KK, Lee SS. Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: Down-regulation of cox-2 and inos through suppression of nf-kappa b activation. Mutat Res. 2001:480–481. 243–268. doi: 10.1016/s0027-5107(01)00183-x. [DOI] [PubMed] [Google Scholar]
- 15.Strauss KI, Barbe MF, Marshall RM, Raghupathi R, Mehta S, Narayan RK. Prolonged cyclooxygenase-2 induction in neurons and glia following traumatic brain injury in the rat. J Neurotrauma. 2000;17:695–711. doi: 10.1089/089771500415436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pahl HL. Activators and target genes of rel/nf-kappab transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 17.Harting MT, Jimenez F, Adams SD, Mercer DW, Cox CS., Jr Acute, regional inflammatory response after traumatic brain injury: Implications for cellular therapy. Surgery. 2008;144:803–813. doi: 10.1016/j.surg.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kong Y, Le Y. Toll-like receptors in inflammation of the central nervous system. Int Immunopharmacol. 2011;11:1407–1414. doi: 10.1016/j.intimp.2011.04.025. [DOI] [PubMed] [Google Scholar]
- 19.Hyakkoku K, Hamanaka J, Tsuruma K, Shimazawa M, Tanaka H, Uematsu S, Akira S, Inagaki N, Nagai H, Hara H. Toll-like receptor 4 (tlr4), but not tlr3 or tlr9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience. 2010;171:258–267. doi: 10.1016/j.neuroscience.2010.08.054. [DOI] [PubMed] [Google Scholar]
- 20.Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115:1599–1608. doi: 10.1161/CIRCULATIONAHA.106.603431. [DOI] [PubMed] [Google Scholar]
- 21.Shakeri M, Boustani MR, Pak N, Panahi F, Salehpour F, Lotfinia I, Meshkini A, Daghighi S, vahedi P, Khani M, et al. Effect of progesterone administration on prognosis of patients with diffuse axonal injury due to severe head trauma. Clin Neurol Neurosurg. 2013;115:2019–2022. doi: 10.1016/j.clineuro.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 22.Shahrokhi N, Haddad MK, Joukar S, Shabani M, Keshavarzi Z, Shahozehi B. Neuroprotective antioxidant effect of sex steroid hormones in traumatic brain injury. Pak J Pharm Sci. 2012;25:219–225. [PubMed] [Google Scholar]
- 23.Barha CK, Ishrat T, Epp JR, Galea LA, Stein DG. Progesterone treatment normalizes the levels of cell proliferation and cell death in the dentate gyrus of the hippocampus after traumatic brain injury. Exp Neurol. 2011;231:72–81. doi: 10.1016/j.expneurol.2011.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Nicola AF, Coronel F, Garay L, Gargiulo-Monachelli G, Y DE, Gonzalez SL, Labombarda F, Meyer M, Guennoun R, Schumacher M. Therapeutic effects of progesterone in animal models of neurological disorders. CNS Neurol Disord Drug Targets. 2013;12:1205–1218. [PubMed] [Google Scholar]
- 25.Su L, Sun Y, Ma F, Lu P, Huang H, Zhou J. Progesterone inhibits toll-like receptor 4-mediated innate immune response in macrophages by suppressing nf-kappab activation and enhancing socs1 expression. Immunol Lett. 2009;125:151–155. doi: 10.1016/j.imlet.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 26.Wang Z, Zuo G, Shi XY, Zhang J, Fang Q, Chen G. Progesterone administration modulates cortical tlr4/nf-kappab signaling pathway after subarachnoid hemorrhage in male rats. Mediators Inflamm. 2011;2011:848309. doi: 10.1155/2011/848309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ishrat T, Sayeed I, Atif F, Hua F, Stein DG. Progesterone is neuroprotective against ischemic brain injury through its effects on the phosphoinositide 3-kinase/protein kinase b signaling pathway. Neuroscience. 2012;210:442–450. doi: 10.1016/j.neuroscience.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Espinoza TR, Wright DW. The role of progesterone in traumatic brain injury. J Head Trauma Rehabil. 2011;26:497–499. doi: 10.1097/HTR.0b013e31823088fa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong R, Renton C, Gibson CL, Murphy SJ, Kendall DA, Bath PM. Progesterone Pre-Clinical Stroke Pooling Project C. Progesterone treatment for experimental stroke: An individual animal meta-analysis. J Cereb Blood Flow Metab. 2013;33:1362–1372. doi: 10.1038/jcbfm.2013.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wright DW, Yeatts SD, Silbergleit R, Palesch YY, Hertzberg VS, Frankel M, Goldstein FC, Caveney AF, Howlett-Smith H, Bengelink EM, et al. Very early administration of progesterone for acute traumatic brain injury. N Engl J Med. 2014;371:2457–2466. doi: 10.1056/NEJMoa1404304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skolnick BE, Maas AI, Narayan RK, van der Hoop RG, MacAllister T, Ward JD, Nelson NR, Stocchetti N the STI. A clinical trial of progesterone for severe traumatic brain injury. N Engl J Med. 2014 doi: 10.1056/NEJMoa1411090. [DOI] [PubMed] [Google Scholar]
- 32.Margulies S, Hicks R. Combination therapies for traumatic brain injury: Prospective considerations. J Neurotrauma. 2009;26:925–939. doi: 10.1089/neu.2008.0794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hua F, Tang H, Wang J, Prunty MC, Hua X, Sayeed I, Stein DG. Tak-242, an antagonist for toll-like receptor 4, protects against acute cerebral ischemia/reperfusion injury in mice. J Cereb Blood Flow Metab. 2015 doi: 10.1038/jcbfm.2014.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Y, Liu W, Sun T, Huang Y, Wang Y, Deb DK, Yoon D, Kong J, Thadhani R, Li YC. 1,25-dihydroxyvitamin d promotes negative feedback regulation of tlr signaling via targeting microrna-155-socs1 in macrophages. J Immunol. 2013;190:3687–3695. doi: 10.4049/jimmunol.1203273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thota C, Farmer T, Garfield RE, Menon R, Al-Hendy A. Vitamin d elicits anti-inflammatory response, inhibits contractile-associated proteins, and modulates toll-like receptors in human myometrial cells. Reprod Sci. 2013;20:463–475. doi: 10.1177/1933719112459225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Atif F, Yousuf S, Sayeed I, Ishrat T, Hua F, Stein DG. Combination treatment with progesterone and vitamin d hormone is more effective than monotherapy in ischemic stroke: The role of bdnf/trkb/erk1/2 signaling in neuroprotection. Neuropharmacology. 2013;67:78–87. doi: 10.1016/j.neuropharm.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hua F, Reiss JI, Tang H, Wang J, Fowler X, Sayeed I, Stein DG. Progesterone and low-dose vitamin d hormone treatment enhances sparing of memory following traumatic brain injury. Horm Behav. 2012;61:642–651. doi: 10.1016/j.yhbeh.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang H, Hua F, Wang J, Sayeed I, Wang X, Chen Z, Yousuf S, Atif F, Stein DG. Progesterone and vitamin d: Improvement after traumatic brain injury in middle-aged rats. Horm Behav. 2013;64:527–538. doi: 10.1016/j.yhbeh.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cutler SM, Cekic M, Miller DM, Wali B, VanLandingham JW, Stein DG. Progesterone improves acute recovery after traumatic brain injury in the aged rat. J Neurotrauma. 2007;24:1475–1486. doi: 10.1089/neu.2007.0294. [DOI] [PubMed] [Google Scholar]
- 40.Chen G, Shi J, Jin W, Wang L, Xie W, Sun J, Hang C. Progesterone administration modulates tlrs/nf-kappab signaling pathway in rat brain after cortical contusion. Ann Clin Lab Sci. 2008;38:65–74. [PubMed] [Google Scholar]
- 41.Grossman KJ, Goss CW, Stein DG. Effects of progesterone on the inflammatory response to brain injury in the rat. Brain Res. 2004;1008:29–39. doi: 10.1016/j.brainres.2004.02.022. [DOI] [PubMed] [Google Scholar]
- 42.Pettus EH, Wright DW, Stein DG, Hoffman SW. Progesterone treatment inhibits the inflammatory agents that accompany traumatic brain injury. Brain Res. 2005;1049:112–119. doi: 10.1016/j.brainres.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 43.Si D, Li J, Liu J, Wang X, Wei Z, Tian Q, Wang H, Liu G. Progesterone protects blood-brain barrier function and improves neurological outcome following traumatic brain injury in rats. Exp Ther Med. 2014;8:1010–1014. doi: 10.3892/etm.2014.1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gilmer LK, Roberts KN, Scheff SW. Efficacy of progesterone following a moderate unilateral cortical contusion injury. J Neurotrauma. 2008;25:593–602. doi: 10.1089/neu.2007.0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takeda K, Akira S. Tlr signaling pathways. Semin Immunol. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 46.Ellis EF, Police RJ, Rice LY, Grabeel M, Holt S. Increased plasma pge2, 6-keto-pgf1 alpha, and 12-hete levels following experimental concussive brain injury. J Neurotrauma. 1989;6:31–37. doi: 10.1089/neu.1989.6.31. [DOI] [PubMed] [Google Scholar]
- 47.Foley LM, Hitchens TK, Melick JA, Bayir H, Ho C, Kochanek PM. Effect of inducible nitric oxide synthase on cerebral blood flow after experimental traumatic brain injury in mice. J Neurotrauma. 2008;25:299–310. doi: 10.1089/neu.2007.0471. [DOI] [PubMed] [Google Scholar]
- 48.Sinz EH, Kochanek PM, Dixon CE, Clark RS, Carcillo JA, Schiding JK, Chen M, Wisniewski SR, Carlos TM, Williams D, et al. Inducible nitric oxide synthase is an endogenous neuroprotectant after traumatic brain injury in rats and mice. J Clin Invest. 1999;104:647–656. doi: 10.1172/JCI6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huttemann M, Lee I, Kreipke CW, Petrov T. Suppression of the inducible form of nitric oxide synthase prior to traumatic brain injury improves cytochrome c oxidase activity and normalizes cellular energy levels. Neuroscience. 2008;151:148–154. doi: 10.1016/j.neuroscience.2007.09.029. [DOI] [PubMed] [Google Scholar]
- 50.Sharma N, Kress Y, Shafit-Zagardo B. Antisense map-2 oligonucleotides induce changes in microtubule assembly and neuritic elongation in pre-existing neurites of rat cortical neurons. Cell Motil Cytoskeleton. 1994;27:234–247. doi: 10.1002/cm.970270305. [DOI] [PubMed] [Google Scholar]
- 51.Ryu JK, Cho T, Wang YT, McLarnon JG. Neural progenitor cells attenuate inflammatory reactivity and neuronal loss in an animal model of inflamed ad brain. J Neuroinflammation. 2009;6:39. doi: 10.1186/1742-2094-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alfonso-Loeches S, Pascual M, Guerri C. Gender differences in alcohol-induced neurotoxicity and brain damage. Toxicology. 2013;311:27–34. doi: 10.1016/j.tox.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 53.Cafferty WB, Yang SH, Duffy PJ, Li S, Strittmatter SM. Functional axonal regeneration through astrocytic scar genetically modified to digest chondroitin sulfate proteoglycans. J Neurosci. 2007;27:2176–2185. doi: 10.1523/JNEUROSCI.5176-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Myer DJ, Gurkoff GG, Lee SM, Hovda DA, Sofroniew MV. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain. 2006;129:2761–2772. doi: 10.1093/brain/awl165. [DOI] [PubMed] [Google Scholar]