

Abstract
“Gene amplification causes overexpression” is a longstanding and well-accepted concept in cancer genetics. However, raking the whole literature, we find only statistical analyses showing a positive correlation between gene copy number and expression level, but do not find convincing experimental corroboration for this notion, for most of the amplified oncogenes in cancers. Since an association does not need to be an actual causal relation, in our opinion, this widespread notion still remains a reasonable but unproven assumption awaiting experimental verification.
Keywords: Cancer, gene amplification, overexpression, translocation
INTRODUCTION
Cancers often manifest increased expression of many oncogenes and amplification of some.[1,2,3] Oncogene amplification is found in a broad spectrum of tumors and is considered to play important roles not only in formation of cancer, but also in its later progression to states of metastasis and therapy resistance.[1,4,5] The amplified copy or copies may appear on the same chromosome as the parental alleles, but may also be translocated to other chromosome(s) [Figure 1a] or even to extra-chromosomal acentric elements.[1,4,6]
Figure 1.
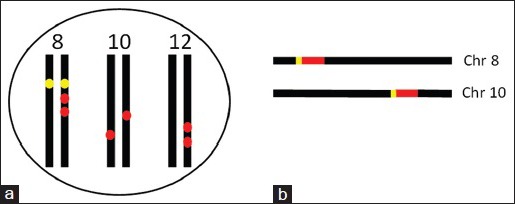
Illustration of the relationship between the parental alleles and the amplified ones of an oncogene. (a) In a cancer cell, a gene that locates at chromosome 8 (yellow dots) has two amplified copies located on one of the two 8th chromosomes, either the paternally or the maternally derived one. Moreover, the gene also has an amplified copy on each of the two 10th chromosomes and two copies on one of the two 12th chromosomes (red dots). Aneuploid chromosomes with amplified oncogenes often appear in cancer, but are not illustrated here. Although the cell shows increased expression of this gene, relatively to the normal cells, it is difficult to pinpoint which one or ones of these eight (two normal and six amplified) alleles are responsible for the excess of the RNA transcripts. (b) If both of the gene (red bar) and its promoter (short yellow bar) at chromosome 8 are amplified and the amplified copy is translocated to one of the 10th chromosomes, the co-amplified promoter may allow the amplified allele to be activated, and thus overexpressed, in a way similar to the activation of the parental allele(s)
Gene expression is known to be regulated not only by various epigenetic changes[7,8,9,10] and regulatory RNAs,[11,12,13,14,15] but also by genetic mechanisms, including gene dose, i.e., copy number of the gene. There are many occasions in which overexpression of one oncogene is not associated with detectable amplification of the gene and thus is likely due to overactivation of the paternally or/and maternally derived alleles of the gene. For instance, in one study of glioblastomas, the MET oncogene is amplified in only 5.1% of cases, but is overexpressed in 13.1% of the cases,[16] indicating that in many cases, the increase in the expression is not due to amplification. Similarly, in a study of esophageal carcinoma, MDM2 copy number is not correlated with its expression level.[17] Overexpression of epidermal growth factor receptor (EGFR) in ependymoma is common and not correlated to gene copy number either.[18] For some genes such as the human epidermal growth factor receptor 2 (HER2),[19] overexpression is more related to gene mutation, and mutation and amplification may occur in different patients. Moreover, gene amplification shows topographic heterogeneity in a given tumor, just like the well-recognized heterogeneity of the expression level. In fact, heterogeneity of gene amplification is omnipresent.[20] However, the copy number and the expression level are often not correlated topographically.[21,22]
There are even some occasions that seem to be counterintuitive, as in which gene amplification is actually associated with decreased expression.[23] One of the possible explanations for this phenomenon is that not only the amplified allele(s) are inactive, but also one or both of the parental alleles are inactivated. In fact, some cancer cases show decreased expression and deletion of some oncogenes, such as decreased c-MYC expression[24] and deletion of the c-MYC oncogene.[25] In our musing, this is reasonable because most genes actually have dual functions, being an oncogene in some situations but being a tumor suppressor gene in the others[26,27,28,29] and because rearrangement is a mechanism to inactivate tumor suppressor genes.[28] EGFR gene is amplified in about 50% of the newly diagnosed cases of glioblastoma, but the amplified allele(s) are often lost during primary culture,[30] suggesting that the excessive copy or copies may no longer be needed for, or may even be detrimental to, the survival of glioblastoma cells in culture and thus are trimmed away. Mechanistically, this occurs likely via DNA recombination during cell division and presumably because the cells are no longer under the selective pressure in the culture,[30] and such removal of the amplified copy or copies is more often seen, and can be much easily verified in bacteria.[31] Therefore, although it is still not quite clear why some cancer cases show decreases in the expression and the gene dose of some oncogenes, it may be related to growth advantage,[3,32,33,34] which may mechanistically vary among cases. It is also possible that the paternally and maternally derived alleles are actually suppressed while the amplified alleles are activated and overexpressed, which may end up with a net result of little change in the expression level.
Relative to the above-described occasions, it is much more often that amplification of an oncogene is associated with increased gene expression,[35] as has been shown by ample statistical analyses on the correlation between the copy number of the gene and its expression level.[36,37] For instance, some studies show that EGFR overexpression is correlated with its amplification in nonsmall-cell lung cancer,[38,39] and a similar correlation is recently reported for the SQLE gene in breast cancer.[40] In fact, most studies conclude that the increase in the expression is ascribed to the gene amplification, i.e., the increase in the gene copy number, making this conclusion a longstanding and well-accepted concept in cancer genetics.[36,37,41] For example, amplification of HER2 gene is considered a major mechanism for its overexpression in breast cancer.[42] However, an association shown in these studies does not need to be an actual cause–result relationship. We have carefully scrutinized the literature but have not found convincing evidence, other than a positive statistical correlation, for this concept of “amplification causes overexpression” for many amplified oncogenes in cancers. In fact, different results and conclusions have been reported occasionally, such as a recent report showing that TOP2A overexpression in hepatocellular carcinoma was not secondary to gene amplification.[43] The fact that one or both of the parental alleles of the oncogene are also activated and overexpressed in many cancer tissues or cell lines, as aforementioned, complicates the situation. For example, the c-MYC oncogene is overexpressed in roughly 50% of the cases across all cancer types.[44,45,46] Amplification of the c-MYC gene also occurs often,[47] but the frequency is still much lower than the overexpression rate in the same cancer types,[44,47] indicating that c-MYC overexpression is not associated with amplification in most cases of almost all cancer types. In other words, overactivation of one or both of the normal c-MYC alleles at the 8q24.21 of the paternally and maternally derived chromosome 8 is responsible for the overexpression in most cases.
It is a very reasonable assumption that the amplified copies or alleles are activated, even when they are translocated to other chromosomes because the regulatory region of the gene, often referred to as “promoter,” is very likely to be co-amplified along with the oncogene to allow the excessive copies to be co-activated along with their parental ones [Figure 1b] However, this assumption may not always be real because an excessive copy at a new location of either the same chromosome or another chromosome is very likely to be under the sway of the local regulatory DNA sequence as well. The result could be a suppression of the promoter, which may be one possible mechanism for the aforementioned suppression of the amplified allele(s). Since in most cases, one or both of the parental alleles are also overexpressed, it is difficult to determine whether the excessive RNA transcripts are derived solely from the parental allele(s), solely from the amplified allele(s), or from both [Figure 1a]. In other words, the observation that in most cases one or both of the parental alleles are activated and overexpressed muddles things up. In fact, to our knowledge, few attempts have hitherto been made to differentiate these three scenarios and differentiate the transcriptional activities between the excessive allele(s) and the parental ones. This is in part because such studies are technically difficult, unless the excessive allele(s) have mutations while the parental ones remain to be the wild type or the excessive alleles have different mutations from those in the parental alleles so that sequencing the complementary DNA can result in information on whether there are any RNAs transcribed from the amplified allele(s). If the amplified alleles have a deletion or insertion or have a mutation that changes the transcript splicing, resulting in a difference in the mRNA size as frequently reported in cancers,[26,48,49,50,51,52] the difference may be identified more easily by using reverse transcription and ensuing polymerase chain reactions to determine the sizes of the mRNA variants.
Most studies of DNA amplification have been focused on the expression of the protein-coding gene in the DNA amplicon, without giving it enough attention that amplification of a genomic region is not the same thing as amplification of the gene in the amplicon. This is because only about 1.5% of the human genome encodes proteins[26,27,50] and because not only protein-coding mRNAs, but also different noncoding RNAs as regulatory factors can be expressed from the DNA amplicon.[26] In fact, some genomic regions that harbor only noncoding RNAs, such as the PVT-1,[53,54] are also amplified in cancers. Probably, in many cases, it is a noncoding transcript produced from the DNA amplicon as a regulatory RNA, but not an mRNA and its protein product of the annotated gene that elicits a biological function to confer a survival or growth advantage on the cancer cell and thus, the cell does not require the mRNA or its protein product to be overexpressed. This not only is one possible explanation for why on many occasions the expression level is not correlated with the gene copy number, but may also explain such occasions as that in which amplification of the MYCN gene is a good prognostic marker for neuroblastoma, but the significance of MYCN overexpression in prognosis is unclear.[55] In a nutshell, noncoding regulatory RNAs derived from the DNA amplicon in particular are much underexplored and deserve many more studies, albeit noncoding RNAs have been extensively studied in cancer in general.
CONCLUSION
The widespread concept that “gene amplification is a cause of increased expression” still remains as a reasonable assumption, but has not yet been a convincingly proven fact for most, if not all, oncogenes in cancers. Therefore, experimental evidence is still required for establishing this assumable causal relationship. A caveat needs to be given that gene rearrangement is a different type of genetic alteration although it is often accompanied by gene amplification. Rearrangement may cause formation of fusion gene(s) that produce fusion mRNAs and fusion proteins,[50,56,57] but their chimeric sequences can easily distinguish themselves from the transcripts from the parental alleles.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
AUTHORS PROFILE
Yuping Jia: Shandong Academy of Pharmaceutical Sciences, Ji’nan, Shandong 250101, China
Lichan Chen: Hormel Institute, University of Minnesota, Austin, MN 55912, USA
Qingwen Jia: Shandong Academy of Pharmaceutical Sciences, Ji’nan, Shandong 250101, China
Xixi Dou: Shandong Academy of Pharmaceutical Sciences, Ji’nan, Shandong 250101, China
Ningzhi Xu: Laboratory of Cell and Molecular Biology, Cancer Institute, Chinese Academy of Medical Science, Beijing 100021, China
Dezhong Joshua Liao: Clinical Research Center, Guizhou Medical University Hospital, Guizhou, Guiyang 550004, P.R. China
Acknowledgment
We would like to thank Dr. Fred Bogott at Austin Medical Center, Austin of Minnesota, for his excellent English editing of this manuscript.
Contributor Information
Yuping Jia, Email: jiayupingygs@163.com.
Ningzhi Xu, Email: xuningzhi@cicams.ac.cn.
Dezhong Joshua Liao, Email: djliao@gmc.edu.cn.
REFERENCES
- 1.Matsui A, Ihara T, Suda H, Mikami H, Semba K. Gene amplification: Mechanisms and involvement in cancer. Biomol Concepts. 2013;4:567–82. doi: 10.1515/bmc-2013-0026. [DOI] [PubMed] [Google Scholar]
- 2.Taylor EM, Lindsay HD. DNA replication stress and cancer: Cause or cure? Future Oncol. 2016;12:221–37. doi: 10.2217/fon.15.292. [DOI] [PubMed] [Google Scholar]
- 3.Zhang J, Lou X, Zellmer L, Liu S, Xu N, Liao DJ. Just like the rest of evolution in Mother Nature, the evolution of cancers may be driven by natural selection, and not by haphazard mutations. Oncoscience. 2014;22(1):580–90. doi: 10.18632/oncoscience.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bagci O, Kurtgöz S. Amplification of cellular oncogenes in solid tumors. N Am J Med Sci. 2015;7:341–6. doi: 10.4103/1947-2714.163641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim S. New and emerging factors in tumorigenesis: An overview. Cancer Manag Res. 2015;7:225–39. doi: 10.2147/CMAR.S47797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mondello C, Smirnova A, Giulotto E. Gene amplification, radiation sensitivity and DNA double-strand breaks. Mutat Res. 2010;704:29–37. doi: 10.1016/j.mrrev.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 7.Paluch BE, Naqash AR, Brumberger Z, Nemeth MJ, Griffiths EA. Epigenetics: A primer for clinicians. Blood Rev. 2016 doi: 10.1016/j.blre.2016.02.002. pii: S0268-960X00011-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thingholm LB, Andersen L, Makalic E, Southey MC, Thomassen M, Hansen LL. Strategies for integrated analysis of genetic, epigenetic, and gene expression variation in cancer: Addressing the challenges. Front Genet. 2016;7:2. doi: 10.3389/fgene.2016.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Minarovits J, Banati F, Szenthe K, Niller HH. Epigenetic Regulation. Adv Exp Med Biol. 2016;879:1–25. doi: 10.1007/978-3-319-24738-0_1. [DOI] [PubMed] [Google Scholar]
- 10.Tuna M, Machado AS, Calin GA. Genetic and epigenetic alterations of microRNAs and implications for human cancers and other diseases. Genes Chromosomes Cancer. 2016;55:193–214. doi: 10.1002/gcc.22332. [DOI] [PubMed] [Google Scholar]
- 11.D’Angelo D, Esposito F, Fusco A. Epigenetic mechanisms leading to overexpression of HMGA proteins in human pituitary adenomas. Front Med (Lausanne) 2015;2:39. doi: 10.3389/fmed.2015.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kushwaha M, Rostain W, Prakash S, Duncan JN, Jaramillo A. Using RNA as molecular code for programming cellular function. ACS Synth Biol. 2016 Mar 29; doi: 10.1021/acssynbio.5b00297. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 13.Kwan JY, Psarianos P, Bruce JP, Yip KW, Liu FF. The complexity of microRNAs in human cancer. J Radiat Res. 2016 doi: 10.1093/jrr/rrw009. pii: rrw009 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Askarian-Amiri ME, Leung E, Finlay G, Baguley BC. The regulatory role of long noncoding RNAs in cancer drug resistance. Methods Mol Biol. 2016;1395:207–27. doi: 10.1007/978-1-4939-3347-1_12. [DOI] [PubMed] [Google Scholar]
- 15.Gomes BC, Rueff J, Rodrigues AS. MicroRNAs and cancer drug resistance. Methods Mol Biol. 2016;1395:137–62. doi: 10.1007/978-1-4939-3347-1_9. [DOI] [PubMed] [Google Scholar]
- 16.Kwak Y, Kim SI, Park CK, Paek SH, Lee ST, Park SH. C-MET overexpression and amplification in gliomas. Int J Clin Exp Pathol. 2015;8:14932–8. [PMC free article] [PubMed] [Google Scholar]
- 17.Michalk M, Meinrath J, Künstlinger H, Koitzsch U, Drebber U, Merkelbach-Bruse S, et al. MDM2 gene amplification in esophageal carcinoma. Oncol Rep. 2016;35:2223–7. doi: 10.3892/or.2016.4578. [DOI] [PubMed] [Google Scholar]
- 18.Friedrich C, von Bueren AO, Kolevatova L, Bernreuther C, Grob T, Sepulveda-Falla D, et al. Epidermal growth factor receptor overexpression is common and not correlated to gene copy number in ependymoma. Childs Nerv Syst. 2016;32:281–90. doi: 10.1007/s00381-015-2981-2. [DOI] [PubMed] [Google Scholar]
- 19.Li BT, Ross DS, Aisner DL, Chaft JE, Hsu M, Kako SL, et al. HER2 amplification and HER2 mutation are distinct molecular targets in lung cancers. J Thorac Oncol. 2016;11:414–9. doi: 10.1016/j.jtho.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stahl P, Seeschaaf C, Lebok P, Kutup A, Bockhorn M, Izbicki JR, et al. Heterogeneity of amplification of HER2, EGFR, CCND1 and MYC in gastric cancer. BMC Gastroenterol. 2015;15:7. doi: 10.1186/s12876-015-0231-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Afzal M, Amir M, Hassan MJ, Hussain MS, Aziz MN, Murad S, et al. Clinical role of HER2 gene amplification and chromosome 17: A study on 154 IHC-equivocal cases of invasive breast carcinoma patients. Tumour Biol. 2016 Jan 6; doi: 10.1007/s13277-015-4657-7. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 22.Onguru O, Zhang PJ. The relation between percentage of immunostained cells and amplification status in breast cancers with equivocal result for Her2 immunohistochemistry. Pathol Res Pract. 2016;212:381–4. doi: 10.1016/j.prp.2016.02.007. [DOI] [PubMed] [Google Scholar]
- 23.Sugimoto Y, Roninson IB, Tsuruo T. Decreased expression of the amplified mdr1 gene in revertants of multidrug-resistant human myelogenous leukemia K562 occurs without loss of amplified DNA. Mol Cell Biol. 1987;7:4549–52. doi: 10.1128/mcb.7.12.4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chung HJ, Levens D. C-myc expression: Keep the noise down! Mol Cells. 2005;20:157–66. [PubMed] [Google Scholar]
- 25.Jensen LB, Bartlett JM, Witton CJ, Kirkegaard T, Brown S, Müller S, et al. Frequent amplifications and deletions of G1/S-phase transition genes, CCND1 and MYC in early breast cancers: A potential role in G1/S escape. Cancer Biomark. 2009;5:41–9. doi: 10.3233/CBM-2009-0570. [DOI] [PubMed] [Google Scholar]
- 26.Jia Y, Chen L, Ma Y, Zhang J, Xu N, Liao DJ. To know how a gene works, we need to redefine it first but then, more importantly, to let the cell itself decide how to transcribe and process its RNAs. Int J Biol Sci. 2015;11:1413–23. doi: 10.7150/ijbs.13436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lou X, Zhang J, Liu S, Xu N, Liao DJ. The other side of the coin: The tumor-suppressive aspect of oncogenes and the oncogenic aspect of tumor-suppressive genes, such as those along the CCND-CDK4/6-RB axis. Cell Cycle. 2014;13:1677–93. doi: 10.4161/cc.29082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Willis NA, Rass E, Scully R. Deciphering the code of the cancer genome: Mechanisms of Chromosome rearrangement. Trends Cancer. 2015;1:217–230. doi: 10.1016/j.trecan.2015.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan C, Xu N, Liao J. Switch of FANCL, a key FA-BRCA component, between tumor suppressor and promoter by alternative splicing. Cell Cycle. 2012;11:3356. doi: 10.4161/cc.21852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liffers K, Lamszus K, Schulte A. EGFR amplification and glioblastoma stem-like cells. Stem Cells Int 2015. 2015:427518. doi: 10.1155/2015/427518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baylay AJ, Ivens A, Piddock LJ. A novel gene amplification causes upregulation of the PatAB ABC transporter and fluoroquinolone resistance in Streptococcus pneumoniae. Antimicrob Agents Chemother. 2015;59:3098–108. doi: 10.1128/AAC.04858-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang G, Chen L, Yu B, Zellmer L, Xu N, Liao DJ. Learning about the importance of mutation prevention from curable cancers and benign tumors. J Cancer. 2016;7:436–45. doi: 10.7150/jca.13832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang J, Lou X, Jin L, Zhou R, Liu S, Xu N, et al. Necrosis, and then stress induced necrosis-like cell death, but not apoptosis, should be the preferred cell death mode for chemotherapy: Clearance of a few misconceptions. Oncoscience. 2014;1:407–22. doi: 10.18632/oncoscience.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amin AD, Rajan SS, Groysman MJ, Pongtornpipat P, Schatz JH. Oncogene overdose: Too much of a bad thing for oncogene-addicted cancer cells. Biomark Cancer. 2015;7(Suppl 2):25–32. doi: 10.4137/BIC.S29326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yabuki N, Sakata K, Yamasaki T, Terashima H, Mio T, Miyazaki Y, et al. Gene amplification and expression in lung cancer cells with acquired paclitaxel resistance. Cancer Genet Cytogenet. 2007;173:1–9. doi: 10.1016/j.cancergencyto.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 36.Micale L, Augello B, Daniele G, Macchia G, L’abbate A, Muehlematter D, et al. Amplification of the G allele at SNP rs6983267 in 8q24 amplicons in myeloid malignancies as cause of the lack of MYC overexpression? Blood Cells Mol Dis. 2011;47:259–61. doi: 10.1016/j.bcmd.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 37.Lockwood WW, Chari R, Coe BP, Girard L, Macaulay C, Lam S, et al. DNA amplification is a ubiquitous mechanism of oncogene activation in lung and other cancers. Oncogene. 2008;27:4615–24. doi: 10.1038/onc.2008.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee HJ, Xu X, Choe G, Chung DH, Seo JW, Lee JH, et al. Protein overexpression and gene amplification of epidermal growth factor receptor in nonsmall cell lung carcinomas: Comparison of four commercially available antibodies by immunohistochemistry and fluorescence in situ hybridization study. Lung Cancer. 2010;68:375–82. doi: 10.1016/j.lungcan.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 39.Suzuki S, Dobashi Y, Sakurai H, Nishikawa K, Hanawa M, Ooi A. Protein overexpression and gene amplification of epidermal growth factor receptor in nonsmall cell lung carcinomas. An immunohistochemical and fluorescence in situ hybridization study. Cancer. 2005;103:1265–73. doi: 10.1002/cncr.20909. [DOI] [PubMed] [Google Scholar]
- 40.Brown DN, Caffa I, Cirmena G, Piras D, Garuti A, Gallo M, et al. Squalene epoxidase is a bona fide oncogene by amplification with clinical relevance in breast cancer. Sci Rep. 2016;6:19435. doi: 10.1038/srep19435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mäkelä TP, Saksela K, Alitalo K. Amplification and rearrangement of L-myc in human small-cell lung cancer. Mutat Res. 1992;276:307–15. doi: 10.1016/0165-1110(92)90017-4. [DOI] [PubMed] [Google Scholar]
- 42.Krishnamurti U, Silverman JF. HER2 in breast cancer: A review and update. Adv Anat Pathol. 2014;21:100–7. doi: 10.1097/PAP.0000000000000015. [DOI] [PubMed] [Google Scholar]
- 43.Panvichian R, Tantiwetrueangdet A, Angkathunyakul N, Leelaudomlipi S. TOP2A amplification and overexpression in hepatocellular carcinoma tissues. Biomed Res Int 2015. 2015:381602. doi: 10.1155/2015/381602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liao DJ, Thakur A, Wu J, Biliran H, Sarkar FH. Perspectives on c-myc, cyclin D1, and their interaction in cancer formation, progression, and response to chemotherapy. Crit Rev Oncog. 2007;13:93–158. doi: 10.1615/critrevoncog.v13.i2.10. [DOI] [PubMed] [Google Scholar]
- 45.Liao DJ, Dickson RB. C-myc in breast cancer. Endocr Relat Cancer. 2000;7:143–64. doi: 10.1677/erc.0.0070143. [DOI] [PubMed] [Google Scholar]
- 46.Wang C, Lisanti MP, Liao DJ. Reviewing once more the c-myc and Ras collaboration: Converging at the cyclin D1-CDK4 complex and challenging basic concepts of cancer biology. Cell Cycle. 2011;10:57–67. doi: 10.4161/cc.10.1.14449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang C, Tai Y, Lisanti MP, Liao DJ. C-myc induction of programmed cell death may contribute to carcinogenesis: A perspective inspired by several concepts of chemical carcinogenesis. Cancer Biol Ther. 2011;11:615–26. doi: 10.4161/cbt.11.7.14688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xing Y. Genomic analysis of RNA alternative splicing in cancers. Front Biosci. 2007;12:4034–41. doi: 10.2741/2369. [DOI] [PubMed] [Google Scholar]
- 49.Zhang J, Lou X, Shen H, Zellmer L, Sun Y, Liu S, et al. Isoforms of wild type proteins often appear as low molecular weight bands on SDS-PAGE. Biotechnol J. 2014;9:1044–54. doi: 10.1002/biot.201400072. [DOI] [PubMed] [Google Scholar]
- 50.Peng Z, Yuan C, Zellmer L, Liu S, Xu N, Liao DJ. Hypothesis: Artifacts, including spurious chimeric RNAs with a short homologous sequence, caused by consecutive reverse transcriptions and endogenous random primers. J Cancer. 2015;6:555–67. doi: 10.7150/jca.11997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang M, Sun Y, Ma L, Wang C, Wu JM, Bi A, et al. Complex alternative splicing of the smarca2 gene suggests the importance of smarca2-B variants. J Cancer. 2011;2:386–400. doi: 10.7150/jca.2.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang M, Wu J, Wu SH, Bi AD, Liao DJ. Splicing of mouse p53 pre-mRNA does not always follow the “first come, first served” principle and may be influenced by cisplatin treatment and serum starvation. Mol Biol Rep. 2012;39:9247–56. doi: 10.1007/s11033-012-1798-2. [DOI] [PubMed] [Google Scholar]
- 53.Guan Y, Kuo WL, Stilwell JL, Takano H, Lapuk AV, Fridlyand J, et al. Amplification of PVT1 contributes to the pathophysiology of ovarian and breast cancer. Clin Cancer Res. 2007;13:5745–55. doi: 10.1158/1078-0432.CCR-06-2882. [DOI] [PubMed] [Google Scholar]
- 54.Colombo T, Farina L, Macino G, Paci P. PVT1: A rising star among oncogenic long noncoding RNAs. Biomed Res Int 2015. 2015:304208. doi: 10.1155/2015/304208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tang XX, Zhao H, Kung B, Kim DY, Hicks SL, Cohn SL, et al. The MYCN enigma: Significance of MYCN expression in neuroblastoma. Cancer Res. 2006;66:2826–33. doi: 10.1158/0008-5472.CAN-05-0854. [DOI] [PubMed] [Google Scholar]
- 56.Mertens F, Tayebwa J. Evolving techniques for gene fusion detection in soft tissue tumours. Histopathology. 2014;64:151–62. doi: 10.1111/his.12272. [DOI] [PubMed] [Google Scholar]
- 57.Mertens F, Johansson B, Fioretos T, Mitelman F. The emerging complexity of gene fusions in cancer. Nat Rev Cancer. 2015;15:371–81. doi: 10.1038/nrc3947. [DOI] [PubMed] [Google Scholar]