

Abstract
Well-defined and reliable clinical outcome assessments are essential for determining whether a drug provides clinically meaningful treatment benefit for patients. In 2015, FDA convened a workshop, “Assessing Neurocognitive Outcomes in Inborn Errors of Metabolism.” Topics covered included special challenges of clinical studies of inborn errors of metabolism (IEMs) and other rare diseases; complexities of identifying treatment effects in the context of the dynamic processes of child development and disease progression; and the importance of natural history studies. Clinicians, parents/caregivers, and participants from industry, academia, and government discussed factors to consider when developing measures to assess treatment outcomes, as well as tools and methods that may contribute to standardizing measures. Many issues examined are relevant to the broader field of rare diseases in addition to specifics of IEMs.
Keywords: Inborn errors, Neurocognitive, Clinical outcome assessment, Natural history studies, Rare diseases, Drug development
1. Introduction
This article summarizes key points discussed among participants at a workshop convened by the U.S. Food and Drug Administration (FDA) in April 2015 entitled, “Assessing Neurocognitive Outcomes in Inborn Errors of Metabolism.” The workshop brought together clinicians, parents/caregivers, and representatives from industry, academia, and government (FDA and National Institutes of Health). Participants presented their perspectives on factors to consider when developing measures to assess clinical outcomes of candidate and approved treatments for diseases resulting from inborn errors of metabolism (IEMs). [Points raised are meant as considerations and should not be interpreted as guidance for drug development. Similarly, discussion of particular scales does not constitute FDA endorsement of these scales for trial endpoints.] Full proceedings of the meeting are available online at http://www.fda.gov/downloads/Drugs/NewsEvents/UCM493766.pdf.
2. Challenges of clinical studies of rare diseases
Clinical studies of rare diseases that affect the brain and neurological systems are challenging by their nature. Developing reliable and valid study endpoints can be difficult due to many factors, including small numbers of patients who are often geographically dispersed; heterogeneity of deficits between patients and within individual patients over time; limited clinical data describing signs and symptoms of disease and its progression; and lack of knowledge about the natural history of many rare diseases, especially in regard to neurocognitive outcomes [1]. The FDA convened the workshop to provide a forum for discussing challenges of and methods for measuring such clinical outcomes in individuals affected by IEMs. While the workshop focused on IEMs, the cases and approaches presented may also be relevant to the assessment of cognitive function in other diseases. This article summarizes meeting presentations and discussions about opportunities for working with FDA to establish well-defined and reliable clinical outcome assessments (COAs); the role of natural history studies in identifying disease and treatment effects; the value of stakeholder collaboration in using and improving neurocognitive assessment tools; lessons learned from clinical studies of rare diseases; and best practices for assessing cognition and behavior to obtain the most useful and comparable data.
3. Establishing clinical neurocognitive outcome assessments for IEMs and other rare diseases
Recent advances in diagnostics and enhanced newborn screening programs have made it possible to identify diseases earlier in life and begin treatment sooner, if treatments are available. Such is the case for many IEMs as well as other rare diseases. Increased understanding of the mechanisms of IEMs has led to development of a substantial number of new treatments. Evaluating outcomes of these treatments, however, requires that researchers distinguish brain changes resulting from treatment effects from those resulting from child development or disease progression (Fig. 1).
Fig. 1.
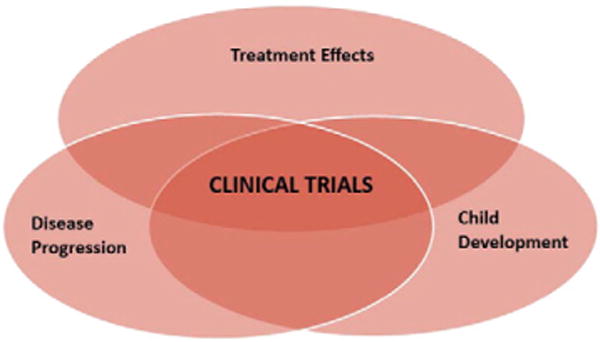
Clinical trials designed to measure treatment outcomes must also take into account child development and disease progression. The dynamic factors of child development and disease progression are likely to have opposing effects. Standardized, well-defined, and reliable measures of neurocognitive outcomes are essential to enable researchers to assess treatment results reliably. Natural history studies aid in identifying factors associated with disease progression in the context of child development.
3.1. Clinical outcome assessments and drug development: a collaborative process
Clinical outcome assessments are an essential part of drug development – they aid in determining whether a drug provides clinically meaningful treatment benefit(s) to patients. FDA’s regulatory standard includes a statement that methods of assessment of subjects’ response should be ‘well defined and reliable’ (21CFR314.126) [2]. In 2009, FDA released Guidance for Industry – Patient-Reported Outcome Measures: Use in Medical Product Development to Support Labeling Claims [3] to aid instrument developers in meeting this regulatory standard. The Guidance provides details on how to establish a COA instrument’s content validity, i.e., the extent to which a COA measures what it purports to measure in a specific context of use. In addition, FDA offers the Clinical Outcome Assessment Qualification Program, which provides specifics on evidence needed and steps to take to qualify COAs for drug development [4]. Stakeholders can work with FDA to evaluate existing measurement tools or develop novel COAs in two ways (described in detail in the full meeting proceedings, http://www.fda.gov/downloads/Drugs/NewsEvents/UCM493766.pdf):
Through an individual drug development program (the traditional method); or
Through FDA’s Center for Drug Evaluation and Research Drug Development Tool Qualification Program, which are designed to produce qualified measures for use across multiple drug development programs.
Although COAs used in clinical trials are not required to be qualified through the COA Drug Development Tool Qualification Program, developing COAs in consultation with FDA can increase the likelihood that the Agency will agree with the content and measurement properties of the COA. In addition, in 2015, FDA issued Guidance for Industry – Critical Path Innovation Meetings [5]. Such meetings, known as CPIMs, are means by which CDER and investigators from industry, academia, patient advocacy groups, and government can communicate to improve efficiency and success in drug development. The goals of CPIMs are to discuss a methodology or technology proposed by the meeting requester and for CDER to provide general advice on how this methodology or technology might enhance drug development.
In many patients with IEMs, measures of cognition, behavior, and activities of daily living may be the factors most relevant to improving symptoms of a disease and thereby have the greatest impact on patients and their families. In addition to direct measurement, parent/caregiver and patient-reported outcomes are essential when developing COAs for diseases resulting from inborn errors of metabolism, and instruments should be developed and validated with these populations. Impact on the functioning of the individual and the family are also important factors to consider when developing COAs for other rare diseases. Reliable and valid measures that are developed through interdisciplinary collaboration and with stakeholder input can lead to better understanding of disease progression and more reliable assessments of treatment efficacy.
4. Natural history studies of rare diseases
Ideally, natural history studies investigate the natural course of a disease from or before inception, through pre-symptomatic, symptomatic, and clinical stages to the point of cure, chronic disease, or death [6]. They are valuable tools for improving understanding of a disease, establishing clinical outcome assessments that aid in identifying treatment effects, and enhancing and accelerating drug development. Natural history studies may: (1) provide a clinical baseline; (2) quantify rate and variability of disease progression; (3) aid in detecting safety concerns; (4) provide context for efficacy evaluation; (5) help identify biomarkers or other surrogate measures and determine correlations with disease; (6) guide dose selection; and (7) help establish the optimal window of intervention. Results of natural history studies are important for designing clinical trials as well as informing benefit-risk analyses and regulatory decision-making, especially for rare and poorly understood conditions. More information about the role of natural history studies in drug development for rare diseases may be found in FDA’s Draft Guidance for Industry – Rare Diseases: Common Issues in Drug Development [7].
4.1. Considerations for conducting natural history studies of disorders that affect the brain
Comprehensive, prospective natural history studies can be essential for identifying sensitive and specific endpoints prior to clinical trials and maximizing the likelihood of generating useful data.[7] Meeting participants identified the following principles to consider when planning and conducting effective natural history studies that measure neurocognition.
Use a developmental model to evaluate changes in ability that is geared toward young and impaired patients.
Determine whether language, physical capabilities, or behavior will limit testing and adjust accordingly by providing any necessary aids (e.g., for vision or hearing), keeping test batteries short, and providing a comfortable and appropriately designed testing environment.
Select neurocognitive tests that can be repeated to acquire multiple data points.
Develop methods and train coordinators to ensure consistent data collection across sites.
Use concise instruments that yield clear, measurable outcomes.
Decide whether a disease-specific measure is needed (e.g., to measure behavior or other outcomes related to quality of life as reported by patients or parents). If so, identify or develop a suitable measurement scale. Natural history studies can help to determine if measures are sufficiently sensitive to identify meaningful changes and whether adjusting or developing new measures is necessary.
Ensure that testers are knowledgeable about the instrument and the disease, can relate to children and parents including those affected by disease, and adhere to protocols while also being sensitive to the needs of the patient.
Determine whether cognitive tests associate with other measures, in order to help validate selected tools and techniques.
Measure factors that have the greatest impact on patients and families in terms of how they feel, function, and survive, as these are likely to improve quality of life.
When possible (e.g., in natural history studies), provide feedback and a summary of information gained from the individual patient and from studies as a whole to parents/caregivers, whether during the course of the study, if possible when relevant to treatment, educational, or other needs, or at the study’s completion.
Participants reported on their experiences with natural history studies for IEMs that have generated substantial data and highlighted keys to success as well as potential pitfalls [8]. These include studies of neurocognitive outcomes in urea cycle disorders, conducted as part of the Longitudinal Study for the Urea Cycle Disorders Consortium, part of the Rare Diseases Clinical Research Network [9]; and natural history studies in children with mucopolysaccharidosis type II (Hunter syndrome) and types IIIA [10] and IIIB (Sanfilippo A and B). These studies are described in detail in the full proceedings of this meeting.
5. Conducting clinical studies of inborn errors of metabolism
5.1. Defining clinically meaningful changes in neurocognition
Presenters noted that clinicians have traditionally been trained to focus on the pathophysiology of disease, the resulting impairments, and associated laboratory measures that can predict the course of disease. For patients with IEMs, however, such measures are often not available and, when they are, may not predict outcomes. While biochemical and other quantitative measures contribute to research on IEMs as well as clinical care, family members and health care providers at the workshop reported that the ability to function cognitively and behaviorally, perform activities of daily living, and participate in society often have the greatest impact on quality of life for patients and families. These factors are not always identified by traditional measures, but are important to take into account when identifying or developing rating scales sufficiently sensitive for clinical research.
5.2. Roles of families of patients with IEMs in research
Meeting participants described how families of patients with IEMs play multiple roles in research: they are subjects of assessments as well as those most greatly affected by the results. The unique and varied perspectives of patients and caregivers provide valuable input for designing clinical research on IEMs and other rare diseases. It is from them that we learn what makes the biggest difference in everyday life. A clinician may document small areas of improvement in a child, yet an important concern may not be ameliorated. Conversely, a change that is barely discernable on a rating scale may enable a patient to cross a threshold to greater independence, such as by having sufficient motor control to hold a cup to drink or to navigate using a wheelchair. Here the use of patient-reported outcome through social media and novel web-based tools can help close the gap [11].
Family members and clinicians explained that many families of patients with IEMs are invested in the ability of research to develop safe and effective treatments, to accurately evaluate benefit-risk, and to support long-term research goals to further ameliorate the effects of IEMs. Researchers and clinicians must consider the impact of research studies on patients and families. Opinions about the impact of repetitive testing on the validity of results vary: some meeting participants indicated that frequent, repetitive testing has little effect on data validity, while others believe it can confound results, particularly in older or more mildly impaired patients who may be subject to learning effects (i.e., “improvement” in test scores due solely to the fact that the subject has been previously exposed to the test). Representatives from families of patients with IEMs at the workshop noted, however, that repetitive testing does have an impact on the child and family, sometimes increasing medical trauma to children who already endure a substantial burden of medical interventions. As such, it is important to offer options to make testing as comfortable and convenient for the family as possible. In addition, testing results provide valuable information to parents and should be provided whenever possible. When clinicians and researchers work with patients and families to fully understand what has the greatest impact on quality of life, they build effective partnerships that are likely to yield the most meaningful clinical and research outcomes.
6. Approaches for assessing cognition and behavior
Meeting participants with expertise in measuring cognition and behavior stated that such assessments for patients with IEMs and other conditions must be based on an understanding of the patient population and the natural history of the disease. When developing outcome measures, it is important to involve patients and caregivers and to pilot test instruments to ensure that they can be reliably and safely completed by patients and are sensitive enough to characterize impairments and detect changes in response to interventions.
6.1. The need for standardized assessments
Developing outcome assessments that are well-defined and reliable is essential for conducting adequate and well-controlled studies that can generate data about treatment effects and be useful for regulatory review [8]. Thus, outcome assessments for clinical trials must be standardized, reliable, and valid [3]. Meeting participants identified the following factors as among those to consider when choosing assessment tools for evaluating treatment outcomes in patients with IEMs and other rare diseases: limited patient populations that are likely to be widely dispersed and may require global research sites; heterogeneous disease manifestations; the need to train assessors to decrease variability; and implementation of quality control measures such as centralized scoring and use of clinical trial monitors to increase accuracy and reduce variability. FDA’s Draft Guidance for Industry – Rare Diseases: Common Issues in Drug Development [7] outlines multiple considerations for selecting appropriate endpoints in clinical trials of treatments for rare diseases.
6.2. Tools and methods for standardizing assessments
6.2.1. Common data elements
An example of ongoing efforts to standardize assessments is the Common Data Elements (CDEs) Project of the National Institute of Neurological Disorders and Stroke (NINDS), part of the National Institutes of Health. This project uses common data elements that apply to multiple fields of research to enable clinical investigators to share information reliably. It facilitates systematic collection, analysis, and sharing of data across research communities. Objectives of the CDE project include: (1) to identify common data elements used in clinical research; (2) to present data elements in standard, widely available formats; (3) to establish common definitions and validate permissible values and ranges to guide researchers in selecting CDEs most applicable to studies; (4) to standardize case report forms and other instruments; and (5) to provide standardized information to researchers for database development. Common data elements can accelerate the pace of clinical research for rare diseases by rendering data more comparable and facilitating multi-center and international efforts [12].
6.2.2. Using remote technology to expand reach of clinical research
Remote assessment – in which the evaluator and individual being evaluated are not located in the same physical space – may offer significant advantages for studies of rare diseases [11]. Remote assessments can reduce burdensome, costly travel for patients and families and increase patient access to research studies [13]. For researchers, remote assessment may facilitate data capture from a larger number of individuals and increase data quality by reducing the number of assessors, increasing measurement expertise, and providing a more controlled environment. Patients who can be assessed in their own homes or other familiar environments may be more motivated to participate in studies with less fatigue that could influence results. In the long term, the ability to use remote technologies may increase patient engagement in research and foster new discoveries and advances in patient care.
Some researchers are using a mixed approach of remote and direct assessments. For instance, the University of Rochester Batten Center (URBC) conducts remote and direct assessments to evaluate clinical features and natural history of juvenile Batten disease, including assessments of behavior and cognition [14,15]. (URBC calculated the average round-trip travel distance for a family attending a single research visit at their site to be more than 1700 miles.) More information about URBC’s use of remote technologies is available in the full meeting proceedings.
6.2.3. The case for disease-specific Scales
Behaviors associated with IEMs are usually related to either specific brain region/functional involvement or the patient’s reaction to the physical and cognitive manifestations of the disease. Examples of disease-specific behavioral problems can be seen in the attention problems that are associated with white matter abnormalities in mucopolysaccharidosis types I and II, the mood and anxiety symptoms seen in Batten disease, and the severe and unique behavioral abnormalities associated with amygdala atrophy in Sanfilippo syndrome Type A. While standard measures of cognition can be used in various IEMs, measures of behavior may require disease-specific scales such as the Unified Batten Disease Rating Scale and the Sanfilippo Behavior Rating Scale [16, 17]. Establishing disease-specific measures can benefit patients and advance understanding of IEMs. Such scales can aid in diagnosis; help to identify root causes of aberrant behaviors; determine the most effective treatments; improve quality of life for patients and their families; and enhance the quality of research.
Researchers studying Sanfilippo syndrome created a disease-specific behavior rating scale by developing detailed descriptions and assessing the significance of specific behaviors; generating, classifying, and pruning items to establish a concrete and detailed scale; and validating the scale with other measures [17]. A step-by-step description of how the Sanfilippo Behavior Rating Scale was developed can be found in the full proceedings of this meeting. The methodology may help researchers establish scales for other diseases with unique behavioral phenotypes.
7. Conclusion
Emerging therapies for IEMs require novel and improved measures for neurocognitive and behavioral endpoints that are easily applied, reliable, and valid. This workshop addressed the significance of such measures for clinical research and the role of natural history studies in developing and evaluating such measures. Meeting participants suggested selecting measures that: (1) are suitable for particular developmental stages, brief, and appropriate to the disability and level of the child; (2) can be administered reliably by trained testers; (3) include common data elements for each disease; and (4) have the greatest impact on quality of life for patients and families. Novel approaches such as remote technologies, computerized Web-based assessment, and development of disease-specific behavioral assessment may enhance the toolbox of researchers studying IEMs. Participants at this workshop identified a number of factors to consider when conducting clinical trials to evaluate safety and efficacy of candidate drugs to treat neurological and neurocognitive manifestations of IEMs. Collaborative efforts among diverse stakeholders: including, industry, academia, and government representatives, advocates, patients, and families, are required to advance safe and effective drugs to treat the brain and allow patients with rare diseases resulting from IEMs to live full and complete lives.
Contributor Information
Elsa Shapiro, Email: shapi004@umn.edu.
Heather R. Adams, Email: Heather_Adams@URMC.Rochester.edu.
Ann J. Barbier, Email: annjbarbier@aol.com.
Teresa Buracchio, Email: teresa.buracchio@fda.hhs.gov.
Peter Como, Email: peter.como@fda.hhs.gov.
Kathleen A. Delaney, Email: kate.delaney@shapiroanddelaney.com.
Florian Eichler, Email: feichler@partners.org.
Jonathan C. Goldsmith, Email: jonathan.goldsmith@fda.hhs.gov.
Melissa Hogan, Email: melissa@savingcase.com.
Sarrit Kovacs, Email: sarrit.kovacs@fda.hhs.gov.
Jonathan W. Mink, Email: Jonathan_Mink@URMC.Rochester.edu.
Joanne Odenkirchen, Email: jo21x@nih.gov.
Melissa A. Parisi, Email: parisima@mail.nih.gov.
Alison Skrinar, Email: ASkrinar@ultragenyx.com.
Susan E. Waisbren, Email: Susan.Waisbren@childrens.harvard.edu.
Andrew E. Mulberg, Email: andrew.mulberg@fda.hhs.gov.
References
- 1.Augustine EF, Adams HR, Mink JW. Clinical trials in rare disease: challenges and opportunities. J Child Neurol. 2013;28:1142–1150. doi: 10.1177/0883073813495959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Section 314.126 – adequate and well-controlled studies, Federal Register, 2012.
- 3.Guidance for Industry Patient-Reported Outcome Measures: Use in Medical Product Development to Support Labeling Claims, Food and Drug Administration, December 2009.
- 4.Food and Drug Administration. Clinical Outcome Assessment Qualification Program. http://www.fda.gov/drugs/developmentapprovalprocess/drugdevelopmenttoolsqualificationprogram/ucm284077.htm2016.
- 5.Guidance for Industry – Critical Path Innovation Meetings. Food and Drug Administration. 2015 [Google Scholar]
- 6.Groft SC, de la Paz MP. Rare diseases – avoiding misperceptions and establishing realities: the need for reliable epidemiological data. Adv Exp Med Biol. 2010;686:3–14. doi: 10.1007/978-90-481-9485-8_1. [DOI] [PubMed] [Google Scholar]
- 7.Guidance for Industry – Rare Diseases. Common Issues in Drug Development. Food and Drug Administration. 2015 Aug; [Google Scholar]
- 8.Delaney K, Rudser K, Yund B, Whitley C, Haslett PJ, Shapiro E. Methods of neurodevelopmental assessment in children with neurodegenerative disease: Sanfilippo syndrome. In: Zschocke J, Gibson KM, Brown G, Morava E, Peters V, editors. JIMD Reports – Case and Research Reports, 13. Springer; Berlin Heidelberg: 2014. pp. 129–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rare Diseases Clinical Research Network. Urea Cycle Disorders Consortium. 2016 http://www.rarediseasesnetwork.org/cms/UCDC.
- 10.Shapiro EG, Nestrasil I, Delaney KA, Rudser K, Kovac V, Nair N, Richard CW, 3rd, Haslett P, Whitley CB. A prospective natural history study of mucopolysaccharidosis type IIIA. J Pediatr. 2016;170:278–287, e274. doi: 10.1016/j.jpeds.2015.11.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dorsey ER, Venuto C, Venkataraman V, Harris DA, Kieburtz K. Novel methods and technologies for 21st-century clinical trials: a review. JAMA Neurol. 2015;72:582–588. doi: 10.1001/jamaneurol.2014.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen MZ, Thompson CB, Yates B, Zimmerman L, Pullen CH. Implementing common data elements across studies to advance research. Nurs Outlook. 2015;63:181–188. doi: 10.1016/j.outlook.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ragbeer SN, Augustine EF, Mink JW, Thatcher AR, Vierhile AE, Adams HR. Remote assessment of cognitive function in juvenile neuronal ceroid lipofuscinosis (Batten disease) – a pilot study of feasibility and reliability. J Child Neurol. 2016;31:481–487. doi: 10.1177/0883073815600863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cialone J, Augustine EF, Newhouse N, Vierhile A, Marshall FJ, Mink JW. Quantitative telemedicine ratings in Batten disease: implications for rare disease research. Neurology. 2011;77:1801–1811. doi: 10.1212/WNL.0b013e3182377e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.University of Rochester Medicine. University of Rochester Batten Center. 2016 https://www.urmc.rochester.edu/neurology/batten-disease-center.aspx.
- 16.Marshall FJ, de Blieck EA, Mink JW, Dure L, Adams H, Messing S, Rothberg PG, Levy E, McDonough T, DeYoung J, Wang M, Ramirez-Montealegre D, Kwon JM, Pearce DA. A clinical rating scale for Batten disease: reliable and relevant for clinical trials. Neurology. 2005;65:275–279. doi: 10.1212/01.wnl.0000169019.41332.8a. [DOI] [PubMed] [Google Scholar]
- 17.Shapiro EG, Nestrasil I, Ahmed A, Wey A, Rudser KR, Delaney KA, Rumsey RK, Haslett PA, Whitley CB, Potegal M. Quantifying behaviors of children with Sanfilippo syndrome: the Sanfilippo Behavior Rating Scale. Mol Genet Metab. 2015;114:594–598. doi: 10.1016/j.ymgme.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]