

Abstract
Lymphoblastoid cell line (LCL) is a common tool to study genetic disorders. However, it has not been fully characterized to what degree LCLs preserve the in vivo status of non-genetic biological systems, such as DNA methylation and gene transcription. We previously reported that DNA methylation in LCLs is highly variable in a data set of ~27,000 CpG dinucleotide sites around transcription start site (TSS) and 63 human subjects including healthy controls and probands of genetic disorders. Disease-causing mutations are linked to differential methylation at some CpG sites, but account for a small proportion of the total variance. In this study, we repeated the experiments to ensure that the high variance is not due to technical error and scrutinized the characteristics of DNA methylation and its association with other biological systems. Using sequence information and ChIP-seq data, we conclude that local CpG density and histone modifications not only correlate to baseline methylation level, but also affect the direction of methylation change in LCLs. Integrative analysis of gene transcription and DNA methylation data of the same subjects shows that medium or high methylation around TSS blocks the transcription while low methylation is a necessary, but not sufficient condition of downstream gene transcription. We utilized epigenetic information around TSS to predict active gene transcription via logistic regression models. The multivariate model using DNA methylation, eight histone modifications, and two regulatory protein complexes (CTCF and cohesin) as predictors has better performance (accuracy = 95.1%) than any univariate models of single predictors. Linear regression analysis further shows that the transcriptional levels predicted by epigenetic markers have significant correlation to microarray measurements (p = 2.2e-10). This study provides new insights into the epigenetic systems of LCLs and suggests that more specifically designed experiments are needed to improve our understanding on this topic.
1. Introduction
Status of cytosine methylation at CpG dinucleotide sites is a key component of epigenetic regulation of gene activity. It influences gene transcription by adjusting the accessibility of chromosomal regions, and controls various biological processes such as X inactivation [1], gene imprinting [2], chromatin remodeling [3], and pathogenesis [4]. De novo methylation is an essential element of cell differentiation during development [5, 6] while in somatic cells, methylation status is believed to remain stable throughout cell division, so studies of tissue-specific methylation often use cultured cells as source material [6–8].
The distribution of the ~30 million CpG sites in the human genome is not even. They are generally under-represented and hyper-methylated in intergenic regions while a large number of unmethylated CpG sites, called CpG islands (CGIs), cluster around transcription start sites (TSS) [9, 10]. Perturbed DNA methylation at gene promoters has been linked to a number of human disorders [11–15]. Technology for quantitative measurement of methylation at all CpG sites is available but costly. A recent study that investigated the dynamics of DNA methylation during cell differentiation used massively parallel sequencing technology to obtain 542 million sequencing reads on average from three samples [6]. Although the reads averagely cover the whole human genome by nine folds, more than 20% of the CpG sites are mapped by less than three reads, making them unsuitable for quantitative comparison between samples. Limiting measurement to regions of higher biological relevance, such as promoters and 5′-UTRs, would lower the experimental cost and increase the practical number of samples of individual studies. Weber et al. compared the methylation at >12,000 CpG islands between normal fibroblasts and SW48 cancer cells via microarray technology and identified over 200 hypermethylated loci in cancer cells [16]. Ehrich et al. used mass spectrometry technology to quantify methylation patterns of >400 cancer-related genes in 59 cancer cell lines and discovered that a large portion of the tested genes have altered methylation in cancer cells [7]. Koga et al. utilized tiling microarrays to measure methylation at promoters of all RefSeq genes in normal melanocytes and eight melanoma cell strains and revealed the diagnostic value of DNA methylation information [17]. These studies demonstrate the usefulness and feasibility of identifying chromosomal regions that exhibit differential methylation under varying biological conditions.
Lymphoblastoid cell lines (LCLs) are established by transforming lymphoblasts with Epstein-Barr virus (EBV) [18]. It is a renewable source of genetic information and a common tool for studying human disorders [19–22]. GM12878, an LCL generated from a female donor, is a model cell line used by the International HapMap [23] and ENCODE [24] Projects. ChIP-seq data of histone modifications in GM12878 are publically available [25], along with DNA methylation data generated by both microarray and Methyl-seq technologies [8].
LCLs usually have normal diploid karyotypes and stable DNA sequences. Its estimated mutation rate is 2 – 30 × 10−7 mutations per cell division [26]. However, the viral transformation and continuous cell culturing and storage may lead to more substantial alterations in the epigenetic, transcriptional, and translational systems. Altered and destabilized DNA methylation was recently reported in LCLs [27–29], which suggests that the in vivo status of epigenetic systems is not fully preserved. Nevertheless, LCLs are a valuable resource for investigating mutationally defined genetic disorders. It is easier to isolate and evaluate the consequence of DNA mutations in cultured cells grown under a controlled environment than in fresh cells whose status can be confounded by numerous environmental and clinical factors. Therefore, a comprehensive characterization of non-genetic biological systems and their association with each other in LCLs would provide valuable information for future studies.
We previously used microarray-based technology to quantify DNA methylation at 27,578 CpG sites in LCLs generated from 22 healthy controls, two Roberts syndrome (RBS) probands and 39 Cornelia de Lange syndrome (CdLS) probands [30]. CdLS is a dominant congenital multisystem disorder with craniofacial, cardiac, gastrointestinal, genitourinary, skin and other system involvement as well as delays in growth and intellectual development. Disease-causing mutations of CdLS have been identified in genes NIPBL, SMC1A, and SMC3, all of which have been associated with cohesin complex [31]. Our comparative analysis identified 152 CpG sites whose methylation was different between the control and the CdLS groups with a high degree of confidence (p<0.001). This number is much smaller than the number of genes whose transcription is significantly altered in CdLS as measured by gene expression microarray studies [19]. Therefore, the disease state is unlikely a major factor of between-sample variation in this data set.
In this study, we used the same 63 samples for a generalized characterization of DNA methylation in LCLs. We repeated the microarray experiments to estimate the contribution of measurement errors to the total variance. Instead of comparing the control and CdLS samples, data analysis in this study is focused on between-sample variation independent of disease state, age, gender and other known clinical variables. Integrative analysis of various existing and new data sets identified distinctive patterns of associations between histone modifications, DNA methylation, and gene transcription. The results of this study will lead to a better understanding of the non-genetic biological systems in LCLs.
2. Material and methods
2.1. Sample preparation of methylation assays
Cell culture, DNA isolation, and bisulfite treatment were performed as described previously [30]. In summary, lymphoblastoid cell lines (LCLs) of 63 human subjects were cultured anonymously and processed in random order; DNA was isolated using the DNA purification kit from Gentra Systems; and 500ng purified DNA from each sample was conversed using the EZ DNA methylation kit from Zymo Research. The bisulfate conversion changed the unmethylated C to T, but made no change at the methylated CpG sites. The 63 prepared LCLs as well as 3 universally methylated and 6 universally unmethylated controls were randomly assigned to 6 Infinium HumanMethylation27 BeadChips (Illumina, Inc.). All human subjects were included in this study under an IRB-approved protocol of informed consent at The Children’s Hospital of Philadelphia and the Misakaenosono Mutsumi Developmental, Medical, and Welfare Center and their detailed description is available as GSE18458 series within Gene Expression Omnibus database.
2.2. Processing of methylation data
Each HumanMethylation27 BeadChip carries beads measuring DNA methylation at 27,578 CpG sites located around 14,495 unique Entrez genes. Each site is measured by two types of beads; one measures the methylated (M) allele and the other measures the unmethylated (U) allele. After the prepared DNA samples were hybridized to the beads and fluorescently stained, the BeadChips were scanned by BeadArray Reader (Illumina, Inc.) and the scanned data were processed by BeadStudio Methylation Module (Illumina, Inc.). Background-subtracted signal intensities of both alleles and a detection p value of each CpG site were exported from BeadStudio and imported into R statistical environment (http://www.r-project.org) for further processing and statistical analysis. The intensities of methylated and unmethylated alleles were normalized separately across 63 LCLs using the quantile spline method [32] of affy package in R. Since CpG sites on X and Y chromosomes can have very different methylation between males and females, those sites were normalized separately in male and female groups. The methylation level at each CpG site in each LCL is represented as β = M/(M+U); where M and U are normalized intensities of methylated and unmethylated alleles. Therefore, β value indicates the fraction of methylated alleles in a cell population. We considered β value less than 0.1 or greater than 0.9 as low or high methylation level corresponding, and the β value between 0.1 and 0.9 as medium methylation level. The whole processed data set is a 63 by 27,578 data matrix of β values ranging between 0 and 1.
2.3. Bioinformatics analysis
UCSC Genome Browser tracks were downloaded using the Table Browser tool. The “HAIB Methyl27” track provided the DNA methylation data of two technical replicates of GM12878. We called a CpG site in low quality if its β value was 0 in either replicate or the β value difference between replicates was greater than 0.05. Enrichment of pre-defined gene sets in significant genes was analyzed via the functional annotation tools of DAVID (Database for Annotation, Visualization and Integrated Discovery) [33]. The functions/packages used for statistical analysis are cor/stats for correlation analysis, aov/stats for ANOVA analysis, t.test/stats for Student’s t test, prcomp/stats for principal components analysis, performance/ROCR) for ROC analysis, glm/stats for logistic regression and lm /stats for linear regression analysis. More information about the data analysis is available in Supplemental Methods.
3. Results
3.1. General characterization of DNA methylation around TSS
We used the Infinium HumanMethylation27 microarray platform to measure the methylation levels of 27,587 CpG sites close to the TSS in 63 lymphoblastoid cell lines (LCLs) after treating extracted DNA with bisulfite conversion. To evaluate the technical errors introduced by microarray experiments, all samples were measured twice at the same location (Wistar Institute). The detailed batch comparison is described in Supplemental File 1. In summary, we found that when the measurements have the best detection p value, 1) all samples except one have high correlation between the duplicated measurements (average Pearson’s r = 0.991); 2) outliers are common, but they are mostly consistent between batches; 3) while about one-eighth of the duplicated measurements have β value difference greater than 0.05, the batch difference of a given CpG site is generally consistent across samples; and 4) CpG sites with medium methylation level (0.1 < β < 0.9) are less affected by batch effect than sites with very high or low methylation. We then concluded that measurements with the best detection p value are precise and repeatable. Since the proportion of measurements having the best quality is substantially higher in the second batch than in the first (97.7% vs. 45.9%), we only used the data of the second batch throughout the rest of this study.
Starting from 27,587 CpG sites and 63 samples, we filtered the data by removing eight samples with over 1,000 less-than-the-best quality measurements, and further excluded CpG sites having any less-than-the-best quality measurements or extreme outliers (more than 20 interquartile ranges from the first or third quartile). The filtering substantially reduced the proportion of technical errors. The remaining data includes 24,952 CpG sites and 55 samples of 19 gender and race matched healthy controls, 2 Roberts syndrome probands, and 34 CdLS probands (21 severe and 8 mild cases with NIPBL mutations, 4 mild cases with SMC1A mutations, and 1 mild case with SMC3 mutation [31]).
Since most CpG sites measured by the microarray platform are located in CpG islands (CGIs) around transcription start sites (TSSs), the distribution of their methylation is skewed to the hypomethylation end (Supplemental Figure 1) and the global average of β values, which indicate the fraction of methylated alleles in all cells, is 0.22. The average β values of 59.5% CpG sites are less than 0.1 and only 20.3% sites are hypermethylated (average β > 0.5). We associated each CpG site to its nearest TSS according to the “UCSC Genes” track of UCSC Genome Browser. Approximately 98% of sites are located within the −1.5 to 1.5 kb region of any TSS. Consistent with previous studies (Figure 4A in [6] and Figure 1E in [7]), sites closer to the TSS are generally less methylated, especially those in CGIs (Figure 2A). About 43% of sites are located within CGIs according to the “CpG Islands” track of UCSC Genome Browser. The average β values of CGI and non-CGI sites are 0.077 and 0.334 respectively. CGI sites close to the ends of CGI generally have higher methylation than sites in the middle (Supplemental Figure 2) and sites close to potential transcription factor binding sites (TFBS) have slightly lower methylation than sites in the flanking regions (Supplemental Figure 3). The TFBS information was downloaded from the UCSC “TFBS conserved” track, which includes human-mouse-rat conserved loci matching to the consensus binding motifs of 258 transcription factors. Methylation at TFBSs differs dramatically between those motifs (Supplemental Table 2). For example, the average β values around loci matching to V$HNF1_01 and V$NRF2_01 are 0.340 and 0.052 respectively.
Figure 2.
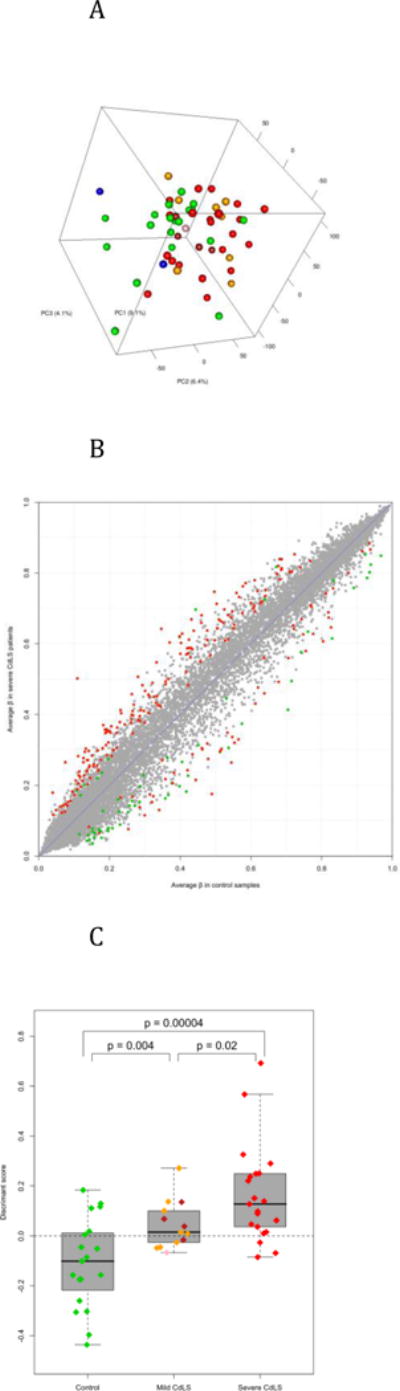
Differential methylation. A) Principal components analysis of LCLs using all measured CpG sites on autosomes. Colors indicate disease status (green = control; red = severe CdLS; orange = mild CdLS with NIPBL mutation; brown = CdLS with SMC1A mutation; pink = CdLS with SMC3 mutation; and blue = Roberts Syndrome). B) Differential methylation between control and severe CdLS samples. Each dot represents one autosomal site. Significant sites (p < 0.01 and | Δβ | > 0.05) were highlighted (green = CGI sites and red = non-CGI sites). C) The control, mild CdLS, and severe CdLS samples could be distinguished according to their methylation pattern. The y-axis indicates the discriminant score that is corresponding to the relative similarity of each sample to the centroids of control and severe CdLS groups. The scores of controls and severe patients were obtained via a leave-one-out procedure, and the scores of mild patients were based on the 283 sites differentially methylated between controls and severe patients (details in Methods). By default, samples with score > 0 would be classified as CdLS. Each diamond represents a sample (colored as in Figure 2A). The p values are the results of Student’s t test.
About two CpG sites were measured around each TSS on average. We queried whether individual sites act independently or adjacent sites are regulated concordantly. The answer to this query will tell us to what degree a single CpG site represents the overall methylation status of surrounding region. We identified 10,878 adjacent pairs of CpG sites located within 1 kb of each other and evaluated the differential methylation of paired sites. The β value difference of adjacent sites is 0.128 on average while the global average of β value differences between any two autosomal sites is 0.288. The average difference is further reduced to 0.041 when sites are within 10 bases of each other (Supplemental Figure 3). We next evaluated the co-regulation of adjacent sites by calculating their correlation across all 55 samples. The average Pearson’s r is only 0.114. Pairs located in the same CGI have slightly higher correlation (average r = 0.124). The correlation between pairs generally increases as their distance becomes shorter (Figure 1B, blue line). Supplemental File 1 shows that the methylation measurements have low sensitivity to subtle change at highly methylated or unmethylated sites. Therefore, when we only use the pairs having medium methylation at both sites, the average r increases to 0.219. Furthermore, sites within 10 bases of each other have a much higher average r of 0.809. We compared the sequence features, such as GC content and TFBS frequency, around correlated and uncorrelated adjacent sites, but were unable to recognize notable differences between these two types of pairs. These results suggest that the methylation of most adjacent CpG sites is not closely co-regulated.
Figure 1.
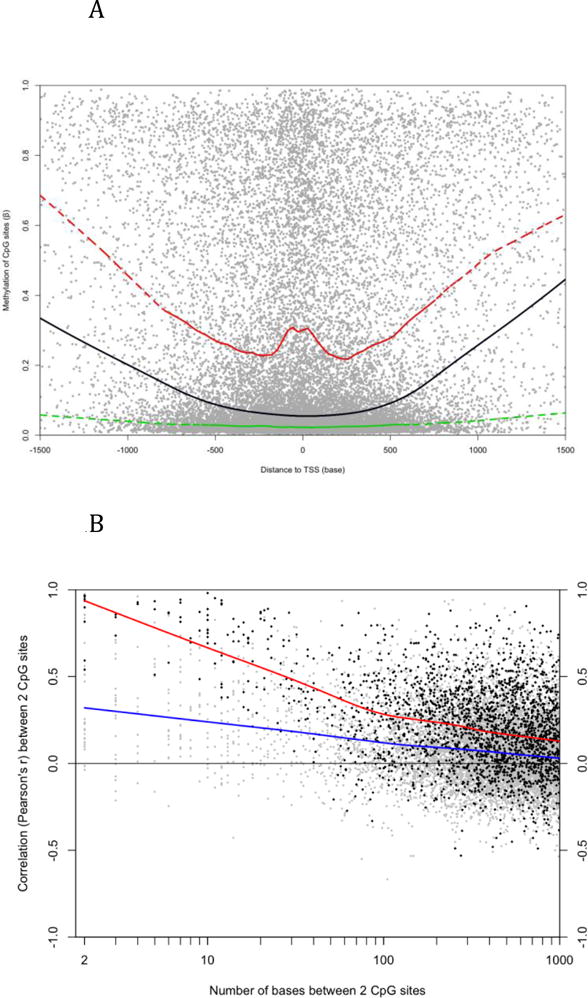
Characteristics of DNA methylation in LCL. A) Each dot represents a CpG site on autosomes. The X-axis indicates the distance to the nearest TSS and the y-axis is the average β value of 55 LCLs. The lines were generated by Lowess smoothing (black: all sites; green: CGI sites; red: non-CGI sites). Non-CGI sites have higher methylation than CGI sites in general no matter their distance to TSS. B) Each dot represents a pair of CpG sites on autosomes. The x-axis is the distance between the two sites and the y-axis indicates their Pearson’s correlation coefficient across 55 LCLs. The lines were generated by Lowess smoothing (blue: all pairs; red: pairs whose average β values are between 0.1 and 0.9 at both sites).
3.2. Differential methylation
DNA methylation at CpG sites around TSSs is highly variable between samples, especially when the β values are between 0.1 and 0.9 (Supplemental Figure 5). To test whether such high variability is also present in other cell types, we performed a meta-analysis of twelve DNA methylation data sets generated from different cell types, but using the same microarray platform (Supplemental File 2). After calculating the correlation of methylation levels between each pair of samples in the same disease or treatment group, we compared the distribution of correlation coefficients of different cell types. LCLs have higher between-sample variation than all the other cell types (T cells, monocytes, whole blood, and colon mucosa) with the exception of colorectal cancer cells.
Outliers are common in the data set as 535 autosomal CpG sites have β values ranged from less than 0.1 to more than 0.9. Principal components analysis (PCA) using all autosomal sites was unable to clearly separate samples by their disease status (Figure 2A). The top three principal components only account 20% for less than of the total variance, suggesting that DNA methylation in LCLs is affected by many factors, including, but not limited to, disease status, gender, genetic background, developmental stage and cell culture condition.
Two-way ANOVA analysis of gender and disease status as two interacting factors identified 283 CpG sites differentially methylated between 19 controls and 21 severe CdLS patients. These sites have ANOVA p values less than 0.01 and β value differences greater than 0.05. Among the 699 sites significantly different between females and males, 610 and 3 are located on chromosomes X and Y respectively, suggesting that gender has little impact on autosomal methylation. We took a closer look at the gender difference on the X chromosome, and noticed that sites in CpG islands are generally unmethylated in males but mostly have higher methylation in females (Supplemental Figure 6). This result was indeed anticipated because increased methylation of CpG islands plays an essential role in X inactivation [34]. An unexpected observation is that the 193 X chromosome sites having greater than 0.5 β values in both genders are significantly more methylated in males than in females (average β = 0.77 vs. 0.71, p = 1.7e-20, paired t test). It is unlikely that such a dramatic difference was caused by technical bias or data processing. We postulate that it is more difficult for females to maintain hypermethylation at those sites on both X chromosomes due to the need to methylate CpG islands. However, a valid interpretation of this observation requires further investigation.
Among the 283 sites differentially methylated between control and severe CdLS samples, 177 are up-methylated and 106 are down-methylated in CdLS. The corresponding false discovery rate (FDR) is 0.24 according to a permutation procedure that shuffled the sample labeling of disease status, but not gender. According to DAVID functional annotation [33], a number of pre-defined gene sets are significantly enriched in the genes downstream to those differentially methylated sites. Some of those gene sets are evidently related to CdLS (Supplemental Table 1). For example, four genes (TBX5, MSX1, MBNL1, and SALL4) involved in embryonic limb morphogenesis, which is one of the most common features of CdLS, have down-regulated CpG sites around their TSS.
The sites differentially methylated between controls and severe patients have two noteworthy features. First, all 283 sites have medium methylation (0.1 < β < 0.9) in at least one group although 57.8% of the total sites have low methylation (β < 0.1) in both groups. We then limited the remaining analysis of this section to the 9,776 sites having medium methylation. Second, the 177 sites up-methylated in CdLS include significantly lower percentage of CGI sites than the 106 down-methylated sites (5.6% vs. 49.1%, p = 4.6e-17, proportional test) while 18.7% of the unchanged sites are located in CGIs (Supplemental Figure 7). This result suggests that the likelihood and direction of methylation change in CdLS are related to local CpG density.
We previously used transcriptional microarray data to generate a diagnostic index of CdLS by comparing healthy controls and severe patients. It was shown that this index could be used to discriminate controls and CdLS patients as well as CdLS subtypes [19]. We then evaluated if methylation information could be used for the same purpose via a combination of nearest centroid classification and leave-one-out validation (details in Supplemental Methods). A methylation-based index classified control samples and severe patients with significant accuracy. In addition, this index can discriminate mild CdLS cases from both controls and severe patients (Figure 2C). However, the leave-one-out validation misclassified six control samples and three severe patients (accuracy = 77.5%) while the index of transcriptional data correctly classified 90.1% testing samples (Figure 1C in [19]). The area under ROC curves of methylation- and transcription-based prediction is respectively 0.860 and 0.985 (Supplemental Figure 8). Therefore, DNA methylation pattern is a less powerful diagnostic index of CdLS than gene transcription pattern.
3.3. The association of DNA methylation with other epigenetic features
GM12878 is a model LCL from a female donor. A variety of genomic data generated from GM12878 are available through the ENCODE (ENCyclopedia Of DNA Element) project [24]. We downloaded three sets of GM12878 data from the UCSC Genome Browser tracks: “HAIB Methyl27” (DNA methylation data generated from Infinium microarrays), “UW DNaseI HS” (DNaseI hypersensitivity data generated by deep sequencing), and “Broad Histone” (CTCF binding and eight histone modification data generated by ChIP-seq experiments).
We were able to directly compare the average β values of two GM12878 replicates and the female samples in our data set since both were generated on the same microarray platform. The Pearson’s r of all autosomal sites between the two vectors of average β values is 0.895 (Supplemental Figure 9). After low quality measurements were removed, the between-data set correlation was improved to 0.947 while the average r value of all female sample pairs in our data set is 0.960. We concluded that GM12878 is compatible with our samples in terms of DNA methylation and it is possible to associate our methylation and gene expression data with data generated from GM12878.
DNaseI hypersensitivity is a sequence feature related to DNA accessibility [35]. More than 100,000 short DNaseI hypersensitivity regions were identified from each of two GM12878 replicates. We mapped the CpG sites in our data to those regions and found that sites located within those regions have significantly lower β values than the other sites (0.045 vs. 0.262, p < 1e-300). Since CGIs and DNaseI hypersensitivity regions are often overlapped, we asked whether CpG density and DNaseI hypersensitivity affect DNA methylation independently. Sites located in DNaseI hypersensitivity region only have slightly lower average β than sites located in CGIs only (0.075 vs. 0.089, p = 0.0001), and the average β of sites located in overlapping regions is further reduced to 0.028 (Supplemental Figure 10). Therefore, CpG density and DNaseI hypersensitivity have additive effect on DNA methylation and DNaseI hypersensitivity is probably a stronger indicator of low methylation than CpG density.
Histone modifications are likely more involved in transcriptional regulation than DNA methylation. ChIP-seq data of eight histone modifications and CTCF binding of GM12878 are available at the 24,379 CpG sites measured by this study. We calculated the correlation between β value and tag enrichment at those sites (Figure 3A) and found that DNA methylation has a negative and relatively stronger correlation with euchromatin (decondensed chromatin) marks such as H3K4me3 and H3K9ac [36], and positive but weak correlation with heterochromatin (condensed chromatin) marks such as H3K27me3 and H4K20me1 [37]. This observation is in agreement with previous studies. For example, Brunner et al. reported that H3K4me3 and H3K27me3 signals are respectively correlated to unmethylated and methylated status in human embryonic stem cells [8], and Wu et al. found that DNA methylation has a negative correlation to H3K9ac in a mouse leukemia cell line [38]. These results suggest that the association between DNA methylation and histone modifications has similar pattern in different cell types and species. The association between DNA methylation and histone modifications is not particularly affected by CpG density except H3K27me3 (Supplemental Figure 11).
Figure 3.
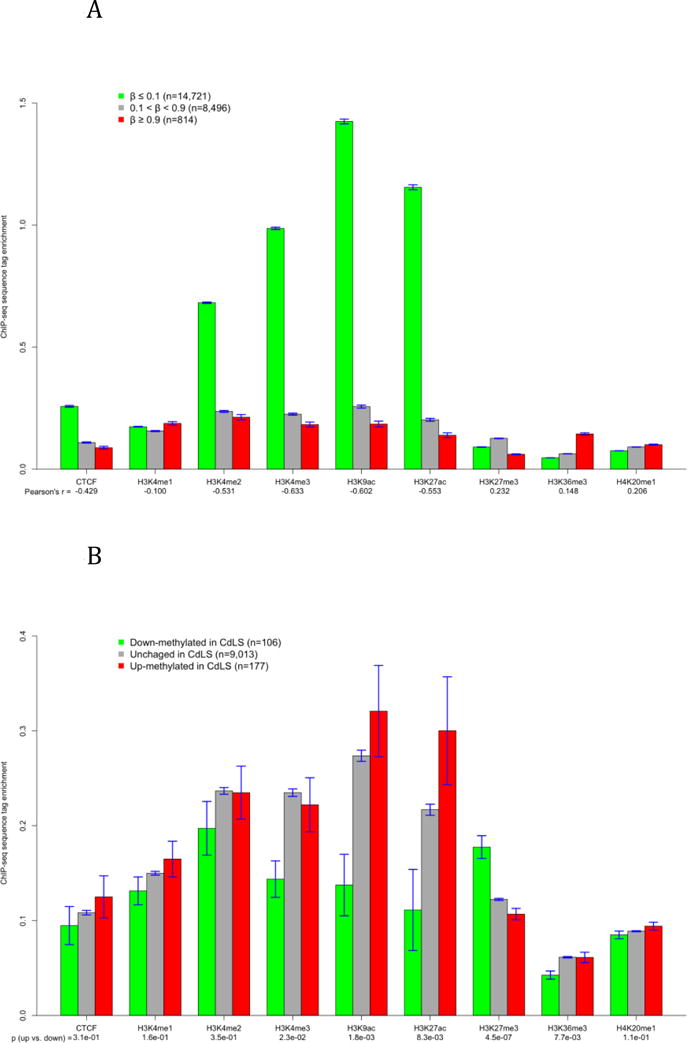
The association between DNA methylation and histone modifications. The y-axis represents the average tag enrichment based on ChIP-seq data. A) The correlation of average methylation to CTCF binding and eight histone marks in GM12878 at all autosomal CpG sites. B) Histone status at CpG sites that were down-methylated, unchanged, and up-methylated in severe patients. The unchanged group include only sites with medium methylation (0.1 < β < 0.9). The p values are results of Student’s t test comparing the down- and up-methylated sites.
Histone modifications are also related to the direction of methylation alterations. CpG sites down-methylated in CdLS have lower tag enrichment of CTCF and all histone modifications except H3K27me3 than up-methylated and unchanged sites (Figure 3B). The exception of H3K27me3 is probably caused by the fact that down-methylated sites include higher percentage of CGI sites than up-methylated and unchanged sites (Supplemental Figure 7) while CGI sites have substantially higher H3K27me3 than non-CGI sites (Supplemental Figure 11). However, CpG density has little effect on other histone modifications. For example, while CGI sites have slightly higher H3K9ac than non-CGI sites, down-methylated sites have significantly lower H3K9ac than other sites. Altogether, these results suggest that histone modifications are not only correlated to baseline DNA methylation, but are also related to the likelihood and direction of methylation alterations in LCL.
3.4. The association between DNA methylation and gene transcription
We previously published a gene expression microarray data set that included LCLs of 39 human subjects: 18 controls, 17 severe CdLS probands, 2 Roberts syndrome probands and 2 Alagille syndrome probands [19]. The DNA methylation of 27 of those subjects (13 controls and 14 severe CdLS patients) were also measured by this study. The probes of Affymetrix U133 Plus 2.0 platform used for the expression experiments were remapped to the current version of NCBI Entrez genes [39] and grouped into 17,726 unique Entrez genes. 10,430 genes measured by at least six mRNA probes and one CpG probe (within −1.5 to 1.5 kb of TSS) was used in the remaining analyses of this section. According to MAS5.0 algorithm [40], 51.7% and 21.8% of these genes were respectively called present and absent in all samples. These genes were considered universally active or inactive in LCL regardless of gender, disease state, or other factors. The details about data processing, gene filtering and annotation mapping are available in Supplemental Methods.
The methylation-expression association of 19,615 pairs of CpG sites and genes is summarized in Figure 4A, which illustrates that the association is dependant on the relative location of CpG sites to the TSSs of downstream genes. In the boxed area of Figure 5A, the overall methylation-expression correlation is negative and highly significant (Spearman’s ρ = −0.54, p = 1.8e-272). However, the correlation was non-linear. When β value is higher than 0.1, methylation level has little impact on gene expression level (ρ = 0.013, p = 0.67). The overall pattern in Figure 5A is consistent with the common perception of how DNA methylation regulates gene expression. Around the TSS and in the 5′-UTR, methylation regulates downstream transcription mainly through physical blocking, so high methylation represses gene expression. In the promoter region, DNA methylation indirectly regulates transcription by adjusting histone and transcription factor accessibility, so its correlation to gene expression could be in either direction. A previous study reported that exons usually have high methylation and the gene body of highly expressed genes is more methylated than the body of inactive genes [6]. This result explains the concurrence of high methylation and high expression in the region beginning approximately 1 kb downstream of the TSS where the coding region of some genes has started.
Figure 4.
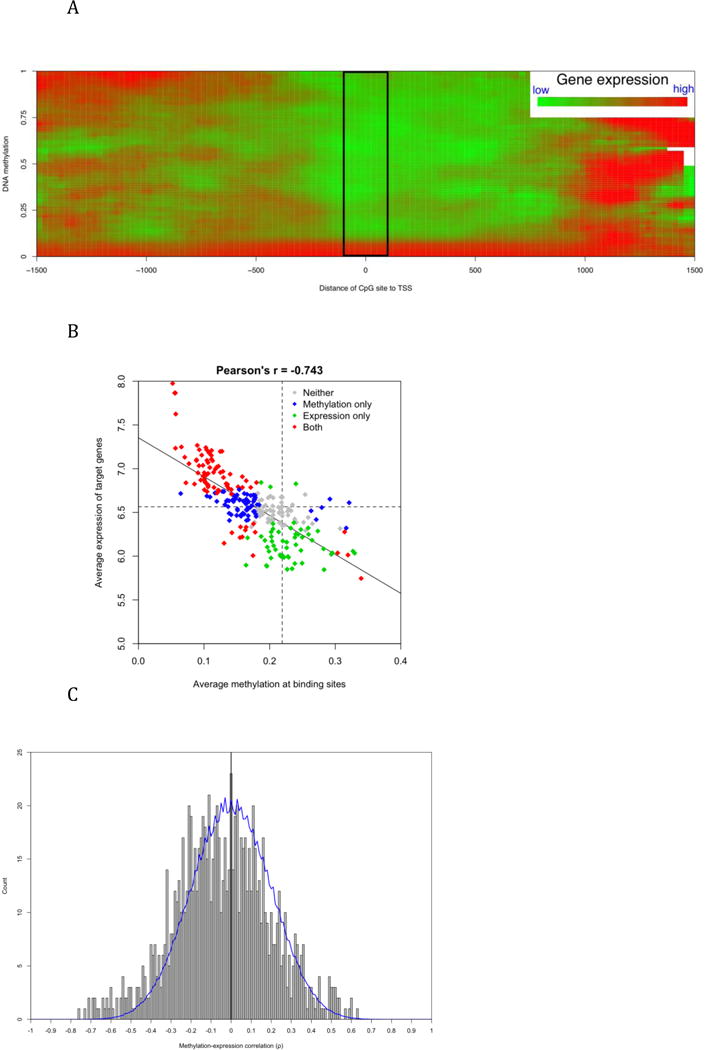
The association between DNA methylation around TSS and downstream gene transcription. A) The distance of the CpG sites to TSS affected the methylation-transcription association based on 19,615 CpG-transcript pairs. Color indicates average transcription level after local smoothing. The black box shows a distinctive pattern of negative correlation when the CpG sites are located within [−100, 100] around TSSs. B) The linearly correlated methylation around TFBS and expression of target genes. Each diamond corresponds to one of 258 TFBS motifs. X-axis represents the average methylation of CpG sites within [−250, 250] around TFBS and y-axis represents average expression of genes with at least one TFBS within [−1500, 1500] of their TSS. Color indicates whether the averages are significantly (p < 0.01) different from the global average. More details are available in Supplemental Table 1. C) The distribution of Spearman’s ρ values of CpG-gene pairs. All pairs include CpG site and gene both having high variance across 27 common samples. Blue line indicates the background distribution generated by 1,000 re-sampling permutations. Among a total of 1,006 pairs, 568 (56.5%) have negative ρ values.
Figure 5. The prediction of gene expression using epigenetic information.
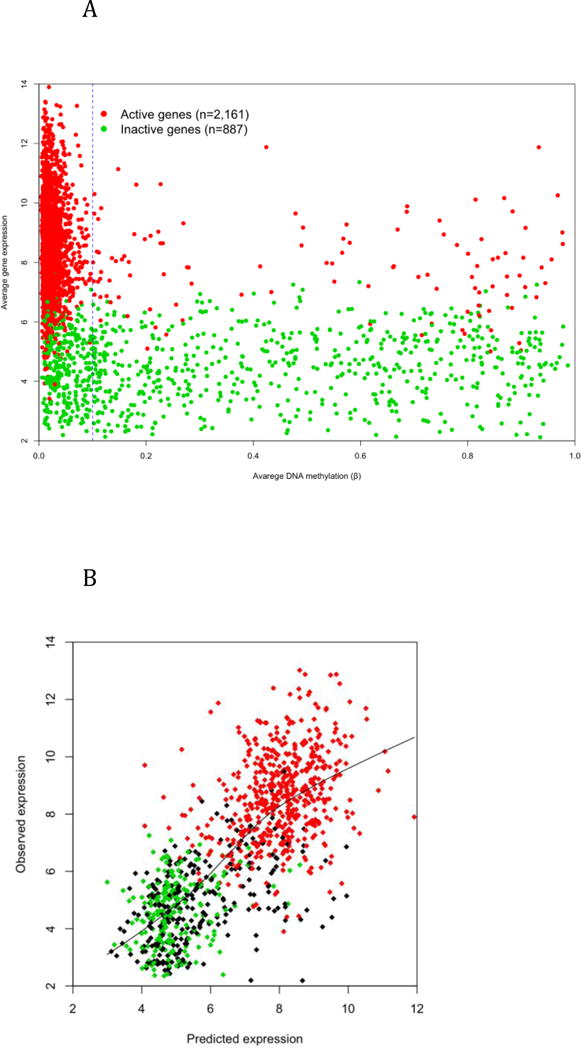
A) The average DNA methylation at TSS and 5′-UTR and the average expression of autosomal genes that are active (red) or inactive (green) in all samples. X chromosome genes are plotted separately in Supplemental Figure 12. B) Predicted vs. observed expression level of 1,000 testing genes (green: inactive, red: active; black partially active). The prediction is based on a linear regression model trained with DNA methylation, cohesin, CTCF and histone modification data of 3,100 genes.
Consistent with earlier results demonstrating that methylation at TFBSs varies dramatically between transcription factors, the transcription of TF target genes also differs significantly in LCL (Supplemental Table 2). The TFBS motif having the lowest average methylation, V$NRF2_01, has the highest average expression of target genes, which is more than 250% of the global average of gene expression. On the other hand, V$HNF1_01 has the highest methylation and lowest target expression. The average methylation and expression have a negative correlation of 0.743 across TFs and change concordantly in a linear pattern (Figure 4B). Interestingly, ten TFBS motifs have both lower methylation and expression than the global average, probably because lower methylation reduces the binding of those TFs or the increased binding of those TFs is repressive to gene expression. Overall, the combined analysis of genomic sequence, DNA methylation, and gene expression information provides a reference of TF activity in LCL. It also indicates that DNA methylation may affect gene expression through regulating TF binding.
We demonstrated in our previous study that changes in DNA methylation contributes little to the overall gene expression variation in CdLS [30]. Although more than one thousand genes demonstrate significantly changed expression in CdLS, the changes usually are of a small magnitude. A deficiency in cohesin or other transcription factors is more likely to be the cause of these subtle expression changes rather than differences in DNA methylation. The latter is probably involved in more dramatic events such as gene activation or inactivation. We hypothesized that a methylation-expression association is more evident when large between-sample variance exists. The methylation-expression correlation across 27 samples common to both studies were calculated for 1,006 CpG-gene pairs on autosomes whose between-sample variance is at the top 25% in both methylation and transcription data. As showed in Figure 4C, the overall correlation is skewed slightly towards the negative side (average Spearman’s ρ = −0.043, p = 2.5e-8). There are 38 pairs having ρ values less than −0.5, corresponding to a permutation FDR of 0.12. Six genes, C21orf56, CIDEB, DDX43, DENND2D, LDHC, and LOXL3, have ρ values less than −0.5 with two CpG sites around their TSSs, indicating that the expression of these genes is more likely to be directly regulated by DNA methylation. There are 17 pairs having ρ values greater than 0.5 (FDR =0.27). On average, CpG sites positively correlated to gene expression are located more upstream than sites negatively correlated to gene expression. (−250 vs. +155 bases of TSS, p = 0.04).
3.5. Prediction of gene transcription based on epigenetic status around the TSS
Figure 4A shows that DNA methylation at TSSs and 5′-UTRs have a relatively consistent association with gene expression in LCLs. We queried whether DNA methylation in those regions could be used to predict the activation of downstream transcription. The analysis was first limited to 3,048 genes that are unanimously active or inactive according to expression microarray data and have at least one measured CpG site located between 100 bases upstream of the TSS and 100 bases downstream of the TSS or the end of 5′-UTR (whichever comes first). These genes were split into two groups of 887 inactive and 2,161 active genes. The vast majority (~95%) active genes have low methylation (β < 0.1), while only about two-thirds inactive genes have non-low methylation (β > 0.1) around their TSS (Figure 5A). Therefore, low methylation is a necessary, but not a sufficient condition of downstream transcription, which also involves histone modifications, TF binding, and polymerase activation. The X chromosome demonstrated an interesting pattern (Supplemental Figure 12). Due to X inactivation in females, the majority of X-linked genes have non-low methylation regardless of whether they are expressed or not; while in males, almost all active genes have low methylation. Among the 102 genes that have active expression but non-low methylation, 79 have no CGI around their TSS, so inhibitory action of high methylation on downstream transcription is weakened when genes have low CpG density around TSS.
We applied a training-testing procedure during which 2,048 genes were randomly selected to train a logistic regression model and the remaining 1,000 genes were used to test model performance. This procedure was repeated 100 times to remove sampling bias. As expected, DNA methylation around the TSS is a highly sensitive, but non-specific, predictor of active transcription and its model outperforms the model using CpG density as predictor (Table 1A). The multivariate model using both CpG density and methylation as predictors has improved performance and predict gene activation with 84% accuracy. Models using our previous cohesin binding data [19] as well as CTCF and histone data of GM12878 as predictors were also created and tested. Remarkably, some histone modifications are strong predictors of gene expression even although the data were generated independently from unrelated sample. For example, H3K9ac alone can predict gene expression with 93.7% accuracy, 93.6% sensitivity and 94.1% specificity. Finally, we integrated all available predictors into one multivariate model, which has better and more balanced performance than univariate models.
Table 1. Predict gene expression with regression models.
Results in the tables are the summary of 100 re-sampling permutations of genes and each permutation randomly 1,000 genes to test the models trained with the other genes. “NA” indicates that the model has no better performance than random prediction. A) Gene expression is represented as a binomial variable (whether a gene is expressed) and logistic regression is used for the modeling. The three best values are highlighted in bold. B). Gene expression is represented as continuous variable (how much a gene is expressed) and so linear regression is used instead. Numbers indicate the Pearson’s correlation coefficients between predicted and observed expression level. Island = within or out of CpG island; Meth = average DNA methylation; AUC = area under ROC curve; ACC = accuracy of prediction; SENS = sensitivity; SPEC = specificity; PPV = positive predictive value; and NPV = negative predictive value. All genes include active, inactive, and partially active genes
| A Prediction of whether genes were active in LCLs by logistic regression models | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Univariate | Multivariate | |||||||||||||
| Island | Meth | Cohesin | CTCF | H3K4mel | H3K4me2 | H3K4me3 | H3K9ac | H3K27ac | H3K27me3 | H3K36me3 | H4K20mel | Island*Meth | Full | |
| AUC | 71.1 | 89.5 | 80.5 | 83.8 | 65.3 | 90.1 | 96.4 | 97.0 | 97.1 | 85.7 | 60.3 | 71.4 | 88.2 | 97.4 |
| ACC | 75.8 | 81.7 | 77.8 | 71.0 | 70.9 | 91.1 | 93.7 | 93.7 | 93.5 | 85.7 | 70.9 | 71.8 | 84.0 | 95.1 |
| SENS | 82.3 | 96.8 | 91.8 | NA | NA | 95.1 | 95.6 | 93.6 | 92.9 | 97.0 | NA | 94.2 | 95.8 | 96.7 |
| SPEC | 59.8 | 44.9 | 43.7 | NA | NA | 31.4 | 89.2 | 94.1 | 94.8 | 58.4 | NA | 17.2 | 55.3 | 91.3 |
| PPV | 83.3 | 81.1 | 80.0 | NA | NA | 92.6 | 95.6 | 97.5 | 97.8 | 85.1 | NA | 73.5 | 83.9 | 96.4 |
| NPV | 58.1 | 85.4 | 68.6 | NA | NA | 87.3 | 89.2 | 85.8 | 84.6 | 88.8 | NA | 55.1 | 84.4 | 91.9 |
| B Prediction of how much genet were expressed by linear regression models | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Univariate | Multivariate | |||||||||||||
| Island | Meth | Cohesin | CTCF | H3K4me1 | H3K4me2 | H3K4me3 | H3K9ac | H3K273C | H3K27me3 | H3K36me3 | H4K20mel | Island*Meth | Full | |
| All genes | NA | 0.40 | 0.31 | 0.16 | 0.10 | 0.47 | 0.65 | 0.66 | 0.61 | 0.48 | 0.65 | 0.19 | 0.44 | 0.73 |
| Active | NA | 0.10 | 0.03 | −0.03 | −0.03 | −0.02 | 0.13 | 0.28 | 0.29 | 0.08 | 0.00 | −0.06 | 0.09 | 0.28 |
| Inactive | NA | −0.04 | 0.01 | −0.02 | 0.06 | 0.05 | 0.10 | 0.13 | 0.10 | 0.13 | −0.04 | −0.11 | −0.02 | 0.16 |
Predicting gene expression level is more challenging than predicting gene activation as epigenetic modifications are not the only regulators of gene expression. Downstream regulators such as transcription factors and miRNAs are probably more important to the tuning of gene expression levels. In addition, gene expression measurements are biased by hybridization efficiency of microarray probes, so they are not exactly correlated to mRNA abundance. To evaluate the predictive ability of epigenetic status on gene expression level, we applied the same training-testing/permutation procedure and used the same predictors to build a series of linear regression models during which 1,052 poised genes (active in part of the 39 samples) were added to make a pool of 4,100 genes in total. While 3,100 genes were randomly selected for training models, the performance of the models was evaluated by the correlation between predicted and observed expression level of the remaining 1,000 genes. The average Pearson’s r of 100 permutations is 0.726 with the multivariate model of all available predictors (Table 1B). However, the correlation is mainly determined by the dramatic difference between inactive and active genes (Figure 6B). When using active genes only, the correlation between expected and observed expression level was substantially decreased, but still significant (average r = 0.272, p = 2.2e-10). The involvement of H3K9ac in gene expression is supported by both types of models and its univariate model has performance close to the full model. The regulatory function of H3K9ac on gene expression has been reported in T cells [41].
4. Discussions
One of the most striking observations of this study is the high variability of methylation at CpG sites while known clinical factors, such as gender and disease-causing DNA mutations, only account for a small portion of the total variance between samples. The total variance is a composite of the following components: 1) measurement error of the microarray experiments; 2) bias introduced by bisulfate treatment or other DNA preparation steps; 3) methylation alteration caused by EBV transformation and cell culturing; and 4) variance inherited from the donors. Since the replicated measurements of the same samples have strong correlation (r = 0.99) between two microarray batches and the fact that we applied a strict filtering procedure to exclude questionable measurements and samples from data analysis, the contribution of measurement error to the total variance is minimized. Replicated samples in the same microarray batch, but processed separately through DNA preparation, also have much better correlation to each other than the correlation of any pairs of different samples (Supplemental File 1). We thus concluded that the majority of the total variance is not from the microarray experiments.
Virus transformation and continuous cell culturing and storage may contribute more to the total variance although our meta-analysis shows that LCLs are more similar to lymphocytes than other cell types (Supplemental File 2). Previous studies have reported altered methylation in LCLs at different chromosomal locations, questioning the fidelity of DNA methylation in LCLs to its in vivo status [27, 29]. Furthermore, the meta-analysis demonstrates that LCLs have a much larger between-sample variance than most types of fresh cells. Similarly, Grafodatskaya et. al. recently observed that LCLs have larger between-sample variance than white blood cells and suggested that methylation alteration in LCLs occurs at random locations [28]. Nevertheless, our results show that GM12878 and LCLs used in this study have highly correlated DNA methylation pattern although as a model cell line, GM12878 has been cultured for many generations (Supplemental Figure 9). Conversely, if methylation alterations take place randomly and accumulatively, we would observe a reduced between-sample correlation over generations.
We postulate that while the biological systems such as the methylome and the transcriptome go through certain alterations during the establishment of LCLs, they will maintain a relatively stable status during cell culturing. Furthermore, the alterations are not random events, so LCLs generated separately from the same donor will have more similar methylation and transcription patterns than those generated from different donors. If proved true, this feature of LCLs will advocate its value in studying genetic disorders. Unlike fresh cells whose status is usually confounded by many uncontrollable factors, cultured cells are more homogeneous and grown under controlled environment. The effect of etiological mutations on biological systems is more isolated and recognizable in cell lines. For example, our previous study used LCLs to identify over one thousand genes significantly dysregulated in CdLS [19] while the magnitude of differential expression is mostly too small to be detected in fresh cells due to their lack of homogeneity. Therefore, LCLs are often a more practical experimental material for studying how etiologic mutations cause abnormalities in downstream systems although it cannot preserve the complexity of in vivo status as fresh or primary cells do. Future experiments that trace methylation alterations throughout LCL culturing and compare methylation patterns before and after EBV transformation in multiple sample groups will more conclusively test these hypotheses.
Although current data cannot directly validate that biological systems in different LCLs will reach and maintain a stable status, this hypothesis is strongly supported by the fact that histone modifications in GM12878 alone can predict gene expression in an unrelated sample set with 95% accuracy (Table 1B). This result also suggests that epigenetic status is the determinant factor of gene activation in LCL. The prediction accuracy is remarkably high considering the existence of a few technical difficulties, such as the small number of samples used for most predictive variables and the possible error of mapping epigenetic status around TSS to 3′-biased expression measurements. Therefore, the actual impact of epigenetic status on transcription activation could be even higher.
This study also suggest that a β of 0.1, or the methylation of 10% of alleles in a cell population, is enough to indicate gene inactivation (Figure 4A). This is unlikely a consequence of biased Cy3/Cy5 measurements because the average β value of all three universally methylated controls is higher than 0.9. This observation brings up a series of questions. What is the cause of such heterogeneity? If a β of 0.1 is enough to inactivate transcription, is it necessary for cells to further increase methylation levels? If maintaining hyper-methylation status around the TSS requires extra energy, does it present an evolutionary disadvantage? In CdLS, there is a trend towards higher methylation levels at non-CGI sites (Figure 2B), which usually has no effect on gene expression since most of the downstream genes are already silenced in control samples. Whether this represents a dysfunctional regulatory system of DNA methylation in CdLS will be one of the topics of our future studies.
Supplementary Material
Highlights.
-
>
DNA methylation is highly variable between LCLs regardless of disease, gender, etc.
-
>
Only very close CpG sites (<=10bp) have co-regulated DNA methylation.
-
>
Histone modifications correlated to baseline methylation and direction of change.
-
>
DNA Methylation around TSS regulateds downstream transcription loosely.
-
>
Regression models use epigenetic data to predict gene activation with 95% accuracy.
Acknowledgments
We are grateful for the participation of the children and families with CdLS and to the CdLS Foundation for their support. The authors also acknowledge support from Dr. Xiaowu Gai and Juan Perin of Center for Biomedical Informatics at CHOP. This study was supported by NIH/NICHD PO1HD052860 (IDK, ZZ), NIH/NICHD R21HD050538 (IDK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mohandas T, Sparkes R, Shapiro L. Reactivation of an inactive human X chromosome: evidence for X inactivation by DNA methylation. Science. 1981;211:393–396. doi: 10.1126/science.6164095. [DOI] [PubMed] [Google Scholar]
- 2.Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362–365. doi: 10.1038/366362a0. [DOI] [PubMed] [Google Scholar]
- 3.Jones P, Veenstra G, Wade P, Vermaak D, Kass S, Landsberger N, Strouboulis J, Wolffe A. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 4.Robertson K. DNA methylation and human disease. Nat Rev Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 5.Okano M, Bell D, Haber D, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 6.Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong C, Low H, Kin Sung K, Rigoutsos I, Loring J, Wei C. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20:320–331. doi: 10.1101/gr.101907.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ehrich M, Turner J, Gibbs P, Lipton L, Giovanneti M, Cantor C, van den Boom D. Cytosine methylation profiling of cancer cell lines. Proc Natl Acad Sci U S A. 2008;105:4844–4849. doi: 10.1073/pnas.0712251105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brunner A, Johnson D, Kim S, Valouev A, Reddy T, Neff N, Anton E, Medina C, Nguyen L, Chiao E, Oyolu C, Schroth G, Absher D, Baker J, Myers R. Distinct DNA methylation patterns characterize differentiated human embryonic stem cells and developing human fetal liver. Genome Res. 2009;19:1044–1056. doi: 10.1101/gr.088773.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 10.Antequera F, Bird A. Number of CpG islands and genes in human and mouse. Proc Natl Acad Sci U S A. 1993;90:11995–11999. doi: 10.1073/pnas.90.24.11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Esteller M, Corn P, Baylin S, Herman J. A gene hypermethylation profile of human cancer. Cancer Res. 2001;61:3225–3229. [PubMed] [Google Scholar]
- 12.Akiyama Y, Maesawa C, Ogasawara S, Terashima M, Masuda T. Cell-type-specific repression of the maspin gene is disrupted frequently by demethylation at the promoter region in gastric intestinal metaplasia and cancer cells. Am J Pathol. 2003;163:1911–1919. doi: 10.1016/S0002-9440(10)63549-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tufarelli C, Stanley J, Garrick D, Sharpe J, Ayyub H, Wood W, Higgs D. Transcription of antisense RNA leading to gene silencing and methylation as a novel cause of human genetic disease. Nat Genet. 2003;34:157–165. doi: 10.1038/ng1157. [DOI] [PubMed] [Google Scholar]
- 14.Nakagawa H, Nuovo G, Zervos E, Martin EJ, Salovaara R, Aaltonen L, de la Chapelle A. Age-related hypermethylation of the 5′ region of MLH1 in normal colonic mucosa is associated with microsatellite-unstable colorectal cancer development. Cancer Res. 2001;61:6991–6995. [PubMed] [Google Scholar]
- 15.Javierre B, Fernandez A, Richter J, Al-Shahrour F, Martin-Subero J, Rodriguez-Ubreva J, Berdasco M, Fraga M, O’Hanlon T, Rider L, Jacinto F, Lopez-Longo F, Dopazo J, Forn M, Peinado M, Carreño L, Sawalha A, Harley J, Siebert R, Esteller M, Miller F, Ballestar E. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 2010;20:170–179. doi: 10.1101/gr.100289.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weber M, Davies J, Wittig D, Oakeley E, Haase M, Lam W, Schübeler D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 2005;37:853–862. doi: 10.1038/ng1598. [DOI] [PubMed] [Google Scholar]
- 17.Koga Y, Pelizzola M, Cheng E, Krauthammer M, Sznol M, Ariyan S, Narayan D, Molinaro A, Halaban R, Weissman S. Genome-wide screen of promoter methylation identifies novel markers in melanoma. Genome Res. 2009;19:1462–1470. doi: 10.1101/gr.091447.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sugimoto M, Tahara H, Ide T, Furuichi Y. Steps involved in immortalization and tumorigenesis in human B-lymphoblastoid cell lines transformed by Epstein-Barr virus. Cancer Res. 2004;64:3361–3364. doi: 10.1158/0008-5472.CAN-04-0079. [DOI] [PubMed] [Google Scholar]
- 19.Liu J, Zhang Z, Bando M, Itoh T, Deardorff M, Clark D, Kaur M, Tandy S, Kondoh T, Rappaport E, Spinner N, Vega H, Jackson L, Shirahige K, Krantz I. Transcriptional dysregulation in NIPBL and cohesin mutant human cells. PLoS Biol. 2009;7:e1000119. doi: 10.1371/journal.pbio.1000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishimura Y, Martin C, Vazquez-Lopez A, Spence S, Alvarez-Retuerto A, Sigman M, Steindler C, Pellegrini S, Schanen N, Warren S, Geschwind D. Genome-wide expression profiling of lymphoblastoid cell lines distinguishes different forms of autism and reveals shared pathways. Hum Mol Genet. 2007;16:1682–1698. doi: 10.1093/hmg/ddm116. [DOI] [PubMed] [Google Scholar]
- 21.Choy E, Yelensky R, Bonakdar S, Plenge R, Saxena R, De Jager P, Shaw S, Wolfish C, Slavik J, Cotsapas C, Rivas M, Dermitzakis E, Cahir-McFarland E, Kieff E, Hafler D, Daly M, Altshuler D. Genetic analysis of human traits in vitro: drug response and gene expression in lymphoblastoid cell lines. PLoS Genet. 2008;4:e1000287. doi: 10.1371/journal.pgen.1000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tuck-Muller C, Narayan A, Tsien F, Smeets D, Sawyer J, Fiala E, Sohn O, Ehrlich M. DNA hypomethylation and unusual chromosome instability in cell lines from ICF syndrome patients. Cytogenet Cell Genet. 2000;89:121–128. doi: 10.1159/000015590. [DOI] [PubMed] [Google Scholar]
- 23.Frazer K, Ballinger D, Cox D, Hinds D, Stuve L, Gibbs R, Belmont J, Boudreau A, Hardenbol P, Leal S, Pasternak S, Wheeler D, Willis T, Yu F, Yang H, Zeng C, Gao Y, Hu H, Hu W, Li C, Lin W, Liu S, Pan H, Tang X, Wang J, Wang W, Yu J, Zhang B, Zhang Q, Zhao H, Zhou J, Gabriel S, Barry R, Blumenstiel B, Camargo A, Defelice M, Faggart M, Goyette M, Gupta S, Moore J, Nguyen H, Onofrio R, Parkin M, Roy J, Stahl E, Winchester E, Ziaugra L, Altshuler D, Shen Y, Yao Z, Huang W, Chu X, He Y, Jin L, Liu Y, Sun W, Wang H, Wang Y, Xiong X, Xu L, Waye M, Tsui S, Xue H, Wong J, Galver L, Fan J, Gunderson K, Murray S, Oliphant A, Chee M, Montpetit A, Chagnon F, Ferretti V, Leboeuf M, Olivier J, Phillips M, Roumy S, Sallée C, Verner A, Hudson T, Kwok P, Cai D, Koboldt D, Miller R, Pawlikowska L, Taillon-Miller P, Xiao M, Tsui L, Mak W, Song Y, Tam P, Nakamura Y, Kawaguchi T, Kitamoto T, Morizono T, Nagashima A, Ohnishi Y, Sekine A, Tanaka T, Tsunoda T, Deloukas P, Bird C, Delgado M, Dermitzakis E, Gwilliam R, Hunt S, Morrison J, Powell D, Stranger B, Whittaker P, Bentley D, Daly M, de Bakker P, Barrett J, Chretien Y, Maller J, McCarroll S, Patterson N, Pe’er I, Price A, Purcell S, Richter D, Sabeti P, Saxena R, Schaffner S, Sham P, Varilly P, Stein L, Krishnan L, Smith A, Tello-Ruiz M, Thorisson G, Chakravarti A, Chen P, Cutler D, Kashuk C, Lin S, Abecasis G, Guan W, Li Y, Munro H, Qin Z, Thomas D, McVean G, Auton A, Bottolo L, Cardin N, Eyheramendy S, Freeman C, Marchini J, Myers S, Spencer C, Stephens M, Donnelly P, Cardon L, Clarke G, Evans D, Morris A, Weir B, Mullikin J, Sherry S, Feolo M, Skol A, Zhang H, Matsuda I, Fukushima Y, Macer D, Suda E, Rotimi C, Adebamowo C, Ajayi I, Aniagwu T, Marshall P, Nkwodimmah C, Royal C, Leppert M, Dixon M, Peiffer A, Qiu R, Kent A, Kato K, Niikawa N, Adewole I, Knoppers B, Foster M, Clayton E, Watkin J, Muzny D, Nazareth L, Sodergren E, Weinstock G, Yakub I, Birren B, Wilson R, Fulton L, Rogers J, Burton J, Carter N, Clee C, Griffiths M, Jones M, McLay K, Plumb R, Ross M, Sims S, Willey D, Chen Z, Han H, Kang L, Godbout M, Wallenburg J, L’Archevêque P, Bellemare G, Saeki K, An D, Fu H, Li Q, Wang Z, Wang R, Holden A, Brooks L, McEwen J, Guyer M, Wang V, Peterson J, Shi M, Spiegel J, Sung L, Zacharia L, Collins F, Kennedy K, Jamieson R, Stewart J, Consortium IH. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weinstock G. ENCODE: more genomic empowerment. Genome Res. 2007;17:667–668. doi: 10.1101/gr.6534207. [DOI] [PubMed] [Google Scholar]
- 25.Mikkelsen T, Ku M, Jaffe D, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim T, Koche R, Lee W, Mendenhall E, O’Donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander E, Bernstein B. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Araten D, Golde D, Zhang R, Thaler H, Gargiulo L, Notaro R, Luzzatto L. A quantitative measurement of the human somatic mutation rate. Cancer Res. 2005;65:8111–8117. doi: 10.1158/0008-5472.CAN-04-1198. [DOI] [PubMed] [Google Scholar]
- 27.Brennan E, Ehrich M, Brazil D, Crean J, Murphy M, Sadlier D, Martin F, Godson C, McKnight A, van den Boom D, Maxwell A, Savage D. Comparative analysis of DNA methylation profiles in peripheral blood leukocytes versus lymphoblastoid cell lines. Epigenetics. 2009;4:159–164. doi: 10.4161/epi.4.3.8793. [DOI] [PubMed] [Google Scholar]
- 28.Grafodatskaya D, Choufani S, Ferreira J, Butcher D, Lou Y, Zhao C, Scherer S, Weksberg R. EBV transformation and cell culturing destabilizes DNA methylation in human lymphoblastoid cell lines. Genomics. 2010;95:73–83. doi: 10.1016/j.ygeno.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 29.Saferali A, Grundberg E, Berlivet S, Beauchemin H, Morcos L, Polychronakos C, Pastinen T, Graham J, McNeney B, Naumova A. Cell culture-induced aberrant methylation of the imprinted IG DMR in human lymphoblastoid cell lines. Epigenetics. 2010;5:50–60. doi: 10.4161/epi.5.1.10436. [DOI] [PubMed] [Google Scholar]
- 30.Liu J, Zhang Z, Bando M, Itoh T, Deardorff M, Li J, Clark D, Kaur M, Tatsuro K, Kline A, Chang C, Vega H, Jackson L, Spinner N, Shirahige K, Krantz I. Genome-wide DNA methylation analysis in cohesin mutant human cell lines. Nucleic Acids Res. 2010 doi: 10.1093/nar/gkq346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu J, Krantz ID. Cornelia de Lange syndrome, cohesin, and beyond. Clin Genet. 2009;76:303–314. doi: 10.1111/j.1399-0004.2009.01271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Workman C, Jensen L, Jarmer H, Berka R, Gautier L, Nielser H, Saxild H, Nielsen C, Brunak S, Knudsen S. A new non-linear normalization method for reducing variability in DNA microarray experiments. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-9-research0048. research0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang dW, Sherman B, Lempicki R. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 34.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 35.Sabo P, Kuehn M, Thurman R, Johnson B, Johnson E, Cao H, Yu M, Rosenzweig E, Goldy J, Haydock A, Weaver M, Shafer A, Lee K, Neri F, Humbert R, Singer M, Richmond T, Dorschner M, McArthur M, Hawrylycz M, Green R, Navas P, Noble W, Stamatoyannopoulos J. Genome-scale mapping of DNase I sensitivity in vivo using tiling DNA microarrays. Nat Methods. 2006;3:511–518. doi: 10.1038/nmeth890. [DOI] [PubMed] [Google Scholar]
- 36.Chambeyron S, Bickmore W. Chromatin decondensation and nuclear reorganization of the HoxB locus upon induction of transcription. Genes Dev. 2004;18:1119–1130. doi: 10.1101/gad.292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 38.Wu J, Wang S, Potter D, Liu J, Smith L, Wu Y, Huang T, Plass C. Diverse histone modifications on histone 3 lysine 9 and their relation to DNA methylation in specifying gene silencing. BMC Genomics. 2007;8:131. doi: 10.1186/1471-2164-8-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dai M, Wang P, Boyd A, Kostov G, Athey B, Jones E, Bunney W, Myers R, Speed T, Akil H, Watson S, Meng F. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33:e175. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu W, Mei R, Di X, Ryder T, Hubbell E, Dee S, Webster T, Harrington C, Ho M, Baid J, Smeekens S. Analysis of high density expression microarrays with signed-rank call algorithms. Bioinformatics. 2002;18:1593–1599. doi: 10.1093/bioinformatics/18.12.1593. [DOI] [PubMed] [Google Scholar]
- 41.Fann M, Godlove J, Catalfamo M, Wood Wr, Chrest F, Chun N, Granger L, Wersto R, Madara K, Becker K, Henkart P, Weng N. Histone acetylation is associated with differential gene expression in the rapid and robust memory CD8(+) T-cell response. Blood. 2006;108:3363–3370. doi: 10.1182/blood-2006-02-005520. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.