

Abstract
Ursolic acid (UA), a well-known natural triterpenoid found in abundance in blueberries, cranberries and apple peels, has been reported to possess many beneficial health effects. These effects include anti-cancer activity in various cancers, such as skin cancer. Skin cancer is the most common cancer in the world. Nuclear factor E2-related factor 2 (Nrf2) is a master regulator of anti-oxidative stress response with anti-carcinogenic activity against UV- and chemical-induced tumor formation in the skin. Recent studies show that epigenetic modifications of Nrf2 play an important role in cancer prevention. However the epigenetic impact of UA on Nrf2 signaling remains poorly understood in skin cancer. In this study, we investigated the epigenetic effects of UA on mouse epidermal JB6 P+ cells. UA inhibited cellular transformation by 12-O-tetradecanoylphorbol-13-acetate (TPA) at a concentration at which the cytotoxicity was no more than 25%. Under this condition, UA induced the expression of the Nrf2-mediated detoxifying/antioxidant enzymes heme oxygenase-1 (HO-1), NAD(P)H:quinone oxidoreductase 1 (NQO1), and UDP-glucuronosyltransferase 1A1 (UGT1A1). DNA methylation analysis revealed that UA demethylated the first 15 CpG sites of the Nrf2 promoter region, which correlated with the re-expression of Nrf2. Furthermore, UA reduced the expression of epigenetic modifying enzymes, including the DNA methyltransferases (DNMTs) DNMT1 and DNMT3a and the histone deacetylases (HDACs) HDAC1, 2, 3, and 8 (Class I) and HDAC6 and 7 (Class II), and HDAC activity. Taken together, these results suggest that the epigenetic effects of the triterpenoid UA could potentially contribute to its beneficial effects, including the prevention of skin cancer.
Keywords: UA, Nrf2, JP6 P+, DNMT, HDAC, skin cancer
1. Introduction
Ursolic acid (UA) is a lipophilic pentacyclic triterpenoid derived from apple peels, basil (Ocimum basilicum), blueberry (Vaccinium spp.), cranberry (Vaccinium macrocarpon), heather flower (Calluna vulgaris), labrador tea (Ledum groenlandicum Retzius), olive (Olea europaea), pear (Pyrus pyrifolia), and rosemary (Rosmarinus officinalis) [1, 2]. UA exerts various biological effects, including anti-inflammatory, anti-atherosclerosis, anti-diabetic, anti-viral, and anticancer activities. Additionally, UA has the ability to decrease reactive oxygen species (ROS) toxicity and increase the activity of antioxidant enzymes [1]. In vivo and in vitro studies have shown that UA inhibits B[a]P- and 7,12-dimethylbenz[a]-anthracene (DMBA)-induced tumor initiating activity, suppresses 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced skin inflammation and tumor promotion in CD-1 and ICR mice, and induces apoptosis in M4Beu human melanoma cells [3-5]. Additionally, UA hinders UVA-induced ROS production, lipid peroxidation, MMP-2 expression, and DNA damage in human keratinocyte HaCaT cells [6]. Recently, studies have revealed that UA protects the brain against cerebral ischemia and protects the liver against CCl4-induced damage in mice via the nuclear factor E2-related factor 2 (Nrf2) pathway [7, 8]. Notably, we have previously shown that dietary phytochemicals, such as apigenin, curcumin, 3,3′-diindolylmethane, γ-tocopherol-rich mixture of tocopherols, sulforaphane, tanshinone IIA, Z-ligustilide and radix angelica, regulate Nrf2 activation via epigenetic modifications [9-16]; however, the effect of UA on the epigenetic regulation of Nrf2 has not been previously examined.
Skin cancer is one of the most prevalent malignant tumors, contributing to the increasing mortality rate of cancer in the US [17]. An imbalance between the production and removal of ROS in the epidermis and dermis may lead to skin tumorigenesis. Exposure to ultraviolet (UV) radiation, ozone layer depletion, excessive time spent outdoors, indoor tanning, and noxious environmental insults induce ROS overproduction [18]. Cells contain a self-defense mechanism that removes ROS through the synthesis of detoxifying/antioxidant enzymes, which include heme oxygenase-1 (HO-1), NAD(P)H:quinone oxidoreductase 1 (NQO1), uridine 5′-diphospho-glucuronosyltransferase (UDP-glucuronosyltransferase, UGT), and glutathione-S-transferase (GST) [19]. Unfortunately, these antioxidant defenses have limited capacity and can be impaired during certain conditions, thereby leading to a redox imbalance that promotes the development of skin cancer.
The genes encoding cytoprotective detoxifying/antioxidant enzymes are controlled by the transcription factor Nrf2. Under homeostatic conditions, Nrf2 is bound to Kelch-like ECH-associated protein 1 (Keap1) in the cytoplasm. Nrf2 is targeted for polyubiquitination and proteasomal degradation through the formation of a Keap1- and Cullin 3-based-E3/Rbx1 ligase complex. Under stress conditions, Nrf2 dissociates from Keap1 and translocates to the nucleus, where it binds to the antioxidant response elements (AREs) of target protective genes and activates transcription [20]. Nrf2 has long been recognized as a pivotal player in the prevention of many diseases, including skin cancer. Nrf2 knockout mice are more susceptible to airway inflammation and asthma, striatal toxicity and behavioral dysfunction, colorectal carcinogenesis, gastric neoplasia, and skin carcinoma upon DMBA/TPA exposure compared with wild-type mice [21-25]. Moreover, a recent study demonstrated that low Nrf2 expression is associated with the oncogenic transformation of mesenchymal stem cells and poor survival in patients with skin cutaneous melanoma, kidney clear cell carcinoma, and prostate cancers [26]. Therefore, understanding the molecular mechanisms by which Nrf2 expression can be altered to slow or prevent the progression of skin cancer is of great importance.
Frequent epigenetic changes during the early stages of tumorigenesis lead to genetic aberrations and promote cancer development [27]. Epigenetics refers to changes in gene expression by DNA methylation and/or post-translational histone modification without alterations of the DNA sequence. The modifications to DNA and histones are driven by DNA methyltransferases (DNMTs) and histone deacetylases (HDACs), respectively [28]. DNA methylation occurs at the 5′ position of cytosines within CpG dinucleotides found in CpG islands. The silencing of tumor suppressor genes by the hypermethylation of CpG islands within promoter regions is a hallmark of cancer. Such methylation in CpG islands impedes the binding of transcription factors and represses transcription. Moreover, protein complexes, such as the methyl-CpG binding domain (MBD) family and HDACs, are recruited to specific loci where they alter the structure of the chromatin and facilitate gene silencing [28-30]. As such, epigenetic modifications as preventive targets have been the focus of numerous studies in cancer, largely due to the notion that epigenetic changes are reversible and affect numerous cellular events in tumorigenesis. The US FDA has approved four epigenetic agents for clinical use: the DNMT inhibitors 5-azacytidine (5-aza, azacytidine) and 5-aza-2′-deoxycytidine (decitabine) and the HDAC inhibitors suberoylanilide hydroxamic acid (vorinostat) and depsipeptide (romidepsin) [31]. However, off-target action, drug resistance and their selective applicability to selective cancers have mitigated their use in treating cancer [31, 32]. As a way of circumventing this challenge, natural compounds found in fruits, vegetables, teas, and medicinal plants have attracted considerable interest due to their ability to overcome oxidative stress and regulate epigenetic events at non-toxic concentrations [33-36]. The aim of this study is to demonstrate the chemopreventive effect of UA and identify UA-induced epigenetic modifications in mouse epidermal cells. We demonstrated that UA activated the Nrf2 pathway by demethylating the Nrf2 promoter and reducing the expression of DNMTs and HDACs, resulting in the inhibition of TPA-induced cell transformation.
2. Materials and Methods
2.1. Reagents and Antibodies
Minimum essential medium (MEM), fetal bovine serum (FBS), penicillin-streptomycin (10,000 U/ml), versene, and Trypsin-EDTA were supplied by Gibco (Grand Island, NY). A Cell-Titer 96 Aqueous One Solution Cell Proliferation Assay Kit was obtained from Promega (Madison, WI). Platinum Taq DNA polymerase was purchased from Invitrogen (Grand Island, NY). Tris-HCl precast gels, turbo transfer buffer, and PVDF membranes were obtained from Bio-Rad (Hercules, CA). Tris-Glycine-SDS running buffer was from Boston BioProducts (Ashland, MA). Super Signal enhanced chemiluminescent substrate, NE-PER Nuclear and Cytoplasmic Extraction Reagents, and BCA Protein Assay Kit were purchased from Thermo (Rockford, IL). Antibodies against Nrf2 (C-20), HO-1 (C-20), NQO1 (H-90), UGT1A1 (V-19), and actin (I-19) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and Abcam (Cambridge, MA). Anti-acetyl histone H3 was from Millipore (Billerica, MA). The protease inhibitor cocktail, radioimmunoprecipitation (RIPA) buffer, and antibodies against HDACs (HDAC1, HDAC2, HDAC3, HDAC4 and HDAC6) were supplied by Cell Signaling Technology (Beverly, MA). The anti-HDAC8 antibody was obtained from Proteintech Group (Chicago, IL), and the anti-HDAC5, -HDAC7, -DNMT3a and -DNMT3b antibodies were from Abcam (Cambridge, MA). Anti-DNMT1 was supplied by Novus Biologicals (Littleton, CO). All other chemicals, unless otherwise noted, were obtained from Sigma (St. Louis, MO).
2.2. Cell culture
JB6 P+ mouse epidermal cells were obtained from the American Type Culture Collection (Manassas, VA). The cells were cultured in MEM supplemented with 5% FBS and penicillin-streptomycin (100 units/ml) at 37°C under a 5% CO2 atmosphere. JB6 P+ cells stably transfected with shMock and shNrf2-knockdown (KD) were maintained in the same medium as JB6 P+ cells and 2 μg/mL puromycin was added.
2.3. Cell viability assay
JB6 P+ cells were seeded at a density of 5 × 104 cells/well into 96-well plates in 5% FBS/MEM. After 24 h, the medium was removed, and the cells were treated with UA (1 and 2.5 μM) in 1% FBS/MEM, where 0.1% DMSO was used as the vehicle control group. The medium containing UA was changed every 2 days for 3 and 5 days. On the day of the assay, 20 μl of Cell Titer 96 Aqueous One Solution in 100 μl of 1% FBS/MEM was added to each well, and the cells were then incubated for 1 h at 37°C in a 5% CO2 incubator. The absorbance was measured at 490 nm.
2.4. Anchorage-independent cell transformation assay
JB6 P+ cells (8 × 103/ml) were suspended in 1 ml of basal medium Eagle (BME) containing 0.33% agar and plated over 3 ml of a solidified BME consisting of 0.5% agar and 10% FBS in 6-well plates in the presence of TPA (20 ng/ml) alone or together with 1 or 2.5 μM UA. The cells were maintained at 37°C in a 5% CO2 incubator for 2 weeks. The cell colonies were photographed using a Nikon ACT-1 microscope (Version 2.20; LEAD Technologies) and counted using Image J (NIH, Bethesda, MD).
2.5. RNA extraction and quantitative real-time PCR (qPCR)
Total RNA was extracted from JB6 P+ cells on days 3 and 5 after treatment using the RNeasy mini kit (Qiagen, Valencia, CA). For cDNA synthesis, 0.5 μg of total RNA was incubated with oligo (dT)16 primers and MultiScribe reverse transcriptase (TaqMan reverse transcription reagents, Applied Biosystems, Grand Island, NY) with the following reaction conditions: 10 min at 25°C, 30 min at 48°C and 5 min at 95°C. The qPCR was performed with an ABI ViiA7 real-time PCR system (Applied Biosystems, Carlsbad, CA, USA) using synthesized cDNA, Power SYBR Green PCR Master Mix (Applied Biosystems, Carlsbad, CA), and a pair of gene-specific primers. β-actin was used as an internal control gene. Each sample was prepared in triplicate and normalized to β-actin. The primers for each qPCR reaction are as follows: Nrf2, 5′-AGCAGGACTGGAGAAGTT-3′ (sense) and 5′-TTCTTTTTCCAGCGAGGAGA-3′ (antisense); HO-1, 5′-CCTCACTGGCAGGAAATCATC-3′ (sense) and 5′-CCTCGTGGAGACGCTTTACATA-3′ (antisense); NQO1, 5′-AGCCCAGATATTGTGGCCG-3′ (sense) and 5′-CCTTTCAGAATGGCTGGCAC-3′ (antisense); UGT1A1, 5′-GAAATTGCTGAGGCTTTGGGCAGA-3′ (sense) and 5′-ATGGGAGCCAGAGTGTGTGATGAA-3′ (antisense); β-actin, 5′-AGAGGGAAATCGTGCGTGAC-3′ (sense) and 5′-CAATAGTGATGACCTGGCCGT-3′ (antisense)
2.6. Western blot analysis
JB6 P+ cells were seeded at a density of 1 × 105 cells in 100-mm dishes with 5% FBS/MEM. After 24 h, the cells were treated with either 0.1% DMSO, 5-azacytidine (5-aza, 250 nM), or each concentration of UA in 1% FBS/MEM. The medium containing each agent was changed every 2 days. The cells incubated with 5-azacytidine (5-aza) serving as a positive control were treated with trichostatin A (TSA, 50 nM) 24 h before harvest. On the day of the harvest, the cells were rinsed with cold PBS and resuspended in 100 μl of RIPA buffer containing a protease inhibitor cocktail and agitated on ice for 30 min. The cells were then centrifuged at 13,000 ×g for 15 min at 4°C, and only a clear supernatant was obtained. The total protein fraction (25 μg of protein) was separated by 4-15% Tris-HCl precast gels. The separated proteins were transferred onto PVDF membranes, which were blocked with PBS containing 0.05% Tween 20 (PBST) and 5% skim milk. After a sequential incubation of the membranes with the primary antibodies and the appropriate secondary antibodies, the immunoreactive bands were detected with the Super Signal enhanced chemiluminescent system and visualized using the Bio-Rad ChemiDoc imaging system (Bio-Rad, Hercules, CA). The band intensity was analyzed using Image J. The protein concentrations were determined using the bicinchoninic acid (BCA) assay.
2.7. DNA isolation and bisulfite genomic sequencing
Genomic DNA was isolated from each group of treated cells using the QIAamp DNA Mini Kit (Qiagen). Then, 500 ng of DNA was subjected to bisulfite treatment using the EZ DNA Methylation Gold Kit (Zymo Research, Irvine, CA). The converted DNA was amplified by touchdown PCR using bisulfite sequencing-specific primers for the first 15 CpG sites of the murine Nrf2 gene. Then, the PCR products were gel extracted using the DNA Extraction Kit (Qiagen) and cloned into pCR4 TOPO vectors (TA cloning kit, Invitrogen). Ten clones per group were sequenced using T7 primers (GeneWiz, South Plainfield, NJ). The sequences for the PCR are as follows: sense, 5′-AGTTATGAAGTAGTAGTAAAAA-3′; anti-sense, 5′-ACCCCAAAAAAATAAATAAATC-3′.
2.8. HDAC activity assay
Nuclear extracts from the treated cells were isolated using NE-PER Nuclear and Cytoplasmic Extraction Reagents. HDAC activity was measured in nuclear extracts using Epigenase HDAC Activity/Inhibition Direct Assay Kit (Epigentek Inc, Farmingdale, NY) following the manufacturer's protocol.
2.9. Statistical analysis
All of the quantitative results are expressed as the mean values ± SD of three independent experiments. Statistical significance was determined by one-way ANOVA and a p value of <0.05 was considered statistically significant in all analysis.
3. Results
3.1 UA inhibits the growth of JB6 P+ cells
We first examined the dose and time-dependent cytotoxicity of UA using mouse epidermal JB6 P+ cells. The cells were treated with six different concentrations of UA (0, 2.5, 0.5, 1, 2.5, 5 or 10 μM, final concentration) dissolved in DMSO (vehicle) for 3 and 5 days. Our previous studies and others have shown that it needs at least 3 days to have cells epigenetically altered [9-16, 37, 38]. UA was not cytotoxic up to 1 μM (Fig. 1); however, at 2.5 μM the cell viability decreased approximately 23% in comparison with vehicle (0.1% DMSO). No difference was observed between 3 and 5 days of treatment, and concentrations greater than 2.5 μM were found to be toxic. Because cell viability was greater than 70% at ≤2.5 μM and cytotoxicity was not time-dependent, the cells were treated with 1 and 2.5 μM UA for 3 days to study the chemopreventive efficacy of UA in the subsequent experiments.
Fig. 1.
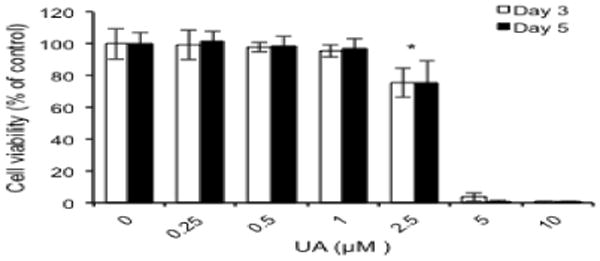
Effects of UA on the growth of JB6 P+ cells. Cells grown in a 96-well plate were treated with the indicated concentrations of UA, and cell viability was analyzed with an MTS assay after 3 and 5 days of treatment. The results are shown as the mean ± SD of triplicate experiments. *p < 0.05 compared with vehicle control (0.1% DMSO).
3.2 UA inhibits TPA-induced transformation of JB6 P+ cells
To determine whether UA exhibits anti-cancer and chemopreventive effects in skin, we studied the effects of UA on the tumor promotion of JB6 P+ cells induced by TPA. The anchorage-independent cell transformation assay is an in vitro system that allows only cells transformed by tumor promoters such as TPA to grow and form colonies. As expected, the cells treated with TPA alone for 2 weeks developed a significant amount of colonies in the soft agar (Fig. 2). The cells treated with TPA and 2.5 μM UA inhibited TPA-induced transformation by 30% compared with the cells treated with TPA alone. Incubation with 1 μM UA did not significantly inhibit transformation (16%). These results demonstrate the chemopreventive effects of UA against TPA-induced transformation in JB6 P+ cells.
Fig. 2.

UA inhibits TPA-induced transformation in JB6P+ cells. Cells (8 × 103/ml) in 1 ml of BME containing 0.33% agar were maintained in the presence of DMSO (control, a), TPA alone (b), UA 1 μM plus TPA (c) and UA 2.5 μM plus TPA. After 2 weeks, the cell colonies were counted. The data are presented as the mean ± S.D. ***p < 0.0001 compared with TPA alone.
3.3 UA upregulates Nrf2 and its downstream detoxifying/antioxidant target genes
TPA-induced ROS production stimulates the neoplastic transformation of JB6 P+ cells [39]. To test whether UA inhibits TPA-induced transformation through the induction of detoxifying/antioxidant enzymes, we investigated the expression levels of HO-1, NQO1 and UGT1A1 at the mRNA and protein levels using qPCR and Western blotting. The cells treated with 2.5 μM UA showed an increase in HO-1, NQO1 and UGT1A1 mRNA expression, whereas 1 μM UA did not (Fig. 3A). Similarly, protein expression was elevated by 2.5 μM UA treatment, but not by 1 μM UA (Fig. 3B).
Fig. 3.
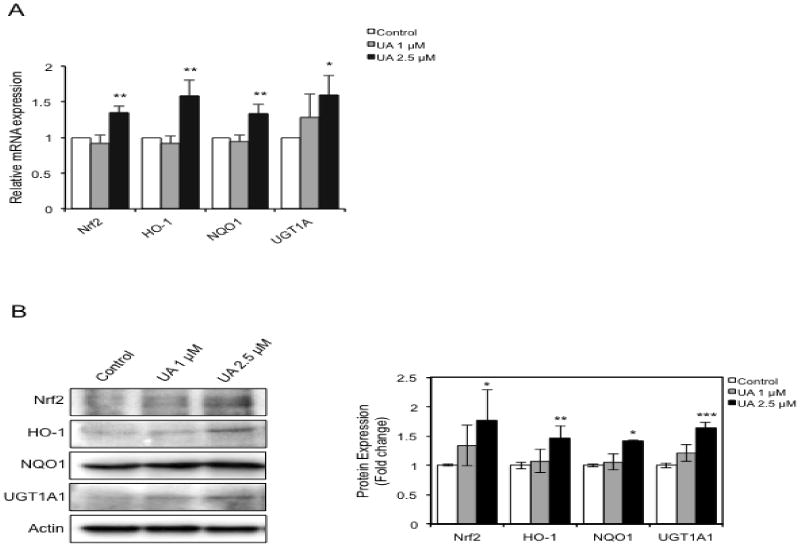
UA upregulates the expression of Nrf2 and its downstream target genes. Cells were treated with each concentration of UA for 3 days, and the total cells were divided for RNA and protein extraction. (A) Total 0.5 μg of RNA was reverse transcribed for cDNA synthesis. The cDNAs were then used to perform qPCR by adding SYBR Green and a pair of gene-specific primers. (B) Western blots and quantification of protein levels. The data shown were normalized to β-actin and expressed as the relative fold change compared with the control. The values are the mean ± SD of three independent experiments. *p < 0.05, **p < 0.001, ***p < 0.0001 compared with vehicle control (0.1% DMSO).
Nrf2 is regarded as an essential regulator of cytoprotective detoxifying/antioxidant enzymes. As such, we then determined whether UA increases Nrf2 expression in JB6 P+ cells. As expected, 2.5 μM UA treatment increased Nrf2 expression; however, 1 μM UA did not result in a significant increase in Nrf2 expression. These results demonstrate that UA inhibits TPA-induced transformation of JB6 P+ cells by, at least in part, augmenting detoxifying/antioxidant enzymes, which is mediated by enhanced Nrf2 expression.
3.4. Expression of Nrf2 downstream target genes by UA is Nrf2 dependent
Next, we clarified whether Nrf2 is required for induction of cytoprotective detoxifying/antioxidant genes by UA treatment. We used Nrf2-Mock and Nrf2-KD stable JB6 P+ cells established in our laboratory [14]. The basal expression of Nrf2 was decreased by about 70% in Nrf2-KD JB6 P+ cells compared with control Nrf2-Mock (Fig. 4). 2.5 μM UA treatment significantly increased protein expression of Nrf2, HO-1, NQO1 and UGT1 A1 in Nrf2-Mock JB6 P+ cells. Conversely, the inducing effects of 2.5 μM UA on the expression of Nrf2 downstream target genes was much smaller in Nrf2-KD compared to those in Nrf2-Mock treated; 30%, 52%, and 51% decrease of HO-1, NQO1, and UGT1A1, respectively. The results indicate that Nrf2 is a direct regulator driving expression of cytoprotective detoxifying/antioxidant genes by UA in JB6 P+ cells.
Fig. 4.
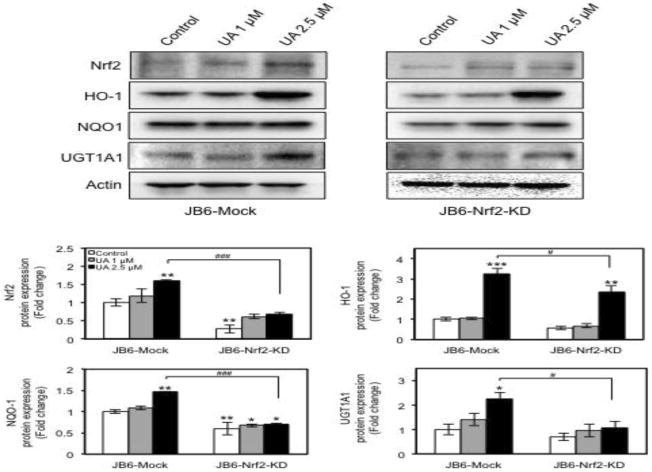
Nrf2 knockdown decreases expression of Nrf2 downstream target genes. Cells were treated with each concentration of UA for 3 days, and whole-cell extracts were prepared as described in Materials and Methods. Then, the proteins were subjected to Western blot to analyze the expression of Nrf2 and its downstream target genes indicated. The protein amounts were normalized to the levels of β-actin and expressed as the relative fold change compared with JB6-Mock control. The values are the mean ± SD of three independent experiments. *p < 0.05, **p < 0.001, and ***p < 0.0001 vs JB6-Mock control. #p < 0.05, ###p < 0.0001 vs JB6-Mock UA 2.5 μM.
3.5 UA decreases Nrf2 promoter methylation
We previously showed that promoter demethylation of Nrf2 is an important epigenetic mechanism underlying Nrf2 activation in prostate cancer TRAMPC1 cells and JB6 P+ cells treated with phytochemicals [9, 10, 12, 14, 15]. To determine whether Nrf2 was epigenetically regulated by UA, we determined the methylation status of the Nrf2 promoter using bisulfite genomic DNA sequencing. The first 15 CpG sites located between -1226 and -863 of the mouse Nrf2 gene promoter relative to the translation start site (+1) were analyzed [40]. As previously reported, the Nrf2 promoter was hypermethylated in JB6 P+ cells (89.3%) (Fig. 5). The cells treated with 5-aza and TSA, well-known inhibitors of DNA methylation and histone deacetylation, respectively, reduced methylation by 46.6%, which is similar to the results from previous studies [14]. Treatment of JB6 P+ cells with 2.5 μM UA decreased methylation by 17% compared with JB6 P+ cells treated with vehicle. Treatment with 1 μM UA resulted in only a 7% decrease in methylation. These results are in accordance with Fig. 3, which shows that the levels of Nrf2 mRNA and protein were increased by 2.5 μM UA treatment, but they were unchanged by 1 μM UA treatment. These findings suggest that UA induces Nrf2 expression by altering the methylation status of the Nrf2 promoter.
Fig. 5.

UA decreases Nrf2 promoter DNA methylation in JB6 P+ cells. The cells were treated with each concentration of UA for 3 days, and then the genomic DNA was isolated for bisulfite conversion. The methylation status of the first 15 CpG sites, the region between -1226 and -863 relative to the translational start site, within the promoter of Nrf2, was analyzed. Positive control cells were treated with 5-aza (250 nM) for 48 h and TSA (50 nM) for 24 h. Ten individual clones were analyzed. The filled and open dots indicate methylated and unmethylated CpG. The data are expressed as a percentage of the total number of methylated cytosines vs. total 15 CpGs of three independent experiments.
3.6 UA alters the levels of epigenetic modifying enzymes
To understand the mechanisms by which UA decreases Nrf2 promoter methylation in JB6 P+ cells, we determined an impact of UA on DNMTs and HDACs, which are involved in methylation-induced gene silencing [28]. The family of DNMTs consists of three members, DNMT1, DNMT3a, and DNMT3b. Treatment with 2.5 μM UA resulted in a significant reduction in DNMT1 and DNMT3a protein levels (Fig. 6A). In addition, 1 μM UA treatment also slightly decreased both DNMT1 and DNMT3a. No significant difference was found between the effects of treatment with 2.5 and 1 μM UA. The DNMT3b protein levels were unaffected by UA treatment. HDACs are classified into four groups: Class I (HDAC1, 2, 3, and 8), Class II (HDAC4, 5, 6, 7, 9, and 10), Class III (SIRT1-7), and Class IV (HDAC 11). UA has previously been reported to increase histone acetylation by strongly inhibiting HDAC1, 3, 4, 5, and 6 [41]. We examined the expression levels of HDAC 1, 2, 3 and 8 (Class I) and HDAC 4, 5, 6 and 7 (Class II). The protein expression levels of all HDACs were diminished in the JB6 P+ cells treated with 2.5 μM UA (Fig. 6B and 6C). Among them, HDAC2 and 8 showed a dose-dependent reduction. The expression of HDAC4 was not affected by UA treatment and HDAC5 was not detected. The decrease of HDAC expression confirmed the inhibition of HDAC activity, whereas the levels of acetylated histone H3 (H3ac), an epigenetic marker for active genes, was increased by 2.5 μM UA treatment (Fig. 6D). Taken together, these results indicate that UA-induced demethylation of the Nrf2 promoter is mediated by the negative regulation of epigenetic modification enzymes.
Fig. 6.
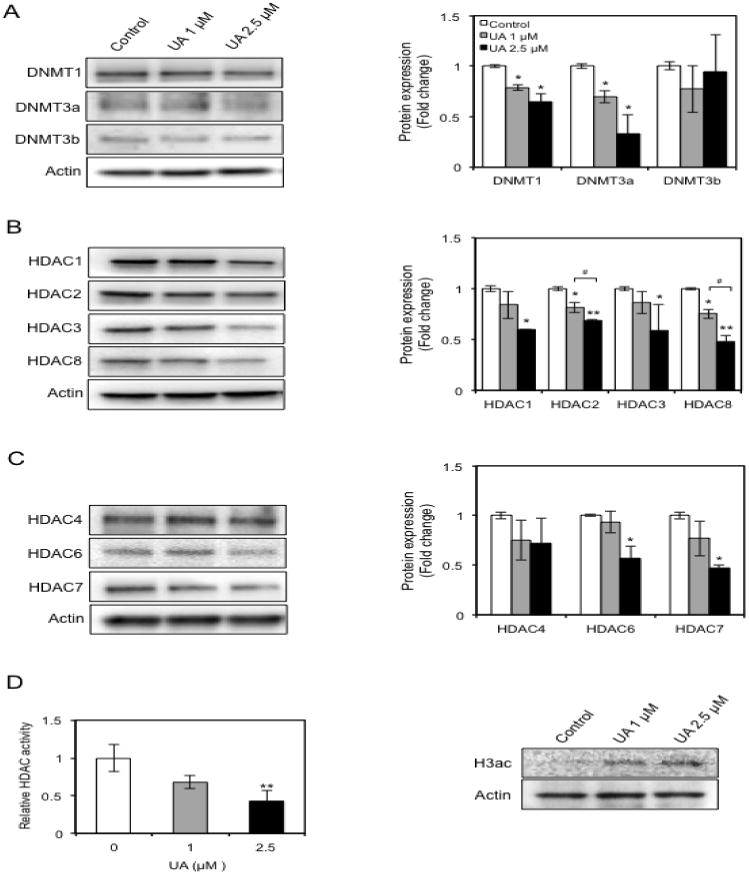
UA decreases the expression of DNMTs (A), Class I and II HDACs (B & C), inhibits HDAC activity, and increases H3ac levels (D) in JB6 P+ cells. The cells were treated with the indicated concentration of UA for 3 days, and the total cell lysates and nuclear proteins were harvested at the end of the treatment. Total protein (25 μg per lane) was separated by SDS-PAGE, and the levels of each protein of interest were determined by Western blot analysis. The isolated nuclear extracts from each group were used to determine total HDAC activity. The protein amounts in Western blot analysis were normalized to the levels of β-actin and data are expressed as the relative fold change compared with the control. The values are the mean ± SD of three independent experiments. The figure H3ac is a representative of three individual experiments. *p < 0.05 and **p < 0.001 vs control. #p < 0.05 vs UA 1 μM.
4. Discussion
A variety of external stimuli continuously make contact with the skin and accelerate the formation of ROS, impairing cellular metabolism, signal transduction, and genomic stability, and ultimately contributing to the development of skin cancer [42]. Therefore, inhibiting and/or reducing oxidative stress by ROS is crucial in preventing skin cancer. Many dietary phytochemicals eliminate ROS toxicity by inducing detoxifying/antioxidant enzymes via Nrf2 activation, which has led to a decrease in cancer development [34]. We have previously shown that curcumin, 3,3′-diindolylmethane, and a γ-tocopherol-rich mixture of tocopherols, sulforaphane, Z-ligustilide and radix angelica regulate Nrf2 activation through an epigenetic pathway in a prostate cancer model [10-13, 16]. Moreover, studies have shown that a variety of natural compounds interact with epigenetic regulators [30]. These studies suggest that natural dietary compounds that are able to epigenetically regulate gene expression are promising chemopreventive agents. Our findings demonstrate that UA, a naturally occurring triterpenoid in fruits and plants, restores the expression of the epigenetically silenced Nrf2 gene by demethylating CpG islands of the Nrf2 promoter, leading to upregulated Nrf2 expression. As a result, the expression of its target genes increases. Subsequently, this results in the inhibition of the TPA-induced neoplastic transformation in JB6 P+ cells.
JB6 P+ mouse epidermal cells, unlike P- cells, are susceptible to tumor promoter-induced transformation and are a suitable in vitro model to study progression in carcinogenesis and the molecular mechanisms of cancer chemoprevention [43]. Previous studies have used JB6 P+ cells to investigate whether dietary agents have the capacity to suppress transformation induced by tumor promoters [14, 15, 44-46]. Thus, we treated JB6 P+ cells with UA to test the chemopreventive potential of UA in TPA-induced transformation. The inhibitory effects of UA on tumor promotion by TPA and B[a]P or DMBA/TPA have been described in mouse skin [3, 4, 47]. Consistent with these reports, we found that UA was effective in inhibiting the transformation-inducing effects of TPA in JBP+ cells at a concentration in which the cytotoxicity was no more than 25% (Fig. 1 and 2). The cumulative ROS production is detected in TPA-induced transformation [43]. In addition, ROS inhibition by detoxifying/antioxidant enzymes attenuates TPA-induced transformation of JB6P+ cells [14, 48]. Conversely, several reports indicate that UA remarkably reduces oxidative stress and increases the activity of antioxidant enzymes [49-51]. We observed that the expression of HO-1 (antioxidant), NQO1, and UGT1A1 (detoxification) noticeably increased at both the mRNA and protein levels in the JB6 P+ cells treated with UA; however, TPA activated AP-1, NF-κB, and ERK 1/2 as well [43, 52]. Moreover, UA targets AP-1, NF-κB, and ERK 1/2 [2]. Hence, our observations suggest that the inhibition of TPA-induced transformation of JB6 P+ cells by UA is partially reliant on ROS reduction through the accumulation of antioxidative/detoxifying enzymes. How UA alters the expression and activity of AP-1, NF-κB, and ERK in TPA-induced transformation remains to be elucidated.
The production of phase II detoxifying/antioxidant enzymes is an innate cellular event that provides protection against deleterious endogenous and exogenous substances. In general, the genes encoding such cytoprotective enzymes are postulated to be regulated in an Nrf2-dependent manner. Thus, Nrf2 is central to the prevention of deleterious diseases, such as skin cancer. We have provided evidence that TPA-induced cell transformation is increased in Nrf2-KD JB6 P+ cells. Furthermore, the inhibitory effect of sulforaphane on TPA-induced cell transformation is blocked upon Nrf2-KD [14]. Many cancer chemopreventive agents acting via Nrf2 activation are phytochemicals. Some examples include carnosol, curcumin, epigallocatechin-3-gallate (EGCG), phenethyl isothiocyanate (PEITC), sulforaphane, and resveratrol [53, 54]. In our study, UA elevated the levels of Nrf2 mRNA and protein (Fig. 3). Additionally, Nrf2 deficiency in Nrf2-KD JB6 P+ cells lowered the effects of UA on the protein expression of detoxifying/antioxidant genes (Fig. 4). These results imply that UA is a chemopreventive dietary phytochemical that targets Nrf2. Our data are strongly supported by recent findings demonstrating that UA-driven activation of Nrf2 protects mice from neuronal defects induced by cerebral ischemia, and hepatotoxicity and fibrosis caused by CCl4. [7, 8].
Furthermore, an isomer of UA, oleanolic acid, and the synthetic oleanane triterpenoid CDDO (2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid), and its methyl (CDDO-Me) and imidazolide (CDDO-Im) derivatives have been shown to be potent Nrf2 inducers [55]. Upregulated Nrf2 expression can in part be achieved by the increased half-life of Nrf2, which is mediated by the reduction of Keap1-dependent ubiquitin/proteasome degradation of Nrf2 [56]. Keap1 is a suppressor protein of Nrf2. Under normal circumstances, Keap1 binds to Nrf2 and causes rapid Nrf2 degradation via polyubiquitination. By contrast, upon high oxidative stress, a subset of cysteine residues in Keap1 are modified, which perturbs the Keap1/Nrf2 interaction and hinders Nrf2 ubiquitination. This enables the accumulation and translocation of Nrf2 into the nucleus where it triggers the transcription of various phase II cytoprotective genes [53]. Sulforaphane modifies cysteine 151 within the BTB (Broad complex, Tramtrack, and Bric-a-brac) domain of Keap1, which results in lowered Nrf2 ubiquitination/degradation and increased stabilization [57]. Moreover, a previous study revealed that Keap1 allows common inducers of phase II genes to alter its cysteine sulfhydryl groups regardless of the inducers' structures [58]. As such, UA might modify cysteine residues in Keap1, resulting in an increase of Nrf2, which facilitates detoxifying/antioxidant expression by binding to the AREs in the promoters of its target genes.
DNA hypermethylation is the most common epigenetic modification in degenerative diseases such as cancer. This modification influences the depression of tumor suppressor genes. To date, many genes have been shown to be silenced by CpG hypermethylation within the promoter region during tumor progression. For example, in skin cancer, 14-3-3sigma (cell cycle), MGMT (DNA repair), RASSF1 (signal transduction), PTEN (apoptosis), and others have been shown to be hypermethylated [29]. Thus, discovering compounds that are able to reduce hypermethylation is an attractive strategy for the prevention of skin cancer. Studies by our group and others have revealed that Nrf2 expression is altered by methylation of CpG sites in the Nrf2 promoter region [9-16, 59]. These studies suggest that the epigenetic modulation of Nrf2 is likely to be a critical mechanism for Nrf2 activation. The present study demonstrates that UA decreased the methylation of the Nrf2 promoter in JB6 P+ cells. Although the effects were not comparable with those of the well-known epigenetic inhibitors 5-aza and TSA in combination, 2.5 μM UA treatment showed similar efficacy to that of 2.5 μmol/L sulforaphane and 6.25 μM apigenin in JB6 P+ cells (20% decrease compared with control in both). Notably, UA induces the expression of SHP-1, a tyrosine-specific protein phosphatase silenced by methylation in leukemias and lymphomas, in human multiple myeloma U266 cells [60]. These results suggest that UA has the potential to modulate DNA methylation, which is implicated in carcinogenesis. Concomitantly, we found that UA decreased the protein levels of DNMT1 and DNMT3a. DNMT1 preserves DNA methylation patterns across generations, whereas DNMT3a and 3b act as de novo methyltransferases [28]. The levels of DNMT1, DNMT3a, and DNMT3b are upregulated in UVB-induced murine skin tumors, and DNMT3a and DNMT3b are increased in stage III and IV cutaneous melanoma patients [61, 62]. Hence, our observations indicate that UA functions as a natural DNMT inhibitor to reduce DNA methylation in the skin. In cancer cells, DNMT1 and DNMT3b collaborate to maintain hypermethylation in the CpG islands of promoters [63]; however, UA did not have a significant effect on DNMT3b expression in JB6 P+ cells. This result may account for the weaker than expected inhibitory effect of UA on TPA-induced transformation and methylation of the Nrf2 promoter.
Hypermethylation in promoter regions provides binding sites for MeCP2, one of the MBD proteins, which subsequently recruits HDACs. HDACs remove acetyl groups from histones, mainly histone H3 and H4. This removal accelerates the formation of a compact chromatic structure, which drives the repression of transcription and causes gene silencing [28, 29]. Because HDACs such as HADC1, HDAC2, HDAC3, HDAC6, and HDC8 are overexpressed in many cancers [64], the discovery of selective HDAC inhibitors has had significant implications for cancer therapy. As natural HDAC inhibitors for skin cancer, EGCG and grape seed proanthocyanidins have been reported to decrease the level of HDAC1 and HDAC activity, accompanied by reduced expression and activity of DNMTs in squamous cell carcinoma [29, 37]. Recently, sulforaphane has been shown to reduce the protein levels of HDAC1-4 and 6 in human keratinocytes [65]. Interestingly, UA from Microtropis japonica significantly decreases the protein levels of HDAC1, 3, 4, 5, and 6 in HL-60 myeloid leukemia cells [41]. Similarly, in our experiments, UA downregulated all Class I HDACs, including HDAC1, 2, 3 and 8, and two from Class II HDACs, HDAC6 and 7 in JB6 P+ cells. Although HDAC4 expression did not decrease, similar results were found in JB6 P+ cells when treated with apigenin, sulforaphane, and tanshinone IIA [9, 14, 15]. A decrease of HDAC expression was linked to a reduced HDAC activity and a dramatic increase of H3ac (Fig. 6). Thus, UA-induced HDACs reduction results in a reduction of HDAC activity and, in turn, an enhanced acetylation of histone, which leads to epigenetic gene activation. Further, these data, together with the DNMTs results, imply that UA prevents DNA hypermethylation through the regulation of DNMTs and HDACs, unlike 5-aza and TSA, which are only specific for the inhibition of DNA methylation and histone deacetylation.
In summary, we demonstrate for the first time that UA restores the expression of Nrf2 by demethylating CpG islands in the Nrf2 promoter in mouse epidermal cells. This alteration is mediated by the reduced expression of enzymes involved in DNA methylation and histone deacetylation and the increased level of histone acetylation. The response to epigenetic alterations of Nrf2 by UA induced an increase in the expression of cytoprotective detoxifying/antioxidant enzymes, which resulted in the suppression of tumor promoter-induced cell transformation. Collectively, our data provide new insight into the function of UA as an epigenetic regulator for the prevention of skin cancer.
Acknowledgments
This work was supported in part by institutional funds.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ikeda Y, Murakami A, Ohigashi H. Ursolic acid: an anti- and pro-inflammatory triterpenoid. Molecular nutrition & food research. 2008;52:26–42. doi: 10.1002/mnfr.200700389. [DOI] [PubMed] [Google Scholar]
- 2.Shanmugam MK, Dai X, Kumar AP, Tan BK, Sethi G, Bishayee A. Ursolic acid in cancer prevention and treatment: molecular targets, pharmacokinetics and clinical studies. Biochemical pharmacology. 2013;85:1579–87. doi: 10.1016/j.bcp.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 3.Huang MT, Ho CT, Wang ZY, Ferraro T, Lou YR, Stauber K, et al. Inhibition of skin tumorigenesis by rosemary and its constituents carnosol and ursolic acid. Cancer research. 1994;54:701–8. [PubMed] [Google Scholar]
- 4.Tokuda H, Ohigashi H, Koshimizu K, Ito Y. Inhibitory effects of ursolic and oleanolic acid on skin tumor promotion by 12-O-tetradecanoylphorbol-13-acetate. Cancer letters. 1986;33:279–85. doi: 10.1016/0304-3835(86)90067-4. [DOI] [PubMed] [Google Scholar]
- 5.Harmand PO, Duval R, Delage C, Simon A. Ursolic acid induces apoptosis through mitochondrial intrinsic pathway and caspase-3 activation in M4Beu melanoma cells. International journal of cancer Journal international du cancer. 2005;114:1–11. doi: 10.1002/ijc.20588. [DOI] [PubMed] [Google Scholar]
- 6.Soo Lee Y, Jin DQ, Beak SM, Lee ES, Kim JA. Inhibition of ultraviolet-A-modulated signaling pathways by asiatic acid and ursolic acid in HaCaT human keratinocytes. European Journal of Pharmacology. 2003;476:173–8. doi: 10.1016/s0014-2999(03)02177-0. [DOI] [PubMed] [Google Scholar]
- 7.Li L, Zhang X, Cui L, Wang L, Liu H, Ji H, et al. Ursolic acid promotes the neuroprotection by activating Nrf2 pathway after cerebral ischemia in mice. Brain research. 2013;1497:32–9. doi: 10.1016/j.brainres.2012.12.032. [DOI] [PubMed] [Google Scholar]
- 8.Ma JQ, Ding J, Zhang L, Liu CM. Protective effects of ursolic acid in an experimental model of liver fibrosis through Nrf2/ARE pathway. Clinics and research in hepatology and gastroenterology. 2014 doi: 10.1016/j.clinre.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 9.Paredes-Gonzalez X, Fuentes F, Su ZY, Kong AN. Apigenin reactivates Nrf2 anti-oxidative stress signaling in mouse skin epidermal JB6 P + cells through epigenetics modifications. The AAPS journal. 2014;16:727–35. doi: 10.1208/s12248-014-9613-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khor TO, Huang Y, Wu TY, Shu L, Lee J, Kong AN. Pharmacodynamics of curcumin as DNA hypomethylation agent in restoring the expression of Nrf2 via promoter CpGs demethylation. Biochemical pharmacology. 2011;82:1073–8. doi: 10.1016/j.bcp.2011.07.065. [DOI] [PubMed] [Google Scholar]
- 11.Wu TY, Khor TO, Su ZY, Saw CL, Shu L, Cheung KL, et al. Epigenetic modifications ofNrf2 by 3,3′-diindolylmethane in vitro in TRAMP C1 cell line and in vivo TRAMP prostate tumors. The AAPS journal. 2013;15:864–74. doi: 10.1208/s12248-013-9493-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang Y, Khor TO, Shu L, Saw CL, Wu TY, Suh N, et al. A gamma-tocopherol-rich mixture of tocopherols maintains Nrf2 expression in prostate tumors of TRAMP mice via epigenetic inhibition of CpG methylation. The Journal of nutrition. 2012;142:818–23. doi: 10.3945/jn.111.153114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang C, Su ZY, Khor TO, Shu L, Kong AN. Sulforaphane enhances Nrf2 expression in prostate cancer TRAMP C1 cells through epigenetic regulation. Biochemical pharmacology. 2013;85:1398–404. doi: 10.1016/j.bcp.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Su ZY, Zhang C, Lee JH, Shu L, Wu TY, Khor TO, et al. Requirement and epigenetics reprogramming of Nrf2 in suppression of tumor promoter TPA-induced mouse skin cell transformation by sulforaphane. Cancer prevention research (Philadelphia, Pa) 2014;7:319–29. doi: 10.1158/1940-6207.CAPR-13-0313-T. [DOI] [PubMed] [Google Scholar]
- 15.Wang L, Zhang C, Guo Y, Su ZY, Yang Y, Shu L, et al. Blocking of JB6 cell transformation by tanshinone IIA: epigenetic reactivation of Nrf2 antioxidative stress pathway. The AAPS journal. 2014;16:1214–25. doi: 10.1208/s12248-014-9666-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Su ZY, Khor TO, Shu L, Lee JH, Saw CL, Wu TY, et al. Epigenetic reactivation of Nrf2 in murine prostate cancer TRAMP C1 cells by natural phytochemicals Z-ligustilide and Radix angelica sinensis via promoter CpG demethylation. Chemical research in toxicology. 2013;26:477–85. doi: 10.1021/tx300524p. [DOI] [PubMed] [Google Scholar]
- 17.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA: a cancer journal for clinicians. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 18.Leiter U, Eigentler T, Garbe C. Epidemiology of skin cancer. Advances in experimental medicine and biology. 2014;810:120–40. doi: 10.1007/978-1-4939-0437-2_7. [DOI] [PubMed] [Google Scholar]
- 19.Chun KS, Kundu J, Kundu JK, Surh YJ. Targeting Nrf2-Keap1 signaling for chemoprevention of skin carcinogenesis with bioactive phytochemicals. Toxicology letters. 2014;229:73–84. doi: 10.1016/j.toxlet.2014.05.018. [DOI] [PubMed] [Google Scholar]
- 20.Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox biology. 2013;1:45–9. doi: 10.1016/j.redox.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, et al. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. The Journal of experimental medicine. 2005;202:47–59. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Calkins MJ, Johnson DA, Townsend JA, Vargas MR, Dowell JA, Williamson TP, et al. The Nrf2/ARE pathway as a potential therapeutic target in neurodegenerative disease. Antioxidants & redox signaling. 2009;11:497–508. doi: 10.1089/ars.2008.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khor TO, Huang MT, Prawan A, Liu Y, Hao X, Yu S, et al. Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer prevention research (Philadelphia, Pa) 2008;1:187–91. doi: 10.1158/1940-6207.CAPR-08-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, et al. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:3410–5. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu C, Huang MT, Shen G, Yuan X, Lin W, Khor TO, et al. Inhibition of 7,12-dimethylbenz(a)anthracene-induced skin tumorigenesis in C57BL/6 mice by sulforaphane is mediated by nuclear factor E2-related factor 2. Cancer research. 2006;66:8293–6. doi: 10.1158/0008-5472.CAN-06-0300. [DOI] [PubMed] [Google Scholar]
- 26.Funes JM, Henderson S, Kaufman R, Flanagan JM, Robson M, Pedley B, et al. Oncogenic transformation of mesenchymal stem cells decreases Nrf2 expression favoring in vivo tumor growth and poorer survival. Molecular cancer. 2014;13:20. doi: 10.1186/1476-4598-13-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nature reviews Cancer. 2006;6:107–16. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- 28.Tsai HC, Baylin SB. Cancer epigenetics: linking basic biology to clinical medicine. Cell research. 2011;21:502–17. doi: 10.1038/cr.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saha K, Hornyak TJ, Eckert RL. Epigenetic cancer prevention mechanisms in skin cancer. The AAPS journal. 2013;15:1064–71. doi: 10.1208/s12248-013-9513-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shankar S, Kumar D, Srivastava RK. Epigenetic modifications by dietary phytochemicals: implications for personalized nutrition. Pharmacology & therapeutics. 2013;138:1–17. doi: 10.1016/j.pharmthera.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Helin K, Dhanak D. Chromatin proteins and modifications as drug targets. Nature. 2013;502:480–8. doi: 10.1038/nature12751. [DOI] [PubMed] [Google Scholar]
- 32.Hatzimichael E, Crook T. Cancer epigenetics: new therapies and new challenges. Journal of drug delivery. 2013;2013:529312. doi: 10.1155/2013/529312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vanden Berghe W. Epigenetic impact of dietary polyphenols in cancer chemoprevention: lifelong remodeling of our epigenomes. Pharmacological research : the official journal of the Italian Pharmacological Society. 2012;65:565–76. doi: 10.1016/j.phrs.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 34.Lee JH, Khor TO, Shu L, Su ZY, Fuentes F, Kong AN. Dietary phytochemicals and cancer prevention: Nrf2 signaling, epigenetics, and cell death mechanisms in blocking cancer initiation and progression. Pharmacology & therapeutics. 2013;137:153–71. doi: 10.1016/j.pharmthera.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miceli M, Bontempo P, Nebbioso A, Altucci L. Natural compounds in epigenetics: a current view. Food and chemical toxicology : an international journal published for the British Industrial Biological Research Association. 2014;73:71–83. doi: 10.1016/j.fct.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 36.Schnekenburger M, Dicato M, Diederich M. Plant-derived epigenetic modulators for cancer treatment and prevention. Biotechnology advances. 2014;32:1123–32. doi: 10.1016/j.biotechadv.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 37.Vaid M, Prasad R, Singh T, Jones V, Katiyar SK. Grape seed proanthocyanidins reactivate silenced tumor suppressor genes in human skin cancer cells by targeting epigenetic regulators. Toxicology and applied pharmacology. 2012;263:122–30. doi: 10.1016/j.taap.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang LS, Kuo CT, Cho SJ, Seguin C, Siddiqui J, Stoner K, et al. Black raspberry-derived anthocyanins demethylate tumor suppressor genes through the inhibition of DNMT1 and DNMT3B in colon cancer cells. Nutr Cancer. 2013;65:118–25. doi: 10.1080/01635581.2013.741759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dhar A, Young MR, Colburn NH. The role of AP-1, NF-kappaB and ROS/NOS in skin carcinogenesis: the JB6 model is predictive. Molecular and cellular biochemistry. 2002;234-235:185–93. [PubMed] [Google Scholar]
- 40.Yu S, Khor TO, Cheung KL, Li W, Wu TY, Huang Y, et al. Nrf2 expression is regulated by epigenetic mechanisms in prostate cancer of TRAMP mice. PloS one. 2010;5:e8579. doi: 10.1371/journal.pone.0008579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen IH, Lu MC, Du YC, Yen MH, Wu CC, Chen YH, et al. Cytotoxic triterpenoids from the stems of Microtropis japonica. Journal of natural products. 2009;72:1231–6. doi: 10.1021/np800694b. [DOI] [PubMed] [Google Scholar]
- 42.Bickers DR, Athar M. Oxidative stress in the pathogenesis of skin disease. The Journal of investigative dermatology. 2006;126:2565–75. doi: 10.1038/sj.jid.5700340. [DOI] [PubMed] [Google Scholar]
- 43.Yang HS, Knies JL, Stark C, Colburn NH. Pdcd4 suppresses tumor phenotype in JB6 cells by inhibiting AP-1 transactivation. Oncogene. 2003;22:3712–20. doi: 10.1038/sj.onc.1206433. [DOI] [PubMed] [Google Scholar]
- 44.Bode AM, Ma WY, Surh YJ, Dong Z. Inhibition of epidermal growth factor-induced cell transformation and activator protein 1 activation by [6]-gingerol. Cancer research. 2001;61:850–3. [PubMed] [Google Scholar]
- 45.Dong Z, Ma W, Huang C, Yang CS. Inhibition of tumor promoter-induced activator protein 1 activation and cell transformation by tea polyphenols, (-)-epigallocatechin gallate, and theaflavins. Cancer research. 1997;57:4414–9. [PubMed] [Google Scholar]
- 46.Lu YP, Chang RL, Lou YR, Huang MT, Newmark HL, Reuhl KR, et al. Effect of curcumin on 12-O-tetradecanoylphorbol-13-acetate- and ultraviolet B light-induced expression of c-Jun and c-Fos in JB6 cells and in mouse epidermis. Carcinogenesis. 1994;15:2363–70. doi: 10.1093/carcin/15.10.2363. [DOI] [PubMed] [Google Scholar]
- 47.Kowalczyk MC, Junco JJ, Kowalczyk P, Tolstykh O, Hanausek M, Slaga TJ, et al. Effects of combined phytochemicals on skin tumorigenesis in SENCAR mice. International journal of oncology. 2013;43:911–8. doi: 10.3892/ijo.2013.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shin JW, Ohnishi K, Murakami A, Lee JS, Kundu JK, Na HK, et al. Zerumbone induces heme oxygenase-1 expression in mouse skin and cultured murine epidermal cells through activation of Nrf2. Cancer prevention research (Philadelphia, Pa) 2011;4:860–70. doi: 10.1158/1940-6207.CAPR-10-0354. [DOI] [PubMed] [Google Scholar]
- 49.Shih YH, Chein YC, Wang JY, Fu YS. Ursolic acid protects hippocampal neurons against kainate-induced excitotoxicity in rats. Neuroscience letters. 2004;362:136–40. doi: 10.1016/j.neulet.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 50.Saravanan R, Pugalendi V. Impact of ursolic acid on chronic ethanol-induced oxidative stress in the rat heart. Pharmacological reports : PR. 2006;58:41–7. [PubMed] [Google Scholar]
- 51.Senthil S, Chandramohan G, Pugalendi KV. Isomers (oleanolic and ursolic acids) differ in their protective effect against isoproterenol-induced myocardial ischemia in rats. International journal of cardiology. 2007;119:131–3. doi: 10.1016/j.ijcard.2006.07.108. [DOI] [PubMed] [Google Scholar]
- 52.Saikali M, Ghantous A, Halawi R, Talhouk SN, Saliba NA, Darwiche N. Sesquiterpene lactones isolated from indigenous Middle Eastern plants inhibit tumor promoter-induced transformation of JB6 cells. BMC complementary and alternative medicine. 2012;12:89. doi: 10.1186/1472-6882-12-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bryan HK, Olayanju A, Goldring CE, Park BK. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochemical pharmacology. 2013;85:705–17. doi: 10.1016/j.bcp.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 54.Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes & development. 2013;27:2179–91. doi: 10.1101/gad.225680.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liby KT, Sporn MB. Synthetic oleanane triterpenoids: multifunctional drugs with a broad range of applications for prevention and treatment of chronic disease. Pharmacological reviews. 2012;64:972–1003. doi: 10.1124/pr.111.004846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang DD, Hannink M. Distinct Cysteine Residues in Keap1 Are Required for Keap1-Dependent Ubiquitination of Nrf2 and for Stabilization of Nrf2 by Chemopreventive Agents and Oxidative Stress. Molecular and Cellular Biology. 2003;23:8137–51. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hur W, Gray NS. Small molecule modulators of antioxidant response pathway. Current opinion in chemical biology. 2011;15:162–73. doi: 10.1016/j.cbpa.2010.12.009. [DOI] [PubMed] [Google Scholar]
- 58.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, et al. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:11908–13. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kang KA, Piao MJ, Kim KC, Kang HK, Chang WY, Park IC, et al. Epigenetic modification of Nrf2 in 5-fluorouracil-resistant colon cancer cells: involvement of TET-dependent DNA demethylation. Cell death & disease. 2014;5:e1183. doi: 10.1038/cddis.2014.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pathak AK, Bhutani M, Nair AS, Ahn KS, Chakraborty A, Kadara H, et al. Ursolic acid inhibits STAT3 activation pathway leading to suppression of proliferation and chemosensitization of human multiple myeloma cells. Molecular cancer research : MCR. 2007;5:943–55. doi: 10.1158/1541-7786.MCR-06-0348. [DOI] [PubMed] [Google Scholar]
- 61.Nandakumar V, Vaid M, Tollefsbol TO, Katiyar SK. Aberrant DNA hypermethylation patterns lead to transcriptional silencing of tumor suppressor genes in UVB-exposed skin and UVB-induced skin tumors of mice. Carcinogenesis. 2011;32:597–604. doi: 10.1093/carcin/bgq282. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 62.Nguyen T, Kuo C, Nicholl MB, Sim MS, Turner RR, Morton DL, et al. Downregulation of microRNA-29c is associated with hypermethylation of tumor-related genes and disease outcome in cutaneous melanoma. Epigenetics : official journal of the DNA Methylation Society. 2011;6:388–94. doi: 10.4161/epi.6.3.14056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2002;416:552–6. doi: 10.1038/416552a. [DOI] [PubMed] [Google Scholar]
- 64.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nature reviews Drug discovery. 2006;5:769–84. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 65.Dickinson SE, Rusche JJ, Bec SL, Horn DJ, Janda J, Rim SH, et al. The effect of sulforaphane on histone deacetylase activity in keratinocytes: Differences between in vitro and in vivo analyses. Molecular carcinogenesis. 2014 doi: 10.1002/mc.22224. [DOI] [PMC free article] [PubMed] [Google Scholar]