

Abstract
Ferroptosis is a non-apoptotic form of cell death induced by small molecules in specific tumour types, and in engineered cells overexpressing oncogenic RAS. Yet, its relevance in non-transformed cells and tissues is unexplored and remains enigmatic. Here, we provide direct genetic evidence that the knockout of glutathione peroxidase 4 (Gpx4) causes cell death in a pathologically relevant form of ferroptosis. Using inducible Gpx4−/− mice, we elucidate an essential role for the glutathione/Gpx4 axis in preventing lipid-oxidation-induced acute renal failure and associated death. We furthermore systematically evaluated a library of small molecules for possible ferroptosis inhibitors, leading to the discovery of a potent spiroquinoxalinamine derivative called Liproxstatin-1, which is able to suppress ferroptosis in cells, in Gpx4−/− mice, and in a pre-clinical model of ischaemia/reperfusion-induced hepatic damage. In sum, we demonstrate that ferroptosis is a pervasive and dynamic form of cell death, which, when impeded, promises substantial cytoprotection.
Previously it was assumed that apoptosis was the only regulated form of cell death. Yet, recent years have shown that non-apoptotic cell death pathways are highly heterogeneous processes characterized by morphologically and biochemically distinct events1. Non-apoptotic cell death pathways have gained special attention particularly due to their role in inflammation and as potential therapeutic target in apoptosis-resistant tumours2,3. In this context, at least two cell death pathways are executed in response to detrimental concentrations of partially reduced forms of oxygen (that is, reactive oxygen species4). They include regulated necrosis (that is, necroptosis)5, and an iron-dependent form of cell death, named ferroptosis6. Ferroptosis is a newly discovered cell death pathway that can be induced in a subset of tumour cell types, including cell lines engineered to overexpress oncogenic RAS, by a number of small molecules named ferroptosis-inducing agents7 (FINs). It is characterized by increased levels of lipid hydroperoxides and iron overload, leading to caspase- and necrosome-independent cell death. FINs have been classified into class I FINs that involve cellular glutathione (GSH) depletion and class II FINs that lack this characteristic. Recently, it has been shown that class II FINs trigger ferroptosis through inhibition of glutathione peroxidase 4 (Gpx4; ref. 8).
So far, exploitation of ferroptosis has been proposed for killing tumour cells in response to specific compounds, and as a potential mechanism involved in oxidative glutamate toxicity in cells and brain slices9,10. We report here direct in vivo evidence that this type of cell death is not only limited to specific tumours treated with FIN agents, but that Gpx4 prevents premature death of mice by actively restraining the ferroptotic machinery in kidney tubular cells. In light of the yet-uncharacterized role of lipid hydroperoxides in ferroptosis, we now provide a detailed picture of a complex lipid oxidation signature in response to Gpx4 inactivation and ferroptosis induction. Furthermore, we identified an in vivo ferroptosis inhibitor shown to be efficacious in inducible Gpx4−/− cells and mice, as well as tissues subjected to ischaemia/reperfusion. This class of small-molecule inhibitors will not only serve to define the involvement of ferroptotic cell death in pathological conditions, but will also facilitate characterization of the underlying molecular paradigms that regulate ferroptosis.
RESULTS
Inducible disruption of Gpx4 causes acute renal failure and early death in an Alox15-independent manner
We previously showed that the inducible disruption of Gpx4 in mouse embryonic fibroblast (MEF) cells (Pfa1 cells) leads to a form of cell death with necrotic-like features that involves 12/15-lipoxygenase (Alox15) and apoptosis inducing factor (AIF) activation11. Accordingly, Alox15−/− cells were found to be resistant to the effects of Gpx4 inhibition through depletion of GSH by L-buthionine sulphoximine11 (BSO; Fig. 1a). To extend this initial analysis, inducible Gpx4−/− cells were treated with a series of inhibitors including MJ33, a competitive and reversible inhibitor of calcium-independent phospholipase A2 (aiPLA2), the lipoxygenase inhibitor zileuton, MK886 (Alox5), PD146176 (Alox15) and the dual inhibitor BWA4C (Alox5 and 15), which all prevented cell death in response to Gpx4 deletion (Fig. 1b).
Figure 1.
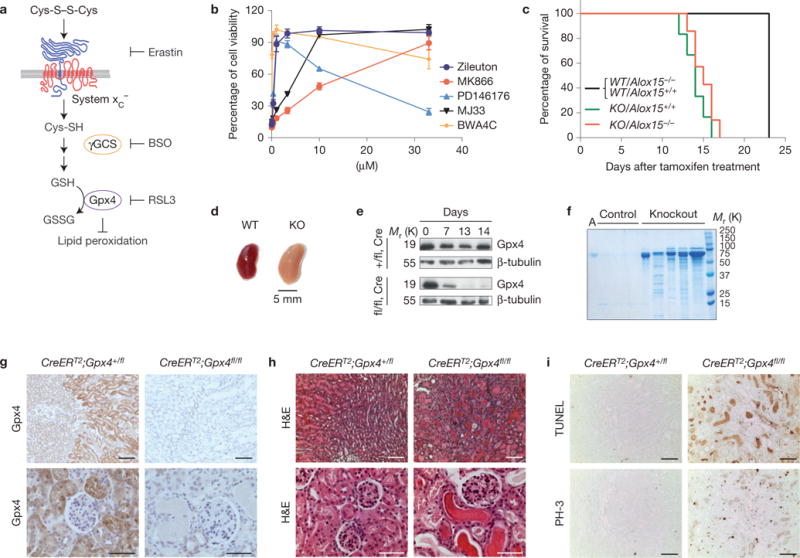
Inducible Gpx4 disruption causes ARF and death in mice. (a) A scheme showing the most important steps of glutathione (GSH) biosynthesis. αToc, α-tocopherol; BSO, L-buthionine sulphoximine; GSSG, oxidized glutathione; γGCS, γ-glutamylcysteine-synthase. (b) Inhibitors against enzymes of arachidonic acid metabolism prevent Gpx4-deletion-induced cell death in a dose-dependent manner. Gpx4 was disrupted in Pfa1 cells by the addition of 1 μM TAM in the presence of increasing concentrations of inhibitors. Cell viability was assessed by using AquaBluer 72 h after knockout induction. Data shown represent the mean ± s.d. of n = 4 of a 96-well plate from a representative experiment wells performed independently four times. (c) Mouse survival after TAM feeding. All induced Gpx4−/− (KO) mice died after approximately 2 weeks of TAM feeding regardless of Alox15 expression. None of the control mice (CreERT2;Gpx4+/fl/Alox15+/+(WT/Alox15+/+), CreERT2;Gpx4+/fl/Alox15−/−(WT/Alox15−/−)) died in the period investigated. Data are percentage of live animals; mean survival of Gpx4-null mice is 13.5 days after the onset of TAM feeding (n = 8 animals for KO/Alox15−/− and WT/Alox15−/− and n = 19 animals for KO/Alox15+/+ and WT/Alox15+/+). Gehan–Breslow–Wilcoxon test: P <0.0001). (d) Overall kidney phenotype of TAM-treated CreERT2;Gpx4fl/fl animals (KO) at time of euthanization. Left, control kidney (TAM-treated CreERT2;Gpx4+/fl, WT); right, enlarged and pale Gpx4−/− kidney. (e) Western blot of whole kidney tissue extracts showing that Gpx4 was efficiently depleted on TAM feeding in CreERT2;Gpx4fl/fl (fl/fl,Cre), but not in control CreERT2;Gpx4+/fl (+/fl,Cre) mice. (f) TAM-inducible CreERT2;Gpx4fl/fl mice present massive albuminuria and unselective proteinuria compared with control mice. Each lane represents one knockout or control animal (A, murine albumin). (g) Immunohistochemical expression analysis of Gpx4 in kidney tissue revealed that Gpx4 was efficiently depleted on TAM treatment of CreERT2;Gpx4fl/fl mice, which is in line with the immunoblot data. Note the high expression of Gpx4 in tubule cells of kidney cortex, whereas glomeruli show only faint Gpx4 expression (bars top row 100 μm and bottom row 50 μm). (h) Histological analysis of kidneys of TAM-treated CreERT2;Gpx4fl/fl animals showed widespread tubular cell death, interstitial edema and proteinaceous casts in distal tubules (bars top row 100 μm and bottom row 50 μm). (i) The number of TUNEL+ cells and mitotic cells (phosho-histone H3 staining, PH-3) is increased in kidneys of symptomatic Gpx4−/− mice (bars 100 μm). Uncropped images of blots are shown in Supplementary Fig. 8.
In light of these and our previous data, we sought to challenge the in vivo relevance of the Gpx4–Alox15 axis in this cell death pathway. We generated and characterized mouse models based on mice with loxP-flanked Gpx4 alleles, along with mice proficient or deficient in Alox15 (ref. 12). After 2–3 breeding steps CreERT2;Gpx4fl/fl/Alox15+/+ and CreERT2;Gpx4fl/fl/Alox15−/− mice were generated, which did not show any abnormalities in the absence of tamoxifen (TAM). On TAM feeding, the overall behaviour of CreERT2;Gpx4fl/fl/Alox15+/+ changed markedly, as indicated by catatonia, hunched posture and loss of body weight, whereupon mice had to be euthanized. Unexpectedly, CreERT2;Gpx4fl/fl/Alox15−/− revealed exactly the same behaviour and had to be euthanized at the same stage as induced CreERT2;Gpx4fl/fl/Alox15+/+ mice (Fig. 1c).
To define the role of Alox15 in cell death, cellular systems were established from CreERT2;Gpx4fl/fl/Alox15−/− mice (PZL cells) and from CreERT2;Gpx4fl/fl/Alox15+/+ mice as control (PZ cells). When Gpx4 deficiency was induced by TAM (Supplementary Fig. 1A), PZL cells died like PZ cells (Supplementary Fig. 1b), which could not be rescued by N-acetylcysteine (NAC) but by α-tocopherol (αToc) as previously reported11 (Supplementary Fig. 1b). As 5-lipoxygenase (Alox5) might compensate for loss of Alox15 (ref. 13), Alox5 was depleted by short hairpin RNA (shRNA) in PZL and PZ cells (Supplementary Fig. 1c,d). Whereas the knockdown of Alox5 in PZL cells partially delayed the cell death process, it did not impact on cell death in PZ cells. From these findings we conclude that no individual lipoxygenase could be identified that is solely responsible for the cell death processes downstream of Gpx4 inactivation.
Inferring that more than one lipid oxygenase is involved in cell death, we endeavoured to characterize in more detail why animals die on Gpx4 disruption. On dissection, grossly, slightly enlarged and pale kidneys were evident in the Gpx4−/− mice (Fig. 1d). In rare cases (<5%), signs of focal liver necrosis were evident. Analysis of Gpx4 expression showed a marked decrease of Gpx4 in the kidney already seven days after TAM treatment (Fig. 1e), which was paralleled by progressive, unselective proteinuria (Fig. 1f) and altered biochemical kidney parameters (Supplementary Table 1).
Immunohistochemistry demonstrated a strong Gpx4 staining in the proximal tubules of wild-type kidney that was lost on TAM administration (Fig. 1g). Histological and ultrastructural analysis as well as terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labelling (TUNEL) revealed widespread cell death of proximal tubular epithelia with progression to effacement of all tubular epithelia and of the parietal epithelial cells of the Bowman’s capsule (Fig. 1h,i and Supplementary Fig. 1f). Proteinaceous casts and cellular debris were observed in affected tubuli (Fig. 1h), and focal signs of tubular regeneration were evident (Fig. 1i). Mononuclear interstitial infiltration and edema were observed in advanced lesions (Fig. 1h and Supplementary Fig. 1g). Notably, CreERT2;Gpx4fl/fl/Alox15−/− mice developed comparable lesions to those of CreERT2;Gpx4fl/fl/Alox15+/+ mice (Supplementary Fig. 1e). Immunohistochemical staining against cleaved caspase-3 revealed faint staining, mainly confined to the lumen of renal tubules, possibly resulting from secondary inflammatory processes (Supplementary Fig. 1g).
Inducible Gpx4 disruption induces Nec1-sensitive ferroptotic cell death
To define the modality of cell death, we used inducible Gpx4−/− cells and chemical inducers for the subsequent studies. Using the pan-caspase inhibitor z-VAD-FMK (zvad) and determining caspase activation, we ruled out an involvement of caspases in cell death induced by Gpx4 loss and FINs, in contrast to parental cells treated with staurosporine or tumour necrosis factor α (TNFα) (Supplementary Fig. 2a–c). This indicates that a non-apoptotic form of cell death is triggered in Gpx4−/− cells.
Therefore, we next addressed whether necroptosis or ferroptosis is activated on Gpx4 inactivation. Indeed, the receptor interacting kinase 1 (Rip1) inhibitor necrostatin-1 (Nec1) and ferrostatin-1 (Fer1), the first described inhibitors of necroptosis and ferroptosis, respectively, rescued Gpx4−/− cells from cell death (Fig. 2a). As ferroptotic cell death entails cellular iron accumulation and lipid peroxidation6, cell death could also be inhibited by the iron chelator deferoxamine (DFO) and by αToc (Fig. 2a; ref. 11).
Figure 2.
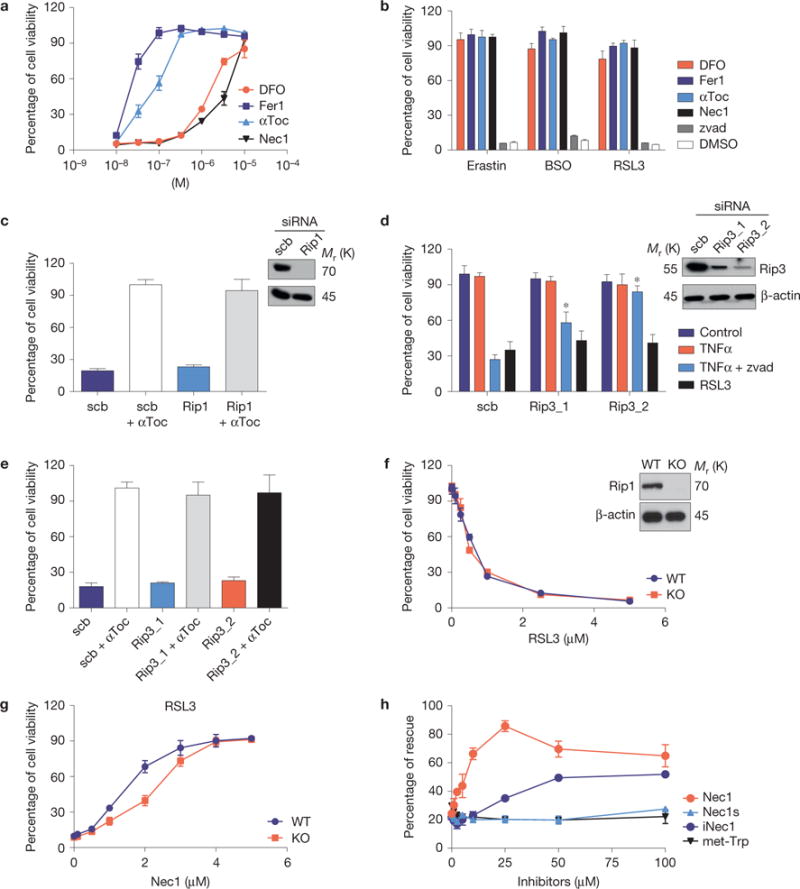
The inducible Gpx4 deletion in immortalized fibroblasts causes ferroptosis. (a) Cell death triggered by the inducible Gpx4 deletion in Pfa1 cells can be rescued by DFO, Fer1, αToc and Nec1 in a dose-dependent manner. Gpx4 was disrupted in Pfa1 cells as described in Fig. 1b (except for DFO, which was added 24 h after knockout induction, all inhibitors were added simultaneously along with TAM). Cell viability was assessed 72 h after knockout induction. (b) Cell death elicited in Gpx4-proficient Pfa-1 cells by the ferroptosis inducing agents (FINs) erastin (2.5 μM), BSO (20 μM) and RSL3 (20 nM) can be prevented by concomitant administration of DFO (10 μM), Fer1 (0.1 μM), αToc (1 μM) and Nec1 (20 μM), but not by the pan-caspase inhibitor Z-VAD-FMK (50 μM, zvad). Cell viability was assessed 24 h thereafter. (c) Rip1 knockdown in Pfa1 cells is unable to protect against cell death induced by Gpx4 deficiency (scrambled, scb). Cells were transfected 6 h after knockout induction and cell viability was assessed 66 h later. (d) Rip3 knockdown attenuates TNFα/Z-VAD-FMK-induced necroptosis, but not RSL3-induced ferroptosis in L929 cells (Scb, scrambled). Knockdown was performed 48 h before addition of the cell death stimuli. Cell viability was assessed 6 h after TNFα (10 ng ml−1), TNFα + zvad (50 μM) or RSL3 (1 μM) treatment. Data shown in c and d represent the mean ± s.d. of n=3 wells of a 12-well plate from a representative experiment performed five times, P = 0.05 (one-way ANOVA). (e) Rip3 knockdown in Pfa1 cells does not rescue cell death induced by Gpx4 knockout (scrambled, scb). Cells were transfected 6 h after knockout induction and cell viability was determined 66 h later. (f) Although the Rip1 inhibitor Nec1 rescued cell death triggered by FINs, Rip1-knockout (KO) cells are equally as sensitive to the Gpx4 inhibitor RSL3 as Rip1 wild-type (WT) cells; cell viability was assessed 24 h after treatment. (g) Nec1 rescues RSL3 (1 μM)-induced cell death in both Rip1 KO and WT cells in a dose-dependent manner. Cells were pre-treated with increasing concentrations of Nec1 for 6 h and viability was assessed 24 h after treatment with FIN agents. (h) Structurally related analogues of Nec1 (Nec1s, iNec) and met-Trp, an inhibitor of indoleamine 2,3-dioxygenase that is also inhibited by Nec1, present different effects on cell death induced by Gpx4 deletion. Rescue was performed by treating cells with increasing concentrations of Nec1 analogues (0–100 μM), and cell viability was assessed 72 h after knockout induction. Data shown represent the mean ± s.d. of n = 3 wells (e,h) or n = 4 wells (a,b,f,g) of a 96-well plate from a representative experiment performed independently at least three times, P= 0.05 (one-way ANOVA followed by Tukey’s multiple comparisons test).
Likewise, depletion of the Gpx4 substrate GSH through BSO or erastin (Fig. 1a and Supplementary Fig. 2d) efficiently triggered rapid cell death (Supplementary Fig. 2e) that could be prevented by the same inhibitors (Fig. 2b and Supplementary Fig. 2f). Furthermore, cell death induced by (1S, 3R)-RSL3 (hereafter RSL3), the first described Gpx4 inhibitor8, could also be prevented by Fer1 and Nec1 (Fig. 2b), indicating that availability of cellular GSH and proper Gpx4 function tightly control ferroptosis. Consistently, all compounds failed to induce caspase activation (Supplementary Fig. 2c), whereas Gpx4 overexpression conferred resistance to FINs (Supplementary Fig. 2g,h; ref. 14).
Although ferroptosis and necroptosis are considered to be two independent forms of non-apoptotic cell death, we were intrigued by the protective effects of Nec1. Therefore, Rip1 was depleted in Pfa1 cells by RNA-mediated interference (RNAi), but no protection was evident in Gpx4−/− cells (Fig. 2c). As Rip1 confers either a pro-death or a pro-survival function5, we next depleted Rip3, the downstream effector of Rip1. As Pfa1 cells express diminutive Rip3 levels (Supplementary Fig. 3a), experiments were performed in L929 cells showing high Rip3 expression (Fig. 2d). Rip3 expression was efficiently decreased in L929 cells by RNAi, sufficient to prevent TNFα/zvad-induced necroptosis, but not RSL3-induced cell death (Fig. 2d), or Gpx4 deletion-induced ferroptosis in Pfa1 cells (Fig. 2e).
To exclude a Rip1 contribution in ferroptosis, we treated Rip1-deficient cells with FINs, showing that Rip1−/− cells were equally sensitive to ferroptosis inducers (Fig. 2f and Supplementary Fig. 3b). Most interestingly, ferroptosis could be prevented in Rip1−/− cells by Nec1 (Fig. 2g and Supplementary Fig. 3b), demonstrating that Nec1 has yet-unrecognized off-target effects in ferroptosis. Indeed, the more specific Nec1-derivative Nec1s did not protect against Gpx4-depletion-induced cell death15 (Fig. 2h).
We next examined ultrastructural changes induced by Gpx4 inactivation and different cell death inducers. These studies revealed that Gpx4−/− cells exhibited swollen mitochondria, a reduced number of cristae with a more lamellar phenotype and an increase in autophagosomes in contrast to parental Pfa1 cells (Supplementary Fig. 3c). Similar to Gpx4−/− cells, BSO-treated Pfa1 cells revealed a reduced number of cristae with more lamellar and tubular structures, whereas autophagosomes could not be detected. Erastin-treated cells presented mitochondria with concentric cristae. Pfa1 cells treated with TNFα showed electron-dense whorled membranes and intermitochondrial bridges, and when treated with staurosporine, swollen mitochondria and a loss of cristae. These data suggest that cell death induced by Gpx4 deletion or GSH depletion is morphologically different from that by classical cell death inducers such as TNFα or staurosporine.
Ferroptosis is triggered by extra-mitochondrial lipid peroxidation and is characterized by the release of oxidized lipid mediators
Lipid hydroperoxides have been described as the driving force of ferroptosis6, although their molecular entity, mode and site of generation remain unclear. Previously, we reported that Gpx4−/− cells exhibit a yet-uncharacterized strong oxidizing component in their lipid compartment as a measure of BODIPY 581/591 C11 oxidation16. In fact, fluorescence-activated cell sorting analysis of Gpx4−/− cells showed that BODIPY 581/591 C11 oxidation could be ameliorated by Fer1 and DFO (Fig. 3a).
Figure 3.
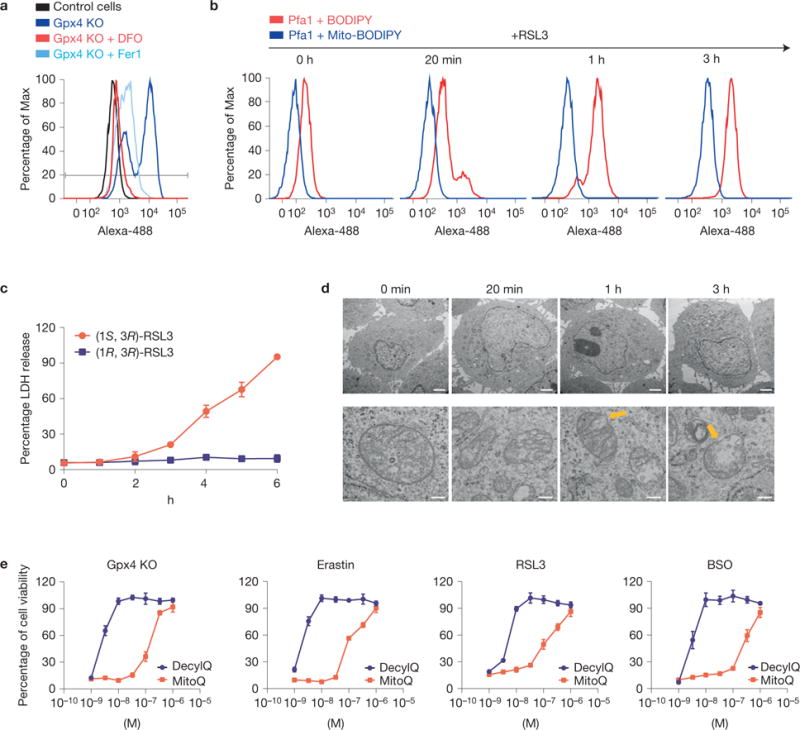
Lipid peroxidation outside the mitochondrial matrix triggers ferroptosis in MEFs. (a) Inducible knockout of Gpx4 leads to increased lipid peroxidation that can be blunted by Fer1 and DFO. Lipid peroxidation was measured 48 h after knockout induction using the redox-sensitive dye BODIPY 581/591 C11. (b) RSL3 causes rapid lipid peroxidation outside the mitochondrial matrix as determined by BODIPY 581/591 C11 versus mitochondrial-targeted BODIPY 581/591 C11 staining; oxidation of the probe was assessed at the indicated time points. (c) Time-dependent increase in LDH release in Pfa1 cells treated with RSL3 (100 nM); supernatants were collected at the indicated time points and assayed for LDH activity. Data shown represent the mean ± s.d. of n = 3 wells of a 12-well plate from an experiment performed independently five times, P =0.05 (one-way ANOVA). (d) Electron micrographs showing a time-dependent OMM rupture (yellow arrows) on ferroptosis induction using RSL3 (50 nM; scale bars 2 μm top row, 200 nm bottom row). (e) The mitochondrial targeted antioxidant MitoQ is by far less efficient in protecting cells against ferroptotic cell death, elicited by Gpx4 knockout (KO), erastin, RSL3 or BSO, compared with the membrane-targeted antioxidant DecylQ. Ferroptosis was induced by Gpx4 knockout in Pfa1 cells by 1 μM TAM or by treatment of non-induced Pfa1 cells with FINs in the presence of increasing concentrations of the inhibitors; cell viability was determined 72 h and 24 h later, respectively. Data shown represent the mean ± s.d. of n=4 wells of a 96-well plate from a representative experiment performed independently at least three times, P =0.05 (one-way ANOVA).
As mitochondrial lipid oxidation seems to be a critical step in the execution of some forms of cell death17, we further addressed whether a ‘lethal lipid signal’ is generated in mitochondria or extra-mitochondrial compartments. Therefore, we comparatively monitored the oxidation of mito- versus non-targeted BODIPY 581/591 C11 in Pfa1 cells treated with RSL3 (Fig. 3b). Interestingly, the non-targeted redox sensor presented a faster oxidation kinetics compared with the mitochondria-targeted one. Furthermore, this oxidation preceded cell lysis (Fig. 3c), indicating that the initial lipid peroxidation step occurs outside the mitochondrial matrix. Moreover, ultrastructural analysis demonstrated that ferroptosis involves outer mitochondrial membrane (OMM) rupture (Fig. 3d), possibly marking the point of no-return in ferroptosis. Surprisingly, cells treated with the lipophilic antioxidant decylubiquinone and its mitochondria-targeted chemical analogue, mitoquinone mesylate (MitoQ), required 100-fold higher MitoQ concentrations to prevent ferroptosis (Fig. 3e), supported by de-localizing the targeted antioxidant with FCCP (Supplementary Fig. 3d)18. This implies that ferroptosis-induced OMM disruption proceeds through a mechanism not amenable to mitochondria-targeted antioxidant therapies.
Reportedly, enriching membranes with arachidonic acid accelerated the cell death process on Gpx4 deletion11. As such, we determined arachidonic acid metabolites released into the cell culture medium on triggering ferroptosis. We now show that the levels of 5-hydroxyeicosatetraenoic acid (HETE), 11-HETE and 15-HETE were increased in the medium of Gpx4−/− cells 48 h after TAM treatment (Fig. 4a), which could be abrogated by zileuton, MK886, PD146176, MJ33, Fer1 and Nec1 (Fig. 4b). In contrast, extracellular 12-HETE levels remained unaffected in response to Gpx4 loss, arguing against an unspecific lipid oxidation (Fig. 4a). MEFs treated with FINs also presented a similar HETE signature (Fig. 4c), demonstrating that lipid mediators are released during ferroptosis. Accordingly, the cell lines described in Supplementary Fig. 1a,b demonstrate that Alox15 deficiency with Alox5 knockdown presented a slower release of lipid oxidation products than parental cells (Supplementary Fig. 4a,b), thus probably explaining their increased resistance to FINs (Supplementary Fig. 4c). Accordingly, treatment of Gpx4−/− cells with 5-, 12- and 15-hydroperoxyeicosatetraenoic acid (HpETEs) accelerated the ferroptotic process, whereas other metabolites, including HETEs and oxoeicosatetraenoic acid, did not influence cell death (Fig. 4d). Equimolar concentrations of H2O2 treatment did not phenocopy the effects of HpETEs, ruling out a general peroxide effect.
Figure 4.
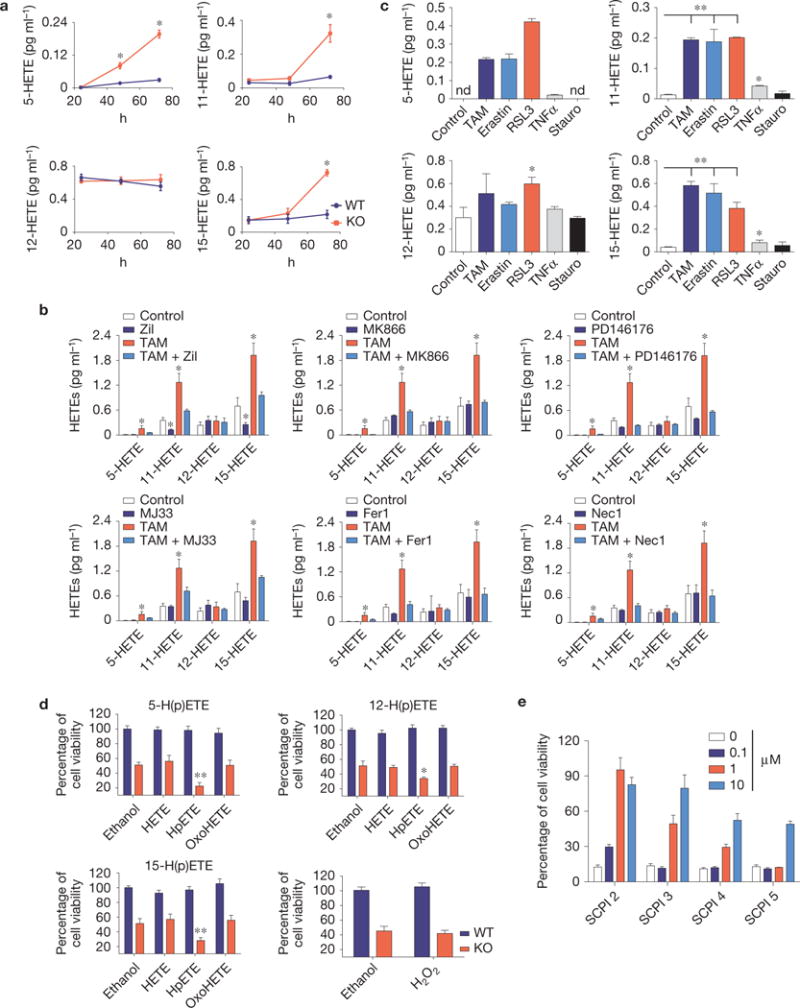
Ferroptosis is characterized by the release of lipid mediators. (a) Quantification of a time-dependent release of arachidonic acid metabolites (5-HETE, 11-HETE, 12-HETE and 15-HETE) in the cell culture media after inducible Gpx4 deletion (KO) using LC–MS/MS analysis. (b) Impact of small-molecule inhibitors on HETE release on Gpx4 deletion. Quantification of released arachidonic acid metabolites was performed by LC–MS/MS. Cells were treated with zileuton (10 μM), MK866 (50 μM), PD146176 (1 μM), MJ33 (10 μM) and Fer1 (0.2 μM) at the time of knockout induction. Analysis was performed in conditioned medium collected 72 h after Gpx4 deletion. (c) Quantification of arachidonic acid metabolites released into the medium 24 h after cell death induction by erastin (2.5 μM), RSL3 (0.75 μM), TNFα (10 ng ml−1) and staurosporine (Stauro; 0.2 μM) using HPLC–MS/MS analysis. In the Gpx4−/− cells, HETE levels were determined 72 h after TAM treatment. Data shown represent the mean ± s.d. of n=3 wells of a 6-well plate from an experiment performed independently three times (a,c) or one time (b). ∗P =0.05, ∗∗P =0.01 (one-way ANOVA, followed by Tukey’s multiple comparisons test (a) or Dunnett’s multiple comparison test (b,c)). (d) Impact of arachidonic acid metabolites on the cell death process elicited by Gpx4 loss. HETEs, HpETEs and OxoHETEs (1 μM) were added to WT or KO Pfa1 cells for Gpx4. Compounds were added 36 h after knockout induction and cell viability was measured 12 h thereafter. H2O2 was used at equimolar concentrations. Data shown represent the mean ± s.d. from n = 4 wells (HpETEs) or n = 6 wells (H2O2) of a 96-well plate from a representative experiment performed three times, ∗P = 0.05, ∗∗P = 0.01, one-way ANOVA followed by Tukey’s multiple comparisons test. (e)=Sterol carrier protein-2 (SCP) inhibitors (SCPI) rescue Gpx4−/− cells from ferroptosis in a dose-dependent manner. Inhibitors were added concomitantly to TAM and cell viability was determined 72 h thereafter. Data shown represent the mean ± s.d. of n = 6 wells of a 96-well plate from a representative experiment performed independently two times, P =0.05 (one-way ANOVA).
Despite the strongly reduced protection of mitochondria-targeted antioxidants (Fig. 3e), it is evident that morphological (Fig. 3d) and chemical (Fig. 4) changes occur in mitochondria and contribute to ferroptosis. Lipid transport proteins, particularly sterol carrier protein 2 (SCP-2), disseminate oxidative stress between compartments19 by preferentially trafficking peroxidized lipids to mitochondria20. In fact, SCP-2 inhibitors21 showed anti-ferroptotic activity (Fig. 4e), which was substantiated by knocking down SCP-2 in Gpx4−/− cells (Supplementary Fig. 4d). Yet, this effect was transient as it only slowed down cell death kinetics (Supplementary Fig. 4e), implying that other translocases may work in parallel with SCP-2.
Oxi-lipidomics analysis reveals a complex lipid oxidation signature of Gpx4-null kidneys
In an untargeted approach, we set out to directly assess lipid oxidation products in Gpx4-null kidneys using lipid extraction and LC–MS analysis22. These investigations were deemed necessary to bypass the use of fluorescence-based redox sensors, whose mode-of-actions are poorly characterized and may lead to potentially misleading conclusions.
Using oxidative lipidomics, we established that in Gpx4−/− kidneys phosphatidylcholine (PC) and phosphatidylethanolamine (PE), as well as the mitochondria-specific phospholipid, cardiolipin (CL), underwent substantial oxidative modifications. To assign specific acyl chain combinations for individual phospholipid molecular species, including PC, we performed MS/MS analysis using the negative ionization mode. For example, the MS/MS pattern of the PC molecular ion with m/z 816.556 [M–H+CH3COOH]− clearly demonstrates the presence of carboxylated ions with m/z 255.232 and m/z 279.232 corresponding to 16:0 and 18:2 fatty acid residues. The potentially confounding carboxylated daughter ions—with m/z 253.232 and m/z 281.232 corresponding to 16:1 and 18:1—were not detected in the MS/MS spectrum. We have presented MS/MS spectra for PC (m/z 816.556), PE (766.539) and CL (1,447.961) in Supplementary Fig. 5.
As Gpx4 catalyses the reduction of double-oxygenated phospholipids into mono-oxygenated phospholipids, we performed LC–MS assessment of these respective species in kidneys from control versus Gpx4−/− animals on day 6 when no manifestations of renal failure were detectable and on day 10 with an overt renal failure phenotype (Fig. 5). To verify the nature of double oxygenated species, we treated lipid extracts with a mild reducing agent, triphenylphosphine, converting hydroperoxy (OOH) to the respective hydroxy (OH) species (Supplementary Fig. 5d). As illustrated in Fig. 5 the di-oxygenated species of phospholipids in Gpx4 mice were represented by OOH-species and their level was significantly higher in PC (Fig. 5a) and PE (Fig. 5b) on day 10 after Gpx4 depletion.
Figure 5.
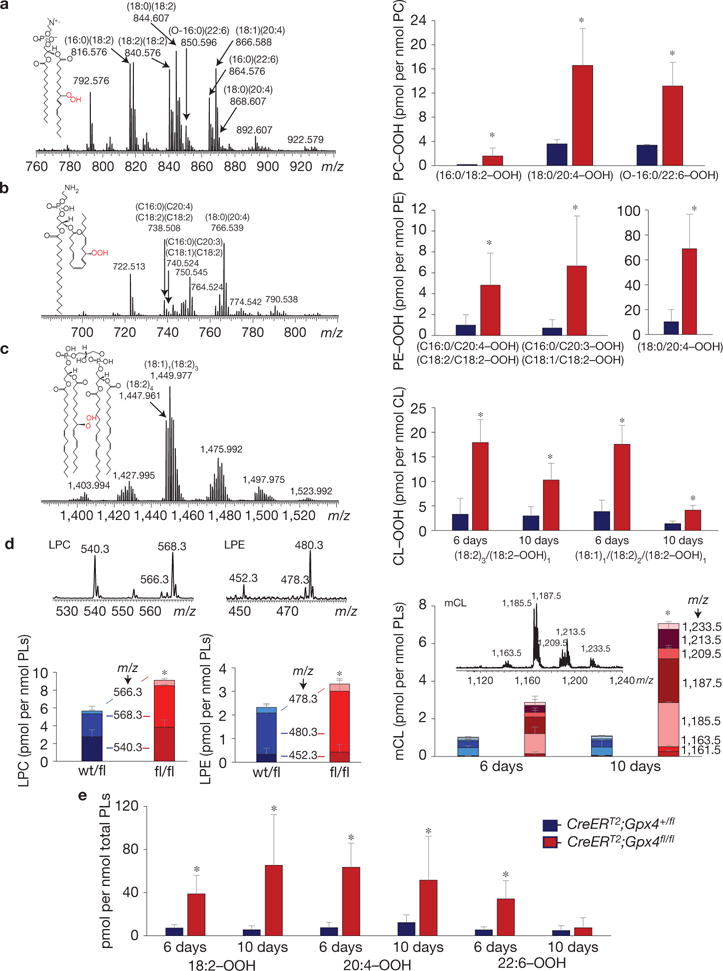
LC–MS characterization of phospholipids and their oxidation products in kidneys of Gpx4-null and wild-type mice. (a) Mass spectrum of PC obtained from kidneys of Gpx4-null mice (left panel) and the levels of hydroperoxy-PC species in Gpx4-null (CreERT2;Gpx4fl/fl) and wild-type (CreERT2;Gpx4+/fl) mice (right panel). Hydroperoxy-PC species (m/z 848.575, 872.575, 876.606, 882.595, 896.575, 898.587 and 900.606) originated from PC species with m/z 816.576 (16:0/18:2), 840.576 (18:2/18:2), 844.607 (18:0/18:2), 850.596 (O-16:0/22:6), 864.576 (16:0/22:6), 866.588 (18:1/20:4) and 868.607 (18:0/20:4). Inset: structural formula of PC–OOH—(16:0/18:2–OOH). (b) Mass spectrum of PE obtained from kidney of Gpx4−/− mice and the content of hydroperoxy-PE species in Gpx4-null mice (right panel). Oxygenated species (m/z 770.507 and 772.522 and m/z 798.538) originated from major PE molecular species with m/z 738.508 (18:2/18:2 or 16:0/20:4), 740.524 (18:1/18:2 or 16:0/20:3) and 766.539 (18:0/20:4). Inset: structural formula of PE–OOH containing hydroperoxy-arachidonic acid—(18:0/20:4–OOH). (c) Mass spectrum of CL obtained from kidney of Gpx4−/− mice (left panel) and levels of hydroperoxy-CLs (m/z 1,479.932 and m/z 1,481.975) originating from species with m/z 1,447.961 (18:2)4 and 1,449.977 18:1/(18:2)3, respectively (right panel). Inset: structural formula of CL–OOH containing hydroperoxy-linoleic acid—(18:2)3/18:2–OOH. (d) Mass spectra and levels of lysophospholipids in samples from mouse kidneys. LPC, lysophosphatidylcholine; LPE, lysophosphatidylethanolamine; mCL, mono-lysocardiolipin. (e) Content of hydroperoxy-derivatives of linoleic (18:2–OOH), arachidonic (20:4–OOH) and docosahexaenoic (22:6–OOH) acids in samples from mouse kidneys. Mass spectra of PC, PE and CL were acquired using a QExactive orbitrap mass spectrometer (ThermoFisher Scientific). The resolution was set up at 140,000 corresponding to 5 ppm deviation in m/z measurement. Mass spectra of LPC, LPE and mCL were acquired by an LXQ ion trap mass spectrometer (ThermoFisher Scientific). Consequently, m/z values are presented to three and one decimal places, respectively. PC molecular ions were detected in a negative ionization mode as acetate adducts [M–H+CH3COOH]−. LPC molecular ions were detected in a negative ionization mode as formate adducts [M–H+HCOOH]−. PE and CL as well as LPE and mCL molecular ions were detected in a negative ionization mode as singly charged ions [M–H]−. Data for hydroperoxy-species are presented as picomoles per nanomole of the parental non-oxidized species. Data shown represent the mean ± s.d. of n = 4 animals of a representative experiment performed independently two times, P =0.05 (one-way ANOVA).
Next, we focused on examining OOH-derivatives of a mitochondria-specific phospholipid, CL, that is recognized as a preferred Gpx4 substrate23. A significantly higher content of CL–OOH species was detected in Gpx4−/− kidneys on day 6 compared with day 10 (Fig. 5c). In addition, for both of these time points the level of CL–OOH was significantly higher compared with control mice. Moreover, we found that on both days, Gpx4-null mice accumulated more mono-lyso-CL (mCL) (Fig. 5c) as well as hydroperoxy-molecular species of free fatty acids, including linoleic, arachidonic and docosahexaenoic acid (Fig. 5e). Significant changes in accumulation of lyso-PC (LPC) and lyso-PE (LPE) in kidneys of Gpx4−/− mice were detectable only on day 10 (Fig. 5d).
As oxidized PLs are preferred substrates for cytosolic and mitochondrial aiPLA2 (ref. 24), which can be inhibited by MJ33 (Fig. 1b), we speculate that the remarkable accumulation of mCL might account for the disrupted morphological changes observed on Gpx4 disruption (Fig. 3d). Moreover, because the mito-targeted redox sensors and antioxidant MitoQ localize to the matrix and the inner leaflet of the mitochondrial inner membrane and because of the finding that Gpx4 resides in the mitochondrial inner membrane space25, we propose a yet-unrecognized oxidation event that may occur at specific submitochondrial locations outside the matrix, which is supported by ultrastructural analysis of cells treated with RSL3 (Fig. 3d). In sum, these data demonstrate that distinctive lipid hydroperoxides accumulate in tissues that are centrally involved in ferroptosis induced by Gpx4 deletion.
A spiroquinoxalinamine derivative inhibits ferroptosis in the kidney
Considering the apparently high modularity of ferroptosis, we sought to identify small-molecule inhibitors with potent anti-ferroptotic properties. We took advantage of the correlation between the in vitro and in vivo phenotype of Gpx4−/− cells and mice, which permitted us to immediately test the in vivo efficacy of screened molecules. We conducted a screening campaign, testing over 40,000 drug-like small molecules in TAM-inducible Gpx4−/− MEFs. This resulted in the discovery of a yet-unrecognized class of spiroquinoxalinamine derivatives, including Liproxstatin-1 (Fig. 6a), which is able to inhibit ferroptosis in the low nanomolar range (Fig. 6b). Liproxstatin-1 prevented BODIPY 581/591 C11 oxidation in Gpx4−/− cells (Fig. 6c). Moreover, Liproxstatin-1 did not interfere with other classical types of cell death, such as TNFα-induced apoptosis and H2O2-induced necrosis (Fig. 6d), and in the bona fide L929 model of TNFα/zvad-induced necroptosis (Fig. 6e). In addition, testing Liproxstatin-1 against a panel of different cell death inducers unrelated to ferroptosis confirmed the specific anti-ferroptotic activity of Liproxstatin-1 (Supplementary Fig. 6). In stark contrast, it showed remarkably potent and specific anti-ferroptotic activity against FIN-triggered cell death (Fig. 6e).
Figure 6.
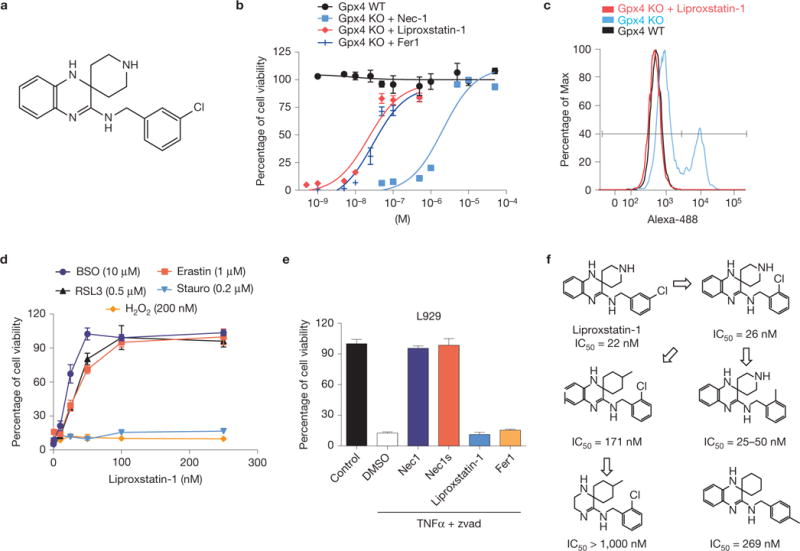
Identification and characterization of ferroptosis inhibitors with in vivo efficacy. (a) Chemical structure of Liproxstatin-1. (b) Dose-dependent rescue of ferroptosis by Liproxstatin-1 in Gxp4−/− cells. On Gpx4 disruption in Pfa1 cells by TAM in the presence of Liproxstatin-1, cell viability was determined 72 h after knockout induction. (c) Liproxstatin-1 (50 nM) completely prevents lipid peroxidation in Gpx4−/− cells. Lipid peroxidation was assessed 48 h after knockout induction using the redox-sensitive dye BODIPY 581/591 C11. (d) Specificity of Liproxstatin-1 towards ferroptosis-inducing triggers. Liproxstatin-1 (200 nM) protects against FINs, such as BSO (10 μM), erastin (1 μM) and RSL3 (0.5 μM), in a dose-dependent manner, whereas it fails to rescue cell death induced by staurosporine (Stauro; 0.2 μM) and H2O2 (200 μM); cell viability was assessed 24 h after treatment. (e) Liproxstatin-1 does not prevent necroptosis. Two hours before triggering necroptosis, L929 cells were treated with Nec1 (10 μM), Nec1s (10 μM), Liproxstatin-1 (1 μM) and Fer1 (1 μM). Necroptosis was induced in L929 cells by a combination of TNFα (5 ng ml−1) and Z-VAD-FMK (50 μM) and cell viability was determined 8 h thereafter using AquaBluer. Data shown represent the mean ± s.d. of n = 4 wells (b,e) or n = 3 wells (d) of a 96-well plate from a representative experiment performed independently at least four times. (f) An initial SAR analysis including IC50 values of Liproxstatin-1 derivatives is shown. IC50 values depicted were calculated from experiments performed with inducible Gpx4−/− cells in 1 well of a 96-well plate, pooled from n=4.
To establish an initial structure–activity relationship study (SAR), we obtained a handful of commercially available analogues of Liproxstatin-1. The compound structures and corresponding IC50 values are depicted in Fig. 6f. In addition, we assessed important ADME (absorption, distribution, metabolism and excretion) parameters (Table 1), indicating a very promising pharmacokinetic profile for Liproxstatin-1. In summary, the initial SAR results provide the rationale for further improvement of Liproxstatin-1 by medicinal chemistry.
Table 1.
ADME-Tox profiling of Liproxstatin-1.
| Cytotoxicity in HepG2 cells | TC50 (μM) | 6 |
| hERG patch clamp assay | IC50 (μM) | >25 |
| CYP450 inhibition | 2C9 IC50 (μM) | >25 |
| 2D6 IC50 (μM) | =4.1 | |
| 3A4 IC50 (μM) | >25 | |
| 1A IC50 (μM) | >25 | |
| 2C19 IC50 (μM) | >25 | |
| Mouse PK (i.v.) 1 mg kg−1 | t1/2 (h) | 4.6 |
| V (ml) | 849 | |
| C (μg ml−1) | 0.4 | |
| Total Cl (ml h−1) | 128 | |
| Total Cl (ml min−1 kg−1) | 66 | |
| MRTINF (h) | 4.9 | |
| Vss (ml) | 629 | |
| Mouse PK (p.o.) 5 mg kg−1 | Cmax (μg ml−1) | 0.08 |
| Tmax (h) | 2 | |
| Bioavailability | (%) | 52 |
An initial ADME-Tox profile for Liproxstatin-1 is provided, including cytotoxicity in HepG2 cells and inhibition of hERG and cytochrome P450 (CYP450) enzymes, as well as basic pharmacokinetic parameters. hERG, ether-à-go-go-related gene; ADME-Tox, absorption, distribution, metabolism and excretion—toxicity; PK, pharmacokinetics; Cl, clearance; MRTINF, mean residence time extrapolated to infinity.
Liproxstatin-1 suppresses ferroptosis in human cells, Gpx4−/− kidney and in an ischaemia/reperfusion-induced tissue injury model
To assess the relevance of these findings in human biology, primary human renal proximal tubule epithelial cells (HRPTEpiCs) were treated with RSL3. As shown in Fig. 7a, RSL3 efficiently triggered cell death in HRPTEpiCs, whereas the inactive analogue ((1R,3R)-RSL3) did not. The specificity of RSL3 was confirmed by Gpx4 overexpression (Supplementary Fig. 7a,d). Supporting the involvement of ferroptosis, treatment with Liproxstatin-1 was able to protect HRPTEpiCs from RSL3-induced cell death (Fig. 7a). Similar findings were obtained in the immortalized human renal proximal tubule epithelial cell line, HK-2 (Supplementary Fig. 7b). Next, we knocked down Gpx4 in HK-2 cells using a pool of siRNAs, revealing a small yet significant decrease in cell viability sensitive to αToc treatment (Supplementary Fig. 7c). Inducing cell death through Gpx4 knockdown, however, turned out to be challenging for the high expression levels of Gpx4 in kidney tubular epithelial cells (Supplementary Fig. 7d). Nonetheless, the Gpx4 knockdown rendered cells more sensitive to ferroptosis-inducing agents (Supplementary Fig. 7e), indicating a Gpx4-regulated ferroptotic machinery in human proximal tubular epithelial cells. Moreover, RSL3-induced BODIPY 581/591 C11 oxidation could be blocked by Liproxstatin-1 (Fig. 7b), demonstrating that Liproxstatin-1 prevents ferroptotic cell death also in humans.
Figure 7.
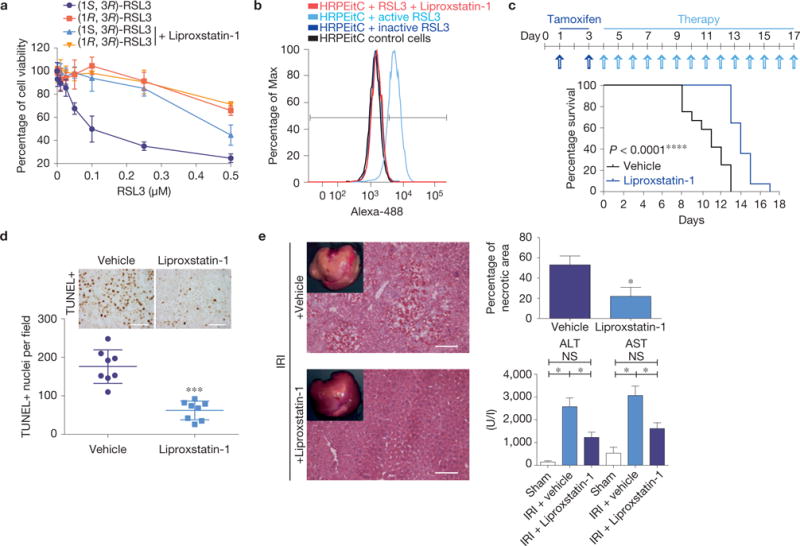
Ferroptosis in human cells and in murine disease models can be targeted by Liproxstatin-1. (a) HRPTEpiCs are susceptible to Gpx4-inhibition-induced ferroptosis, which can be prevented by Liproxstatin-1 (100 nM). Dose-dependent killing of HRPTEpiCs by active (1S,3R)-RSL3 in contrast to inactive (1R,3R)-RSL3. Viability was assessed 24 h after treatment using AquaBluer. Data shown represent the mean ± s.d. of n = 3 wells of a 96-well plate from a representative experiment performed independently three times. (b) Liproxstatin-1 prevents RSL3 (0.2 μM)-induced lipid oxidation in HRPTEpiCs. Lipid peroxidation was assessed 24 h after knockout induction using the redox-sensitive dye BODIPY 581/591 C11. A representative experiment is shown performed independently four times. (c) Liproxstatin-1 retards ARF and death of mice induced by Gpx4 deletion; median survival was calculated to be 11 days for vehicle-treated (n = 12) and 14 days for Liproxstatin-1-treated mice (n = 13), Gehan–Breslow–Wilcoxon test: P <0.0001. Representative experiment shown was performed two times. Mice were injected daily with Liproxstatin-1 (10 mg kg−1, i.p.) during the course of the experiment. (d) Quantification of TUNEL cells in kidneys of vehicle- and Liproxstatin-1-treated animals at 9 +days after TAM administration. Data shown represent the mean ± s.d. of n = 4 comparable anatomical sections from a representative experiment performed two times (scale bars 50 μm). (e) The extent of tissue injury on transient ischaemia/reperfusion in liver of C57BL/6J mice can be ameliorated by the ferroptosis inhibitor Liproxstatin-1 as measured by AST/ALT (n = 17) for vehicle and for Liproxstatin-1 each) and by determining the necrotic area (n = 5). Data represent the mean ± s.e.m.; ∗P = 0.05 or ∗∗∗P = 0.001 (one-way ANOVA) followed by Dunnett’s post-test.
Next, we assessed the in vivo potential of Liproxstatin-1 to prevent the consequences of inducible Gpx4 disruption in animals. On TAM treatment of CreERT2;Gpx4fl/fl mice, mice were injected daily with Liproxstatin-1 intraperitoneally (i.p.) until the mice showed signs of acute renal failure (ARF), at which point they were euthanized (Fig. 7c). Notably, Liproxstatin-1 remarkably extended survival compared with the vehicle-treated group. TUNEL staining at day 9 after TAM treatment showed a strongly reduced number of TUNEL+ cells in Liproxstatin-1 compared with the vehicle-treated group (Fig. 7d), suggesting that Liproxstatin-1 delays ferroptosis in tubular cells. The discrepancy between death of mice due to ARF in Fig. 1c and vehicle-treated animals in Fig. 7c is explained by the mode of TAM administration, feeding versus i.p. injection.
As an independent proof-of-concept, we analysed the in vivo efficacy of Liproxstatin-1 in a bona fide model of hepatic ischaemia/reperfusion injury, providing evidence that Liproxstatin-1 mitigated tissue injury in ischaemia/reperfusion-induced liver injury (Fig. 7e). Hence, these data implicate ferroptosis as a contributor in ischaemia/reperfusion-induced tissue injury and hold great promise for the development of therapeutics to treat related pathologies.
DISCUSSION
Gpx4 is one of the most important antioxidant enzymes in mammals11,26,27. This vital role is mainly credited to its unique function as the only known enzyme able to reduce phospholipid hydroperoxides28. After having shown that loss of Alox15 alone cannot rescue ARF and cell death induced by Gpx4 inactivation, we provide here a more intricate picture where other lipoxygenase isoforms may compensate for Alox15 deficiency. Although sources of reactive oxygen species other than lipoxygenases, such as NADPH oxidases, have been implicated, their contribution seems to be cell type dependent, whereas lipid-oxidizing enzymes may confer a more general impact6,7. In addition, we introduce the release of fatty acids through aiPLA2 and its trafficking possibly through SCP-2 as downstream events involved in ferroptosis execution.
A direct measurement of PL oxidation products by LC–MS analysis in vivo showed higher levels of di-oxygenated species of PC, PE and CL in the kidneys of Gpx4−/− mice versus control animals, indicating that not only extra-mitochondrial compartments but also mitochondria were involved in the deregulated lipid peroxidation caused by Gpx4 deficiency. Yet, rescue of Gpx4−/− cells with mitochondria-targeted antioxidants was far less efficient than with untargeted ones, arguing that the priming lethal lipid signal must be generated outside the mitochondrial matrix.
In accordance with a role for ferroptosis in vivo, we now demonstrate that proper kidney function depends on a functional Gpx4 protein, as the inducible Gpx4 depletion leads to massive cell death of renal tubular epithelia. Interestingly, a recent report using inducible ferritin heavy chain (FtH)-knockout mice in kidney presented a similar phenotype on different challenges that is dependent on a non-haem iron mechanism29, thus highlighting the importance of restraining lipid peroxides and iron in tubular cells as these are expected to fuel the ferroptotic pathway and culminate in ARF. It is worth mentioning that in the FtH-null mouse, no damage was observed under basal conditions, indicating that pure iron overload is not sufficient to elicit ferroptosis. However, the priming effect would be an increase in lipid hydroperoxides to an extent that iron would work only as a catalyst. Nonetheless, we point out that although the iron chelator DFO prevented cell death induced by Gpx4 loss in vitro, the iron dependence of renal cell death in the inducible Gpx4−/− model remains to be formally shown in vivo.
As we provide evidence that ferroptosis plays an important role in ischaemia/reperfusion-induced tissue damage, targeting other non-apoptotic pathways might help pave the way towards improved protection against ischaemia30. It is therefore intriguing to consider how ferroptosis is functionally intertwined with other types of cell death4. We also highlight that because Nec1 has been used previously to define necroptotic cell death, but can also protect against ferroptosis through an unknown target, many earlier studies may now be suspected to encompass ferroptotic cell death. Hence, the finding that the ferroptotic signalling cascade is amenable to in vivo pharmacological intervention (Fig. 7) opens new possibilities for redox-targeted therapies. Last, this study provides the rationale for future investigations concerning the role of how lipid-derived electrophiles translate their message to redox-sensitive proteins triggering cellular demise31.
METHODS
Methods and any associated references are available in the online version of the paper.
METHODS
Chemicals
All chemicals were purchased from Sigma-Aldrich unless stated otherwise. MitoQ was kindly provided by M. P. Murphy (Cambridge, UK). The collection of screening compounds was purchased from ChemDiv, ChemBridge and Enamine. Necrostatin-1 was obtained from Enzo Life Sciences GmbH.
Cell lines
The TAM-inducible Gpx4−/− cells (Pfa1) were described previously11. Rip1-knockout cells were generously provided by J. Silke (Melbourne, Australia)32. Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Human renal proximal tubular epithelial cells (HRPTEpiCs; no. 4100) were purchased from ScienCell and cultured according to the manufacturer’s recommendations. HK-2 cells were cultured in defined keratinocyte-SFM-supplemented bovine pituitary extract and recombinant epidermal growth factor according to ATCC.
Cell viability assays and inhibitor studies
To induce the knockout of Gpx4, cells were seeded onto 96-well plates (1,000 cells per well) and treated with 1 μM 4–OH-tamoxifen (TAM) after plating. Cell viability was assessed at different time points after treatment (usually 72 h, unless stated otherwise) using AquaBluer, unless stated otherwise, as an indicator of viable cells according to the manufacturer’s recommendations (MultiTarget Pharmaceuticals, LLC). Alternatively, cell death was also quantified by measuring released lactate dehydrogenase (LDH) activity using the Cytotoxicity Detection Kit (LDH) according to the manufacturer’s instructions (Roche Diagnostics Deutschland).
Immunoblotting
Western blot analysis of cell as well as tissue extracts was performed essentially as described previously11,33, by using antibodies against Gpx4 (monoclonal rat antibody 1B4 (ref. 11) or 1:1,000, no. ab125066, Abcam), Rip3 (1:1,000, no. hGTX107574, Genetex), Rip1 (1:1,000, no. 3493, Cell Signaling Technologies) and ß-actin (1:1,000, no. A5441, Sigma).
Inducible disruption of Gpx4 in mice
The generation of mice with loxP-flanked (floxed, fl) Gpx4 alleles (Gpx4tm2Marc, in the following referred to as Gpx4fl/fl) was previously outlined in detail11. To bypass early embryonic lethality of Gpx4−/− mice, we took advantage of the TAM-inducible ROSA26–CreERT2 deleter strain (in the following referred to as CreERT2) generously provided by A. Berns (The Netherlands Cancer Institute, Amsterdam, Holland)34. These mice carry a conditional Cre recombinase (CreERT2) allele targeted to the ubiquitously expressed ROSA26 locus. The oestrogen receptor T2 (ERT2) moiety fused to Cre retains the recombinase in the cytosol. Tamoxifen administration releases this inhibition, thus permitting inducible recombination of loxP-flanked alleles. Gpx4fl/fl were first crossed with CreERT2 deleter mice to obtain CreERT2;Gpx4+/fl mice. CreERT2;Gpx4+/fl were then mated to Gpx4fl/fl mice yielding CreERT2;Gpx4fl/fl mice. CreERT2;Gpx4wt/fl mice did not show any pathological alterations and therefore served as controls. To achieve inducible disruption of the loxP-flanked Gpx4 allele(s), mice (>8 weeks of age; male and female) were fed a diet containing tamoxifen citrate as described previously35 (n=19).
Generation of ALOX15/Gpx4 double-knockout mice
To generate Alox15/Gpx4 compound mutant mice, Alox15−/− (Alox15tm1Fun) mice (The Jackson Laboratory) were first crossed with CreERT2;Gpx4fl/fl mice to generate CreERT2;Gpx4+/fl/Alox15+/− mice. Subsequently, CreERT2;Gpx4+/fl/Alox15+/− were mated with Gpx4+/fl/Alox15+/− mice to obtain CreERT2;Gpx4+/fl/Alox15−/− and Gpx4fl/fl/Alox15−/− mice. Finally, intercrossing of CreERT2;Gpx4+/fl/Alox15−/− mice with Gpx4fl/fl/Alox15−/− mice yielded CreERT2;Gpx4fl/fl/Alox15−/− mice. Disruption of the floxed Gpx4 alleles was achieved by feeding mice (>8 weeks of age; male and female) with a diet containing tamoxifen citrate35 (n=8).
Isolation of CreERT2;Gpx4fl/fl/Alox15−/− and CreERT2;Gpx4fl/fl/Alox15+/+ cells
CreERT2;Gpx4fl/fl/Alox15−/− and CreERT2;Gpx4fl/fl/Alox15+/+ mice were euthanized by cervical translocation. After dissection, lungs were minced and directly plated in standard DMEM containing 10% FBS and antibiotics as described previously11. CreERT2;Gpx4fl/fl/Alox15−/− (PZL cells) and CreERT2;Gpx4fl/fl/Alox15+/+ cells (PZ cells) were incubated at 5% oxygen and 5% CO2 in a water-saturated atmosphere for at least 20 passages to obtain spontaneously immortalized lung fibroblasts36.
Histological analysis
Kidneys were removed, trimmed free of adjacent tissues and weighed to the nearest 0.1 mg. The kidneys were cut into parallel slices perpendicular to the longitudinal axis, three 1 mm3 cubes were taken from the kidney cortex and embedded in Epon for transmission electron microscopy, and the remaining tissue was embedded in paraffin or plastic for light microscopy as described previously37.
Immunohistochemistry
Immunostaining was performed using 5-μm-thick sections of 4% paraformaldehyde-fixed paraffin-embedded tissue samples or using 10-μm-thick sections of snap-frozen tissue samples. The following primary antibodies were used on cryosections: rabbit anti-phospho-histone H3 antibody (1:200; no. 9701; Cell Signaling Technology, New England Biolabs GmbH) for proliferating cells, rat anti-F4/80 antibody (1:1,000; BM4007; Acris Antibodies GmbH) for macrophages and rabbit anti-Cleaved Caspase-3 antibody (1:200; no. 9664; Cell Signaling Technology, New England Biolabs GmbH) for detection of apoptotic nuclei. Immunohistochemical staining for Gpx4 expression was performed on paraffin sections using a rabbit anti-Gpx4 antibody (1:250; no. 3649-1; Epitomics). As secondary antibodies a biotinylated rabbit anti-rat IgG (1:200; BA-4000; Vector Laboratories, Linaris, Wertheim-Bettingen) and a biotinylated goat anti-rabbit IgG (1:200; BA-1000; Vector Laboratories) were used. Staining was performed using the Vector ABC kit and Vector DAB kit (Vector Laboratories).
Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labelling (TUNEL)
For the detection of TUNEL-positive cells, the ApopTag Peroxidase In Situ Apoptosis Detection Kit was used according to the manufacturer’s instructions (S7100; Serologicals, Millipore).
Measurement of clinical parameters
Serum samples were analysed for chloride, creatinine and urea using the Architect ci8200 Autoanalyser (Abbott) and reagents supplied by Abbott. The measurements were kindly performed at the City Clinic Munich in Schwabing, Munich, Germany.
Staining for lipid peroxidation in cells by flow cytometry
Cells were plated in 10 cm dishes in DMEM supplemented with 10% FBS and treated for defined times. After incubation, cells were washed and BODIPY 581/591 C11 (Invitrogen) or mito-BODIPY 581/591 C11 (kind gift from M. P. Murphy) was added for 1 h for analysis by flow cytometry. Cells were trypsinized, washed and transferred to FACS tubes in HBSS containing 2% FBS. Flow cytometry was performed using the BD FACS Canto II.
Synthesis of RSL3
Synthesis and characterization of RSL3 stereoisomers were performed according to ref. 8 with modifications for the installation of the chloroacetamide moiety as follows. The corresponding RSL3 intermediate (0.4 g,1.1 mmol, 1.0 equivalent) was dissolved in 5 ml of anhydrous CH2Cl2 and triethylamine (0.17 ml, 1.2 mmol, 1.1 equivalent) was added. The reaction mixture was cooled to 0 °C, after which a 0 °C solution of chloroacetyl chloride (0.1 ml, 1.2 mmol, 1.1 equiv.) in 1.0 ml CH2Cl2 was added dropwise. After 1 h at 0 °C, the reaction was quenched with aqueous saturated NH4Cl. The organic phase was separated, and the aqueous phase was extracted with 3×10 ml CH2Cl2. The organic extracts were combined and washed with 30 ml H2O and 30 ml brine, dried with Na2SO4 and then concentrated to give the crude product as a yellow oil. The compound was purified by dry-loaded silica gel chromatography in a gradient elution from 100% hexane to 50:50 ethyl acetate/hexane. An identical protocol was used to obtain the chloroacetamide derivative of the other three diastereomers. The four diastereomers were obtained in 80 to 90% yield.
Transmission electron microscopy of cells
Cell pellets were fixed in 2.5% electron microscopy grade glutaraldehyde in 0.1 M sodium cacodylate buffer pH 7.4 (Science Services), postfixed in 2% aqueous osmium tetraoxide, dehydrated in gradual ethanol (30–100%) and propylene oxide, embedded in Epon (Merck) and cured for 24 h at 60 °C. Semithin sections were cut and stained with toluidine blue. Ultrathin sections of 50 nm were collected onto 200 mesh copper grids, and stained with uranyl acetate and lead citrate before examination by transmission electron microscopy (Zeiss Libra 120 Plus, Carl Zeiss NTS GmbH). Pictures were acquired using a Slow Scan CCD (charge-coupled device) camera and iTEM software (Olympus Soft Imaging Solutions).
siRNA and transfection protocol
Cells were plated at 25,000 cells per well in a 6-well plate and transfected with Hyperfect according to the manufacturer’s recommendations (Qiagen). The cells were kept in the transfection mixture until the end of the assay. siRNAs for Ripk1, Ripk3 and Scp2 were purchased from Qiagen (no. GS19766, no. GS56532 and no. GS20280, respectively). Gpx4 siRNA was obtained from On-Target Plus SMARTpool human GPx4 siRNA (no. L-011676-00-0005, Thermo Scientific Dharmacon).
Determination of intracellular GSH levels
Cells were seeded on a 6-well plate (1 × 105 cells per well) and cultured for 24 h in the routine culture medium. After treatments with different inhibitors, cells were washed three times with ice-cold PBS, extracted with 5% trichloroacetic acid, and then treated with ether to remove the trichloroacetic acid. Total glutathione content in the aqueous layer was measured using an enzymatic method, based on the catalytic action of glutathione in the reduction of 5,5′-dithiobis (2-nitrobenzoic acid) by the glutathione reductase system38.
Measurement of hydroxyeicosatetraenoic acids by LC–MS/MS
To determine the amount of HETEs released into the medium on Gpx4 deletion, cells were seeded on 6-well dishes with or without treatment. Supernatants were collected at the indicated time points after treatment and analysed for the presence of arachidonic acid oxidation products.
For lipid extraction, PGE2-d4 and 15-HETE-d8 (5 ng) were added to each sample before extraction as an internal standard. Samples were also spiked with butylated hydroxytolulene and DTPA (100 μM final concentration in samples). Lipids were then extracted by adding a solvent mixture (1 M acetic acid, 2-propanol, hexane (2:20:30)) to the sample at a ratio of 2.5 ml solvent mixture to 1 ml sample, vortexing and then adding 2.5 ml of hexane. Following vortexing and centrifugation (1,000g, 5 min), lipids were recovered in the upper hexane layer. The samples were then re-extracted by the addition of an equal volume of hexane followed by further vortexing and centrifugation. The combined hexane layers were then dried under vacuum and analysed for 5-, 8-, 11-, 12- and 15-HETE using LC–MS/MS (as described below).
For free eicosanoid quantification using LC–MS/MS, samples were separated on a C18 Spherisorb ODS2, 5 μm, 150 × 4.6-mm column (Waters) using a gradient of 50–90% B over 20 min (A, water/acetonitrile/acetic acid, 75:25:0.1; B, methanol/acetonitrile/acetic acid, 60:40:0.1) with a flow rate of 1 ml min−1. Products were quantified by LC–MS/MS electrospray ionization on an Applied Biosystems 4000 Q-Trap using parent-to-daughter transitions of m/z 319.2 (HETE, [M-H]−) to m/z 115.1 (5-HETE), 155.1 (8-HETE), 167.1 (11-HETE), 179.1 (12-HETE), 219.1 (15-HETE) and m/z 327.2 to 226.2 for 15-HETE-d8, with collision energies of −20 to −30 V. Products were identified and quantified using synthetic lipid standards run in parallel under the same conditions. Acquisition of product ion spectra was triggered during elution of ions of interest, with the instrument operating in ion trap mode.
Oxidative lipidomics
Phospholipids/oxidized phospholipids were characterized after 2D-HPTLC separation39, followed by LC–MS analysis as described previously40. To assess phospholipid hydroperoxides and fatty acid hydroperoxides, lipid extracts were treated with a reductant, triphenylphosphine (10 mg ml−1, for 20 min at 4 °C in methanol)41, converting hydroperoxy- into respective hydroxy-species42. All mass spectra were acquired in data-dependent negative ionization mode using an inclusion list for CL, PC and PE species, isolation width of 1.0 Da and high-energy collision of 24. The peaks with a signal-to-noise ratio of 3 and higher were taken into consideration. Isotopic correction was performed by entering the chemical composition of each species into the Qual browser of Xcalibur (operating system). PC and LPC molecular ions were detected as acetate adducts and formate adducts. Other phospholipids were detected as singly charged ions. Mass spectra of PC, PE and CL were acquired using a QExactive orbitrap mass spectrometer (ThermoFisher Scientific), whereby the resolution was set up at 140,000 with 5 ppm error. The mass spectra of LPC, LPE and mCL were acquired using an LXQ ion trap mass spectrometer (ThermoFisher Scientific). Thus, the m/z values are presented to three and one decimal places, respectively. Standards for phospholipids and oxygenated fatty acids were from Avanti Polar Lipids and Cayman Chemicals. Standards of mCLs were prepared as described previously43.
Phenotypic screening for ferroptosis inhibitors
In brief, compound seeding onto 96-well plates (1,000 cells per well) was carried out simultaneously with 1 μM TAM administration (leading to Gpx4 inactivation) obviating multiple medium changes, followed by incubation for 72 h. Cell viability was assessed subsequently using the live/dead assay dye AquaBluer. In the primary screening round, all compounds were tested at a single concentration of 10 μM and positive hits were selected from wells with >80% cell viability. To confirm primary hits, compounds were re-screened in the same assay and dose-dependent survival as well as toxicity curves were obtained using concentrations of 0–100 μM. IC50 and TC50 values were calculated using the GraphPad Prism software. Validated hits were then evaluated based on efficacy, selectivity for ferroptosis, therapeutic range and physicochemical properties. In addition, an in silico ADME-Tox screening was implemented to exclude compounds with potential in vivo side effects. To further validate Liproxstatin-1, SAR studies were performed using commercially available derivatives (ChemDiv).
Cytochrome P450 inhibition, hERG channel inhibition and pharmacokinetics studies
All experiments were performed by Cyprotex Discovery, according to established protocols.
HepG2 toxicity measurement
Cells were incubated with increasing concentrations of compound for 72 h and cell viability was assessed after treatment using AquaBluer as an indicator of viable cells according to the manufacturer’s recommendations. TC50 values were calculated using the GraphPad Prism software.
Animal experiments (in vivo rescue experiment)
Animals included in the treatment study of inducible Gpx4−/− mice were equally distributed between sex and weight, with typically 8–10 weeks of age. The average weight within the groups was between 22 and 24 g. Groups were formed to have comparable numbers of females/males of the same age. Animal weight was arranged to have a similar distribution between females and males. For the pharmacological inhibitor experiments, CreERT2;Gpx4fl/fl mice were injected on day 1 and 3 with 0.5 mg TAM dissolved in Miglyol. On day 4, compound treatment was started (Liproxstatin-1: 10 mg kg−1) along with vehicle control (1% dimethylsulphoxide (DMSO) in PBS). Liproxstatin-1 and vehicle control were administered once daily by i.p. injection. Survival analysis was performed using the GraphPad Prism software and statistical analysis was done according to the log-rank (Mantel–Cox) test. The compounds, vehicle and Liproxstatin-1, were both odourless and colourless ensuring no detectable bias. Injections and daily animal assessment were performed in a blinded fashion. When animals showed terminal signs, they were euthanized. No statistical method was used to predetermine sample size for the treatment of the Gpx4−/− mice. Mice were kept under standard conditions with food and water ad libitum (ssniff). All experiments were performed in compliance with the German Animal Welfare Law and have been approved by the institutional committee on animal experimentation and the government of Upper Bavaria.
Animal experiments (liver ischaemia/reperfusion)
Animals (C57BL/6J) for the liver ischaemia/reperfusion study were male mice between 8 and 10 weeks of age, typically between 22 and 24 g and purchased from Charles River. The animals were randomly distributed. Investigators performing the injections/surgeries were blinded. All animals were euthanized 24 h following transient ischaemia/reperfusion. The surgical procedure was performed as described elsewhere44. In brief, C57BL/6J mice were narcotized with xylazine/ketamine and shaved at their front. After opening the abdominal cavity an atraumatic clip was placed across the portal vein, hepatic artery, and bile duct, just above branching to the right lateral lobe. After 90 min of ischaemia, the clamp was removed and the liver was reperfused. Blood and tissues were collected from anaesthetized animals after 24 h. Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured using a Dimension 1500 Vista Analyzer.
Data presentation and statistical analyses
Data are presented as mean ± s.d. unless stated otherwise. As a general rule for the cell-based experiments, the graphs show the mean ± s.d. of n = x wells (x values are given in the figure legends) representative of a single experiment performed independently y times (y value is given in figure legends) for reproducibility. Statistical analysis was performed using GraphPad Prism 5.0 software.
Supplementary Material
Acknowledgments
We are grateful to D. Green for his valuable comments and critical reading of the manuscript. We would also like to thank A. Berns, The Netherlands Cancer Institute, Amsterdam, for providing the ROSA26-CreERT2 mouse line, J. Silke (The Walter and Eliza Hall Institute, Melbourne, Australia) for the Rip1-knockout cells and M. P. Murphy (Medical Research Council, London, UK) for provision of MitoQ and Mito-BODIPY. We are most grateful to L. Pichl for excellent technical assistance. This work was in part supported by the Deutsche Forschungsgemeinschaft (DFG) CO 291/2-3 to M.C., a fellowship from Alexander von Humboldt-Stiftung to J.P.F.A., a fellowship from the Japan Society for the Promotion of Science (JSPS) to S.K., the Human Frontier Science Program (HFSP) RGP0013 to M.C. and V.E.K., HL114453 (NIH) to V.E.K., and by the m4 Award (Bavarian Ministry of Economic Affairs) to M.C. and J.A.S.
Footnotes
Note: Supplementary Information is available in the online version of the paper
AUTHOR CONTRIBUTIONS
J.P.F.A. and M.C. conceived the study. Electron microscopy studies were carried out and analysed by M.A. and A.W. Eicosanoid release and data analysis were performed by V.J.H. and V.B.O. Oxi-lipidomics analysis and interpretation was performed by Y.Y.T., V.A.T. and V.E.K. Optimized RSL3 synthesis was conceived and carried out by S.B.T. and B.R.S. Liver ischaemia/reperfusion experiments were performed and analysed by E.E. and E.K.G. Gpx4/Alox15 double-knockout generation and histopathological analysis was conceived and performed by G.W.B., N.H., H.B., F.N., I.E. and R.W. Small-molecule screen was conceived and analysed by B.P., J.A.S. and M.C. Experiments were performed by J.P.F.A., M.S., N.H., D.B., S.K., T.S., H.F., O.Y. and M.H. Evaluation and interpretation of the in vitro data was performed by J.P.F.A., O.R. and M.C. Paper was written by J.P.F.A., V.E.K. and M.C. All authors read and commented on the paper.
COMPETING FINANCIAL INTERESTS
The authors declare that a patent application has been filed for some of the compounds described in this work.
References
- 1.Galluzzi L, et al. Molecular definitions of cell death subroutines: recommendations of the nomenclature committee on cell death 2012. Cell Death Differ. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol. 2013;14:727–736. doi: 10.1038/nrm3683. [DOI] [PubMed] [Google Scholar]
- 3.Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov. 2012;11:109–124. doi: 10.1038/nrd3627. [DOI] [PubMed] [Google Scholar]
- 4.Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- 5.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 6.Dixon SJ, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2013;10:9–17. doi: 10.1038/nchembio.1416. [DOI] [PubMed] [Google Scholar]
- 8.Yang WS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skouta R, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc. 2014;136:4551–4556. doi: 10.1021/ja411006a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conrad M, Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (−): cystine supplier and beyond. Amino Acids. 2012;42:231–246. doi: 10.1007/s00726-011-0867-5. [DOI] [PubMed] [Google Scholar]
- 11.Seiler A, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8:237–248. doi: 10.1016/j.cmet.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 12.Sun D, Funk CD. Disruption of 12/15-lipoxygenase expression in peritoneal macrophages. Enhanced utilization of the 5-lipoxygenase pathway and diminished oxidation of low density lipoprotein. J Biol Chem. 1996;271:24055–24062. [PubMed] [Google Scholar]
- 13.Gilbert NC, et al. Conversion of human 5-lipoxygenase to a 15-lipoxygenase by a point mutation to mimic phosphorylation at Serine-663. FASEB J. 2012;26:3222–3229. doi: 10.1096/fj.12-205286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mannes AM, Seiler A, Bosello V, Maiorino M, Conrad M. Cysteine mutant of mammalian GPx4 rescues cell death induced by disruption of the wild-type selenoenzyme. FASEB J. 2011;25:2135–2144. doi: 10.1096/fj.10-177147. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi N, et al. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Disease. 2012;3:e437. doi: 10.1038/cddis.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conrad M, et al. 12/15-lipoxygenase-derived lipid peroxides control receptor tyrosine kinase signaling through oxidation of protein tyrosine phosphatases. Proc Natl Acad Sci USA. 2010;107:15774–15779. doi: 10.1073/pnas.1007909107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kagan VE, et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005;1:223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- 18.Cocheme HM, et al. Mitochondrial targeting of quinones: therapeutic implications. Mitochondrion. 2007;7(suppl):S94–S102. doi: 10.1016/j.mito.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 19.Kriska T, Pilat A, Schmitt JC, Girotti AW. Sterol carrier protein-2 (SCP-2) involvement in cholesterol hydroperoxide cytotoxicity as revealed by SCP-2 inhibitor effects. J Lipid Res. 2010;51:3174–3184. doi: 10.1194/jlr.M008342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vila A, Levchenko VV, Korytowski W, Girotti AW. Sterol carrier protein-2-facilitated intermembrane transfer of cholesterol- and phospholipid-derived hydroperoxides. Biochemistry. 2004;43:12592–12605. doi: 10.1021/bi0491200. [DOI] [PubMed] [Google Scholar]
- 21.Kim MS, Wessely V, Lan Q. Identification of mosquito sterol carrier protein-2 inhibitors. J Lipid Res. 2005;46:650–657. doi: 10.1194/jlr.M400389-JLR200. [DOI] [PubMed] [Google Scholar]
- 22.Tyurina YY, et al. Characterization of cardiolipins and their oxidation products by LC-MS analysis. Chem Phys Lipids. 2014;179:3–10. doi: 10.1016/j.chemphyslip.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nomura K, Imai H, Koumura T, Kobayashi T, Nakagawa Y. Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycaemia-induced apoptosis. Biochem J. 2000;351:183–193. doi: 10.1042/0264-6021:3510183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McIntyre TM. Bioactive oxidatively truncated phospholipids in inflammation and apoptosis: formation, targets, and inactivation. Biochim Biophys Acta. 2012;1818:2456–2464. doi: 10.1016/j.bbamem.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liang H, et al. Short form glutathione peroxidase 4 is the essential isoform required for survival and somatic mitochondrial functions. J Biol Chem. 2009;284:30836–30844. doi: 10.1074/jbc.M109.032839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wortmann M, et al. Combined deficiency in glutathione peroxidase 4 and vitamin e causes multiorgan thrombus formation and early death in mice. Circ Res. 2013;113:408–417. doi: 10.1161/CIRCRESAHA.113.279984. [DOI] [PubMed] [Google Scholar]
- 27.Yant LJ, et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med. 2003;34:496–502. doi: 10.1016/s0891-5849(02)01360-6. [DOI] [PubMed] [Google Scholar]
- 28.Brigelius-Flohe R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta. 2013;1830:3289–3303. doi: 10.1016/j.bbagen.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 29.Zarjou A, et al. Proximal tubule H-ferritin mediates iron trafficking in acute kidney injury. J Clin Invest. 2013;123:4423–4434. doi: 10.1172/JCI67867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Linkermann A, et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci USA. 2013;110:12024–12029. doi: 10.1073/pnas.1305538110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang C, Weerapana E, Blewett MM, Cravatt BF. A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat Methods. 2014;11:79–85. doi: 10.1038/nmeth.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong WW, et al. RIPK1 is not essential for TNFR1-induced activation of NF-κB. Cell Death Differ. 2010;17:482–487. doi: 10.1038/cdd.2009.178. [DOI] [PubMed] [Google Scholar]
- 33.Schneider M, et al. Mitochondrial glutathione peroxidase 4 disruption causes male infertility. FASEB J. 2009;23:3233–3242. doi: 10.1096/fj.09-132795. [DOI] [PubMed] [Google Scholar]
- 34.Hameyer D, et al. Toxicity of ligand-dependent Cre recombinases and generation of a conditional Cre deleter mouse allowing mosaic recombination in peripheral tissues. Physiol Genomics. 2007;31:32–41. doi: 10.1152/physiolgenomics.00019.2007. [DOI] [PubMed] [Google Scholar]
- 35.Kiermayer C, Conrad M, Schneider M, Schmidt J, Brielmeier M. Optimization of spatiotemporal gene inactivation in mouse heart by oral application of tamoxifen citrate. Genesis. 2007;45:11–16. doi: 10.1002/dvg.20244. [DOI] [PubMed] [Google Scholar]
- 36.Parrinello S, et al. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003;5:741–747. doi: 10.1038/ncb1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Herbach N, et al. Diabetic kidney lesions of GIPRdn transgenic mice: podocyte hypertrophy and thickening of the GBM precede glomerular hypertrophy and glomerulosclerosis. Am J Physiol Renal Physiol. 2009;296:F819–F829. doi: 10.1152/ajprenal.90665.2008. [DOI] [PubMed] [Google Scholar]
- 38.Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem. 1969;27:502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- 39.Rouser G, Fkeischer S, Yamamoto A. Two dimensional then layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids. 1970;5:494–496. doi: 10.1007/BF02531316. [DOI] [PubMed] [Google Scholar]
- 40.Tyurina YY, et al. A mitochondrial pathway for biosynthesis of lipid mediators. Nat Chem. 2014;6:542–552. doi: 10.1038/nchem.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka R, Hatate H, Ito M, Nakamura T. Elevation of lipid peroxide level and production of hydroxy lipids in cultured Hepa-T1 cells by oxidative stressors. Fisheries Sci. 2006;72:665–672. [Google Scholar]
- 42.Tyurina YY, Tyurin VA, Epperly MW, Greenberger JS, Kagan VE. Oxidative lipidomics of gamma-irradiation-induced intestinal injury. Free Radic Biol Med. 2008;44:299–314. doi: 10.1016/j.freeradbiomed.2007.08.021. [DOI] [PubMed] [Google Scholar]
- 43.Kim J, Hoppel CL. Monolysocardiolipin: improved preparation with high yield. J Lipid Res. 2011;52:389–392. doi: 10.1194/jlr.D010587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eggenhofer E, et al. Unconventional RORgammat+ T cells drive hepatic ischemia reperfusion injury. J Immunol. 2013;191:480–487. doi: 10.4049/jimmunol.1202975. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.