

Abstract
Background
Tuberculosis (TB) is a serious infectious disease in that 90 % of those latently infected with Mycobacterium tuberculosis present no symptoms, but possess a 10 % lifetime chance of developing active TB. To prevent the spread of the disease, early diagnosis is crucial. However, current methods of detection require improvement in sensitivity, efficiency or specificity. In the present study, we conducted a microarray experiment, comparing the gene expression profiles in the peripheral blood mononuclear cells among individuals with active TB, latent infection, and healthy conditions in a Taiwanese population.
Results
Bioinformatics analysis revealed that most of the differentially expressed genes belonged to immune responses, inflammation pathways, and cell cycle control. Subsequent RT-PCR validation identified four differentially expressed genes, NEMF, ASUN, DHX29, and PTPRC, as potential biomarkers for the detection of active and latent TB infections. Receiver operating characteristic analysis showed that the expression level of PTPRC may discriminate active TB patients from healthy individuals, while ASUN could differentiate between the latent state of TB infection and healthy condidtion. In contrast, DHX29 may be used to identify latently infected individuals among active TB patients or healthy individuals. To test the concept of using these biomarkers as diagnostic support, we constructed classification models using these candidate biomarkers and found the Naïve Bayes-based model built with ASUN, DHX29, and PTPRC to yield the best performance.
Conclusions
Our study demonstrated that gene expression profiles in the blood can be used to identify not only active TB patients, but also to differentiate latently infected patients from their healthy counterparts. Validation of the constructed computational model in a larger sample size would confirm the reliability of the biomarkers and facilitate the development of a cost-effective and sensitive molecular diagnostic platform for TB.
Electronic supplementary material
The online version of this article (doi:10.1186/s12859-015-0848-x) contains supplementary material, which is available to authorized users.
Keywords: Tuberculosis, Latent infection, Gene expression, Biomarker
Background
Tuberculosis (TB) is an infectious disease caused by various strains of mycobacteria, with Mycobacterium tuberculosis (Mtb) being the most common causative agent [1]. It is a serious global health threat with one-third of the world’s population estimated to be latently infected with Mtb [2]. Though about 90 % of those infected with Mtb are asymptomatic, possessing only a 10 % lifetime chance of developing active TB [3], even in developing countries with established healthcare systems, TB is still a deadly disease.
In 2006, the World Health Organization launched a “Global Plan to Stop Tuberculosis” that aims to save 14 million lives from TB by 2015. This objective is being hampered by the increase in HIV-associated tuberculosis and the emergence of multiple drug-resistant tuberculosis (MDR-TB) [4]. The only currently available vaccine is bacillus Calmette–Guérin (BCG) [5]. The vaccine is often administered to children, but the effectiveness of protection decreases after about 10 years.
With TB being one of the most common causes of death from infectious diseases, the current challenge is developing a sensitive and efficient method for the detection of latent TB infection (LTBI). The disease begins in the lungs via infection from the blood stream or aerosol droplets [6]. After TB bacteria enter the bloodstream, they can spread throughout the body and infect various tissues [7], such as the heart skeletal muscles, pancreas, or thyroid [8]. However, in LTBI, the bacteria remain dormant for several years before producing active TB. Even after treatment, the affected individual may still be susceptible to reactivation due to immunosuppression, or multiple-drug resistance in TB bacteria [4].
Substantial gene expression studies have revealed differences in the transcriptome between healthy controls and active TB or LTBI patients [9–11]. These findings not only uncovered important genetic signatures indicative of active TB and LTBI, but also identified transcriptionally regulated markers that are diverse in functions. In particular, these candidate genes are responsible for various key biological processes including inflammatory responses, immune defense, cell activation, homeostatic processes, regulation of cell proliferation and apoptosis. Moreover, these studies demonstrated the importance of cytokine and chemokine responses in the progression from latent infection to active disease [12–14]. However, the overall gene expression array results vary due to diverse genetic background of the study population and differences in the study design.
Early diagnosis of TB is crucial for preventing its spread, but the detection of LTBI is a major challenge as the carriers are often asymptomatic. Sputum smear acid-fast staining, though fast and inexpensive, is not the most sensitive and specific diagnostic test. While the tuberculin skin test represents a common diagnostic method, it has a tendency to produce false-positive results in individuals previously inoculated with BCG [15]. Culturing of TB bacteria usually takes time and diagnosis based on the test results is not always accurate. The interferon gamma release assays (IGRA) seem to have the potential of becoming the gold standard for TB test. The assays have been introduced into clinical practice to measure the amount of interferon-gamma (IFN-γ) released by blood cells infected wtih Mtb [16]. Unfortunately, this method is more expensive and requires blood samples with normal levels of viable leukocytes, which is not always possible in immunocompromised individuals. Consequently, an alternative quantitative polymerase chain reaction method was developed to detect the immune response to TB infection [17]. Yet, as most gene expression study results suggest, genetic background may influence the specificity and sensitivity of diagnosis.
Recently, Lu et al. (2011) conducted a gene expression microarray study to investigate the possibility of using mRNAs as biomarkers to differentiate active TB from LTBI [18]. Interestingly, in their study, the expression of IFN-γ, the biomarker used in IGRA, was not significantly different between the active TB and LTBI group [18]. Instead, the combination of three genes, CXCL10 (chemokine C-X-C motif ligand 10), ATP10A (ATPase, class V, type 10A) and TLR6 (toll-like receptor 6) appeared to be effective at distinguishing between active and latent TB infection. In contrast, IL-8 (Interleukin 8), FOXP3 (forkhead box P3), and IL-12β (interleukin 12 beta) were demonstrated to be the best discriminating biomarkers for TB and LTBI by Wu et al. [14]. Discrepancies between the two studies may be attributable to the differences in genetic background. At the same time, these findings suggest that not only are gene expression biomarkers more significant indicators of active TB, but they may also represent a more sensitive detection method for LTBI. Nevertheless, the same combination of genetic markers may not be applicable in another population.
For the present study, we attempted to compare the gene expression profiles in peripheral blood mononuclear cells among individuals with active TB, LTBI, and healthy conditions. We identified a panel of mRNAs that differed among these groups and subsequent validations with independent samples established the potential use of these gene expression biomarkers for the discrimination of LTBI from active TB in the Taiwanese population.
Results
Differentially expressed genes among TB, LTBI, and healthy controls
To identify candidate genes whose expression levels may differentiate among TB, LTBI, and healthy controls, we followed the workflow as illustrated in Fig. 1. The TB, LTBI, and healthy controls recruited for gene expression profiling did not differ significantly in age (One-way ANOVA: F2,18 = 0.21, p = 0.81; Additional file 1). Compared to healthy individuals, 31 and 16 genes were up-regulated in TB and LTBI, respectively (Fig. 2). While a total of 267 genes showed significantly reduced expression in TB patients relative to healthy controls, 111 genes appeared to be expressed at a lower level in those affected with LTBI compared with their healthy counterparts. Between TB and LTBI, 169 genes were differentially expressed, with 103 genes presenting increased abundance and 66 genes exhibiting decreased expression in LTBI relative to TB. Among these differentially expressed genes, three and 11 were also up-regulated and down-regulated, respectively, between LTBI and healthy controls. A list of the differentially expressed genes is provided in Additional file 2.
Fig. 1.
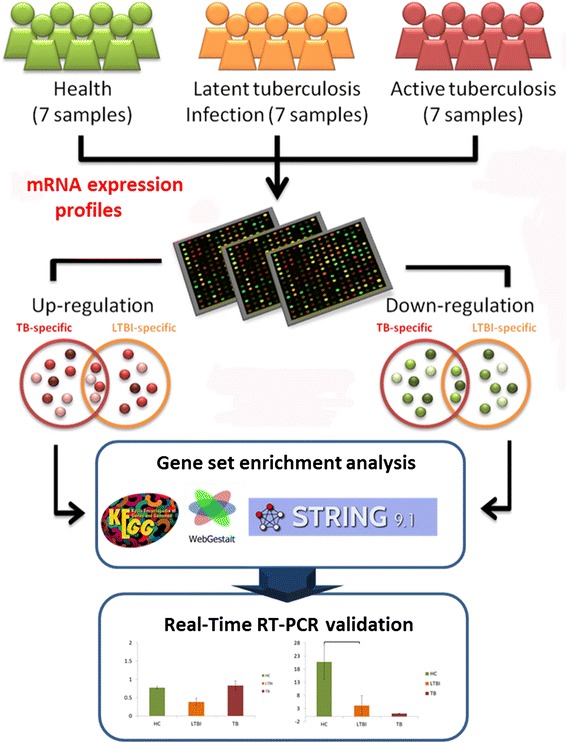
System flow of our analysis
Fig. 2.
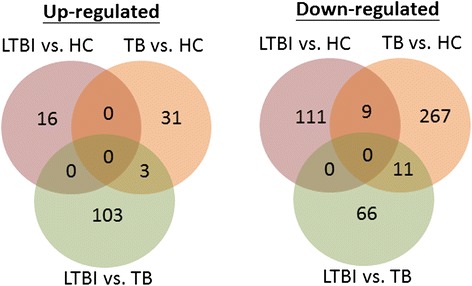
Number of differentially expressed genes among TB, LTBI, and healthy control (HC) group. Significant differential expression is represented by an absolute log2 fold change ≥1, FDR < 0.05
Functions, pathways, and interactions associated with the differentially expressed genes
According to the gene set enrichment analysis, genes differentially expressed among TB, LTBI, and healthy controls were over-represented in different GO categories (Table 1). The detailed lists of GO comparisons can be found in Additional files 3, 4, and 5 for LTBI reltiave to healthy controls, TB relative to healthy controls, and LTBI relative to TB, respectively. Compared with healthy controls, TB-specific gene expression profile appeared to be mostly related to leukocyte differentiation, lymphocyte activation, chemokine receptor activity, and regulation of immune response. In contrast, those latently infected with TB and healthy controls showed differing expression in genes belonging to regulation of metabolism, apoptosis, translation, and signal transduction pathways involving MAP kinase phosphatase and protein tyrosine/threonine phosphatase activities. Between the TB and LTBI group, the differentially expressed genes were not only enriched in immune system associated categories such as immune response activation and regulation, as well as natural killer cell and T-cell differentiation, but these genes were also involved in cellular processes like translation, transcription, and mRNA catabolism.
Table 1.
Gene Ontology categories enriched by differentially expressed genes among TB, LTBI, and healthy control (HC)
| TB vs. HC | LTBI vs. HC | LTBI vs. TB |
|---|---|---|
| 1. Immune response | Regulation of metabolic process | Response to cold |
| 2. Leukocyte differentiation | Regulation of cellular Metabolic process | Immune Response-regulating signaling pathway |
| 3. Immune system process | Regulation of biosynthetic process | Cellular process |
| 4. B cell activation | Apoptotic process | Immune response- activating signal transduction |
| 5. Lymphocyte differentiation | Death | Heterotypic cell-cell adhesion |
| 6. Regulation of immune response | Regulation of gene expression | NK T cell differentiation |
| 7. Positive regulation of response to stimulus | MAP kinase phosphatase activity | Regulation of mRNA catabolic process |
| 8. Lymphocyte activation | Translation factor activity, | Translation regulator activity |
| 9. Leukocyte activation | Translation initiation factor activity | NF-kappaB binding |
| 10. Chemokine receptor activity | Protein tyrosine/threonine phosphatase activity | Translation repressor activity |
WebGestalt setting: multiple test adjustment = Benjamini-Hochberg, significance level = top 10 (Benjamini-Hochberg adjusted p < 0.05); minimum number of genes for a category = 2
Pathway analysis revealed that relative to LTBI and healthy controls, most genes affected by active TB appeared to be involved in the regulation of immune responses (Table 2). For example, many differentially expressed genes between the TB and healthy control group were mapped to pathways associated with cytokine-cytokine receptor interaction, inflammatory responses such as rheumatoid arthritis, graft-versus-host disease and cancer. In addition, the transcriptional profiles that differed between LTBI and TB showed genes concentrated in apoptosis and signaling pathways involving chemokines, Toll-like receptors, and lymphocytes such as B- and T-cells. On the other hand, for genes differentially expressed between LTBI and healthy control, the most enriched pathway belonged to MAPK signaling cascade, followed by adipocytokine signaling modulated inflammatory response and Toll-like receptor signaling mediated innate immunity. The full lists of pathway comparisons for LTBI reltiave to healthy controls, TB relative to healthy controls, and LTBI relative to TB can be found in Additional files 6, 7, and 8, respectively.
Table 2.
Pathways enriched by differentially expressed genes among TB, LTBI, and healthy control (HC)
| KEGG pathway | ID | Genes |
|---|---|---|
| TB vs. HC | ||
| Cytokine-cytokine receptor interaction | 04060 | CCR6, IL7R, CCR7, FLT1, CCR2, TNFSF13B, FASLG, IL2RB, CD27, IL23A, TGFBR2 |
| Rheumatoid arthritis | 05323 | TLR4, IL23A, HLA-DOB, ATP6V0E2, FLT1, TNFSF13B |
| Pathways in cancer | 05200 | MYC, ITGA6, FASLG, RASSF5, HSP90AA1, CASP8, TRAF4, TRAF5, TGFBR2, ETS1 |
| Graft-versus-host disease | 05332 | FASLG, KLRC1, PRF1, HLA-DOB |
| MAPK signaling pathway | 04010 | FASLG, RRAS2, TGFBR2, MYC, RASA2, PRKACB, MAP3K1 |
| LTBI vs. HC | ||
| MAPK signaling pathway | 04010 | TNF, JUN, DUSP2, ATF4, NR4A1, FOS, DUSP5, DDIT3, PDGFRB, DUSP1 |
| Adipocytokine signaling pathway | 04920 | POMC, TNF, STK11, CAMKK2, NFKBIA, SOCS3 |
| Leishmaniasis | 05140 | TNF, JUN, FOS, NFKBIA |
| Toll-like receptor signaling pathway | 04620 | TNF, JUN, FOS, NFKBIA |
| Cytokine-cytokine receptor interaction | 04060 | TNF, CXCL2, TNFSF14, PDGFRB, OSM |
| LTBI vs. TB | ||
| Chemokine signaling pathway | 04062 | GSK3B, CCR6, CXCL16, ARRB2, PIK3CG, NFKBIA, PRKACB |
| Apoptosis | 04210 | CASP8, PIK3CG, NFKBIA, TNFRSF10B, PRKACB |
| T cell receptor signaling pathway | 04660 | GSK3B, PTPRC, FOS, PIK3CG, NFKBIA |
| Toll-like receptor signaling pathway | 04620 | LY96, CASP8, FOS, PIK3CG, NFKBIA |
| B cell receptor signaling pathway | 04662 | GSK3B, FOS, PIK3CG, NFKBIA |
Benjamini-Hochberg adjusted p < 0.05
Protein interaction analysis identified specific interaction network modules for active TB, LTBI, and healthy controls. The network modules were grouped according to their GO annotations and have been cross-validated with the STRING database [19, 20] (Fig. 3). Among the genes differentially expressed between LTBI and healthy controls, protein interactions involved in transcriptional regulation (ATF3, ATF4, JUNB, FOSB, and DDIT3), as well as translation initiation (EIF1 and EIF5) appeared to be the most important. Whereas proteins that regulate interferon-beta production (LY96 and TLR4), apoptotic signaling (HSP90AA1, LRRK2, TGFBR2, FASLG, CASP8,), bacterial invasion (SEPT1 and SEPT6), and Wnt signaling pathway (HIC1 and CTBP2) seemed to represent the underlying variations between TB and healthy controls, the differences between TB and LTBI might be contributed by proteins that modulate transcription (FOS and DDIT3), phagosome formation (TUBA1A and TUBB4B), autophagy (CASP8 and TNFRSF10B), and interferon-gamma signaling (ARRB2, PTAFR, NFKBIA). Additional files 9, 10, and 11 contain detailed lists of enriched protein interaction modules associated with genes differentially expressed in LTBI relative to healthy controls, TB relative to healthy controls, LTBI relative to TB, respectively.
Fig. 3.
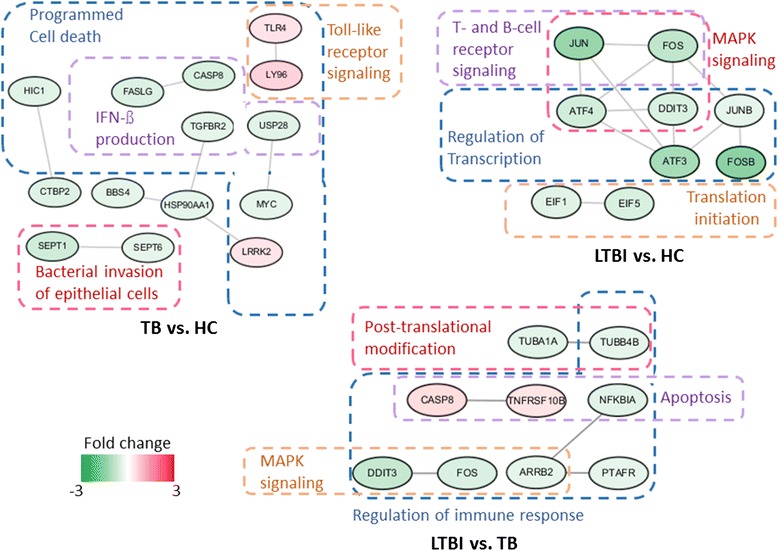
Protein interaction networks of differentially expressed genes among TB, LTBI, and healthy control (HC). Genes are grouped according to their associated pathways and functions
Validation of differentially expressed candidate biomarkers
Many of the identified differentially expressed genes among the TB, LTBI, and healthy control group have also been implicated in TB pathology by other groups. However, as indicated by our analysis, several of these genes play roles in other infections, inflammatory diseases, cancers or even common cold. For real-time RT-PCR validation, we selected genes that are known to be expressed in the lungs and showed clear differences in transcript abundance (fold change ≥1) in at least one of the comparisons; that is, TB versus healthy controls, LTBI versus healthy controls, or LTBI versus TB. Additional volunteers were recruited for gene expression validation. To avoid overlaps with other respiratory tract infections, we chose three differentially expressed genes that may not be directly involved in mediating the immune and inflammatory responses against common respiratory infections. These genes were NEMF (nuclear export mediator factor), ASUN (asunder spermatogenesis regulator), and DHX29 (DEAH (Asp-Glu-Ala-His) box polypeptide 29). Then, we selected PTPRC (protein tyrosine phosphatase, receptor type, C) or CD45, an estalished marker of active TB [21], as a reference standard.
Though the independently recruited participants differed significantly in age (Additional file 1), RT-PCR results successfully verified the array observations in the gene expression array experiment (Fig. 4), indicating that age might not have been a major factor. Subsequent ROC analyses confirmed that PTPRC expression may be able to detect active TB, while ASUN could discriminate TB or LTBI from healthy individuals (Fig. 5). Other than PTPRC, the transcript abundance of DHX29 could also distinguish the differences between TB and healthy controls. In contrast, NEMF did not demonstrate to be a good discriminatory biomarker.
Fig. 4.
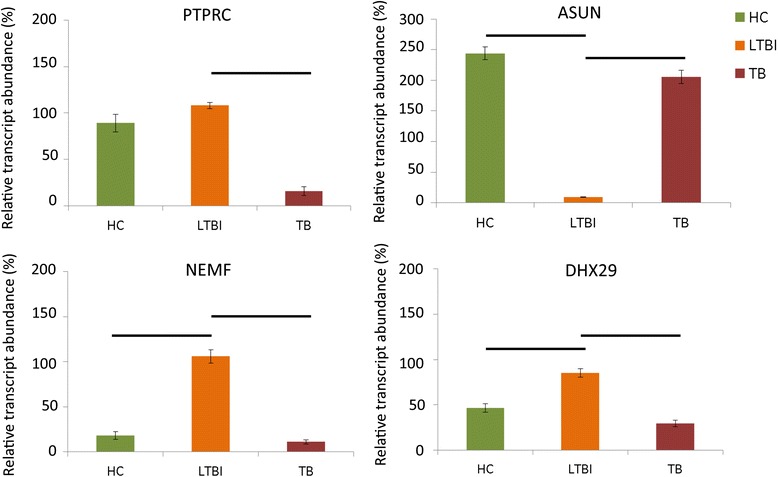
Validation of four differentially expressed genes among TB, LTBI, and healthy controls (HC). Statistical significance (p < 0.05) is represented by a horizontal bar
Fig. 5.
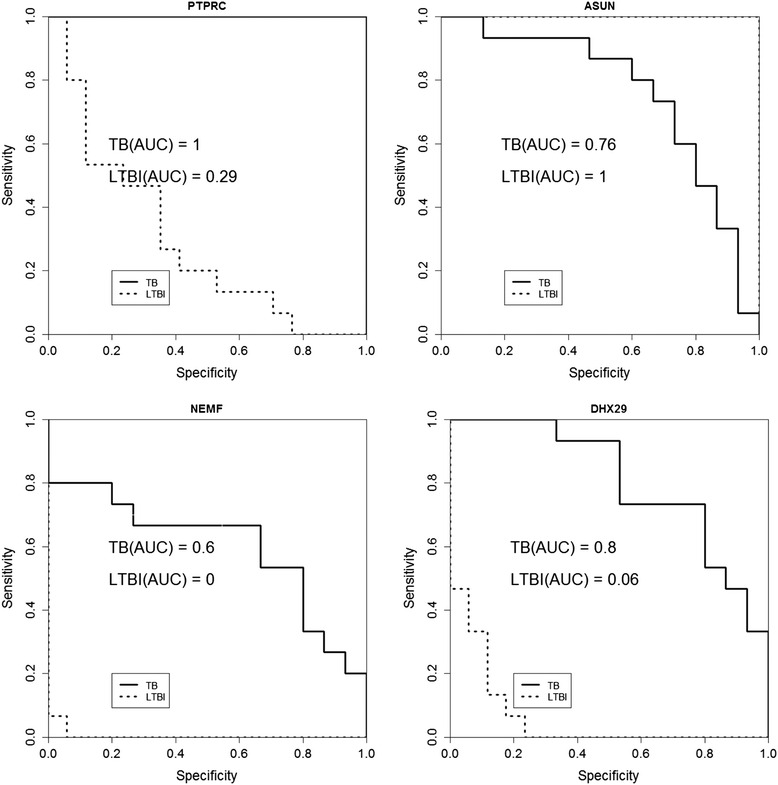
ROC analysis of four differentially expressed genes among TB, LTBI, and healthy controls (HC). AUC represents the area under the curve
Finally, to assess the ability of PTPRC, DHX29, and ASUN in classifying TB, LTBI, and healthy individuals, as a proof of concept experiment, we tested the performance of classification models built with the candidate biomarkers using a sample size of 17 LTBI, 15 TB, and 15 healthy individuals. We utilized four classifiers: decision tree, random forest, support vector machine (SVM), and Naïve Bayes. As evaluated by a 5-fold cross-validation approach, the accuracy, sensitivity, and specificity of the models constructed with single candidate genes were relatively low compared to those built using a combination of biomarkers (Additional file 12 for single gene models; Table 3 for hybrid models). The Naïve Bayes-based model, which was constructed with the expression levels of PTPRC, DHX29, and ASUN as the selected features, yielded the best performance (Table 3).
Table 3.
Performance of diagnostic support models constructed using combinations of candidate biomarkers with various classifiers
| Features | Classifier | Accuracy | Sensitivity | Precision | AUC |
|---|---|---|---|---|---|
| PTPRC+ASUN | Decision tree | 91.49 % | 91.5 % | 97.7 % | 0.943 |
| PTPRC+ASUN+DHX29 | Random Forest | 93.62 % | 93.6 % | 93.6 % | 0.982 |
| PTPRC+ASUN+DHX29 | SVM | 95.74 % | 95.7 % | 96.2 % | 0.969 |
| PTPRC+ASUN+DHX29 | Naïve Bayes | 97.87 % | 97.9 % | 98 % | 0.979 |
Sensitivity: TP/(TP+FN); Precision: TP/(TP+FP); performance was evaluated by 5-fold cross-validation
Discussion
TB is a serious health threat among the young, elderly, and immunocompromised. Variations in the transcriptional profile of human peripheral blood mononuclear cells in the presence of Mtb infection are complex and can be attributed to multiple factors, including age [22], genetic background [14, 23], and study designs. Therefore, identifying distinct genetic signatures of active TB, LTBI, and healthy individuals that is population-specific becomes a challenging task.
In the current study, we examined the transcriptomes of active TB, LTBI, and healthy individuals, and uncovered specific molecular markers and pathways associated with each group. We found that there were more genes showing differential expression between the TB group and healthy controls as compared with LTBI versus healthy controls. GO and pathway enrichment analyses revealed that the transcriptional profiles of TB individuals differed from those of healthy controls in immune system processes such as leukocyte and lymphocyte activation, differentiation, chemokine receptor activity. In contrast, although immune pathway alterations were indeed observed in individuals with LTBI at the transcript level, metabolic processes in these individuals also differed from the healthy controls. On the other hand, between TB and LTBI patients, the most important genes seemed to be mediators of inflammation, immune system responses, and apoptosis.
Our results support findings from other studies in that infection with Mtb triggers a relay of inflammatory signals and immune responses from the host. Upon entry, Mtb are recognized by various host receptors, including Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain-like receptors that are expressed on immune cells [24]. This host-pathogen interaction initiates a cascade of inflammatory responses, whereby alveolar macrophages produce cytokines and chemokines to alert the host of the infection [25]. In response to this signal, T- and B-lymphocytes aggregate around the infected macrophages to form granulomas, a microenvironment to prevent bacterial spread and an isolated region for the lymphocytes to invoke apoptosis of the infected macrophages [26]. The bacteria have also evolved tactics to avoid this fate. After being engulfed by macrophages, the bacteria multiply in the phagosome, causing macrophage necrosis and allowing for their escape from the host defenses to infect other cells [25]. This is evident in our results in which a lot of the differences between TB and healthy controls were associated with mediators of immune responses.
In particular, our protein interaction analysis suggests that Toll-like receptor 4 (TLR4) may mediate the host defense against Mtb. It is known that TLR4 is important in modulating the balance between apoptosis and necrosis in Mtb-infected macrophages [27]. Our analysis showed that TLR4 may interact with LY96 (lymphocyte antigen 96) and S1PR1 (Sphingosine-1-phosphate receptor 1) in TB patients. LY96 is responsible for establishing the binding between the lipopolysaccharide on the bacterial cell membrane and TLR4 on the host cell surface, thus activating a series of immune responses in infected individuals [28].
The bacterial strain Mtb has quite a few tricks to blunt the bactericidal mechanisms of infected macrophages so as to promote their persistence in the host. In latent TB infection, the bacteria manipulate the host antigen presentation processes and enter a non-replicating state [29]. As a result, the bacteria remain dormant inside the phagosomes, and the granuloma became their safe hideout. This is evident in our observation that TB and LTBI differed not only in the regulation of immune responses, but also in the modulation of phagosome, autophagy and apoptosis. In fact, several apoptosis-associated molecules, such as decoy receptor 3, prostaglandin 2, and lipoxin, have been shown to correlate significantly with the status of Mtb infection [30, 31].
Compared with healthy controls, genes involved in cell cycle control seemed to be differentially regulated in individuals with LTBI. Our interaction analysis showed that this difference may be attributable to the interactions between mediators of the MAPK (mitogen-activated protein kinase) signaling pathway, such as JUN (Jun proto-oncogene), JUNB (Jun B proto-oncogene) and FOSB (FBJ murine osteosarcoma viral oncogene homolog B), as well as regulators of translation. MAPK signaling is one of the pathways responsible for modulating the host’s innate immunity [32]. In fact, MAPK activation is important for inducing the expression of genes involved in inflammatory responses, but inactivation of MAPK activity is also important to prevent host tissue damage [32]. On the other hand, our interaction analysis also indicated that translational regulation represent important differences between LTBI and healthy individuals. It is known that bacteria can influence host tranlation in order to immobilize host defenses and promote their own survival [33]. Therefore, our finding supports that MAPK signaling and tranlsational control may underlie the differences between LTBI and healthy conditions.
In addition, alterations in the expression of genes involved in metabolic processes seemed to be a major difference between the LTBI group and healthy controls. Metabolism-related manifestations are known to be associated with TB [34]. It has been demonstrated that Mtb-infected macrophages would become lipid-loaded (foamy) in the granuloma, and the fatty acids accumulated as triacylglycerol represent the vital source of energy for dormant Mtb [35]. Also, certain immune-endocrine-metabolic alterations are thought to exist in TB patients, though molecular studies have yet to reach a consensus as to which molecules are the major players in this process [36–39].
To date, most identified biomarkers for active TB and LTBI are related to inflammatory and immune responses triggered by the pathogen infection [14, 18]. However, TB may be comorbid with various communicable diseases including influenza, bacterial pneumonia, HIV, syphilis, leishmaniasis, as well as non-communicable disorders such as diabetes, alcohol-related diseases, chronic obstructive pulmonary disease, coronary artery disease, cancer, etc. [40]. In fact, our gene set enrichment analysis mapped four genes, TNF, JUN, FOS, NFKBIA, that were differential expressed between LTBI and healthy controls to leshimaniasis-related pathways.
To avoid finding biomarkers that overlap with other diseases, we chose to verify differentially expressed genes that have not been identified as TB or LTBI biomarkers in independently recruited samples. We also included an established active TB marker, PTPRC, as a test reference. As a result, ASUN, DHX29, and NEMF were successfully confirmed to be differentially expressed among active TB, LTBI, and healthy individuals. Subsequent ROC analysis revealed the potential of PTPRC, ASUN, and DHX29 in discriminating among TB, LTBI, and healthy conditions. Further classification experiments also indicated that combinining the three canidate biomarkers may be more effective in achieving accurate identification of the different disease states.
To be a biomarker, the gene should to be related to the pathogenesis of the disease. However, since functional analysis was not performed in this study, we try to postulate how these candidate genes may be involved in TB pathology based on their known functions. Among the validated genes, PTPRC is an essential regulator of host immune response through the modulation of T- and B-cell receptor signal transduction [41]. In the guinea pigs, PTPRC expression appeared to increase after exposure to Mtb and decrease after the infection persisted for a longer period of time [42]. This is in line with our observation that PTPRC transcript level was decreased in active TB compared to LTBI and healthy state. ASUN is critical for the regulation of mitotic cell cycle [43]. As the number of T-cells can determine whether an individual is susceptible to Mtb infection or active TB [44], we suspect that the high level of ASUN expression in LTBI relative to the active TB and healthy controls may be associated with T-lymphocyte differentiation or proliferation. NEFM is a nuclear export mediator that have been implicated as a tumor suppressor in lung cancer [45]. Nuclear export is an important process for the regulation of autophagy [46]. A recent in vivo study performed in mice suggested that autophagy can suppress the progression towards active TB by inhibiting Mtb growth [47]. Finally DHX29 is a helicase protein that participates in translation initiation, and its down-regulation has an inhibitory effect on cancer growth [48], which may be related to the altered cellular processes in TB.
Note that our study is limited by the small sample size and lack of functional studies to determine the roles that the identified candidate biomarkers play in the pathogenesis of TB. This might have also been the reason that we did not observe more genes associated with the TLR signaling pathway, an established mechanism underlying Mtb recognition [49]. As a result, this pathway was not the most enriched in any of the comparisons. Equally likely is that the changes in TLR genes might be dependent on treatment duration. It has been demonstrated that the increase in TLR4 expression level appeared to be more significant compared to TLR2 after 1 month of treatment in TB patients when compared with healthy controls [50]. In our study, samples were collected at the time of diagnosis. More evident changes might be observed if we had examined the temporal expression profiles of TB and LTBI patients during treatment. Also, our experiments were focused on blood cells, instead of lung tissues. Therefore, the results are more relevant to biomarkers associated with TB and LTBI, and perhaps indirectly related to the pathology of the disease in the lungs. Moreover, BCG is known to have an effect on gene expression [51, 52]. In Taiwan, BCG vaccine should be administered to every newborn; therefore, all of our study participants have been inoculated with BCG. This may make our finding specific to not only the Taiwanese people, but also to those who have received this vaccine. Finally, despite the fact that evaluation of the biomarkers yielded relatively good results, due to the limited sample size and the variable prevalence of TB and LTBI in different seasons, we could not eliminate the possibility that other diseases or environmental factors may affect the effectiveness of the candidate biomarkers.
Conclusions
In conclusion, we have performed a genome-wide gene expression study comparing the transcriptional profiles among TB and LTBI patients, as well as healthy controls. Gene set enrichment analyses have identified specific biological processes and pathways associated with genes that are differentially expressed among these groups. We have uncovered novel discriminatory biomarkers for TB and LTBI. Moreover, we have demonstrated, as a proof of concept, that the expression levels of PTPRC, ASUN, and DHX29 may be used as diagnostic biomarkers. Validation of the diagnostic support system in a larger sample size would help confirm the discriminative potential of these biomarkers and facilitate the development of a cost-effective and sensitive molecular diagnostic platform for TB.
Methods
Clinical sample collection
The analytical flow of our study is illustrated in Fig. 1. All procedures were reviewed and approved by the Institutional Review Board of Taoyuan General Hospital, Ministry of Health and Welfare, Taoyuan, Taiwan. Written informed consents were obtained from all participants. For those who had reduced ability to consent (including minors), the carers or guardians gave written informed consents on behalf of these participants. Eligibility for entry into the study was based on clinical signs and symptoms of Mtb infection. All participants were interviewed and examined by a physician. Each subject received the sputum smear test, T-SPOT TB test, and took a chest radiograph. TB subjects were those who showed clinical signs of TB, in addition to having been tested positive on all tests. LTBI subjects were recruited from close contacts (>8 h/day for a total of >40 h of close contact) with active TB patients, tested negative on the smear test, positive on the T-SPOT TB test, appeared normal on their chest radiographs, and exhibited no clinical evidence of active TB. Healthy controls were individuals who had not been in close contact with TB or LTBI patients, obtained negative results on all tests and showed no clinical signs of TB or LTBI. Individuals with allergic diseases, diabetes, cancer, immune-compromised conditions, and co-infections with any types of infectious diseases were excluded. In total, seven healthy individuals, seven patients with active TB, as well as seven subjects with LTBI, were included in the microarray experiment. Additional participants (15 TB, 17 LTBI, and 15 healthy individuals) were recruited as independent testing samples for validation of the expression profiling results. Age and gender information are provided in Additional file 1. In Taiwan, every newborn must be inoculated with BCG; therefore, the BCG inoculation status for every participant was positive.
RNA isolation
RNA was isolated from peripheral blood mononuclear cells (PMBC). The quality of RNA was determined by an optical density (OD) 260/280 ratio ≥1.8, and OD 260/230 ratio ≥1.5 on a spectrophotometer, in addition to the intensity of the 18S and 28S rRNA bands on a 1 % formaldehyde-agarose gel. RNA quantity was detected by a spectrophotometer. RNA integrity was examined on an Agilent Bioanalyzer. RNA with an RNA integrity number (RIN) ≥7.0 and 28S/18S >0.7 was subjected to microarray analysis.
Gene expression analysis
RNA samples were subjected to Human OneArray® v6 (Phalanx Biotech, Hsinchu, Taiwan). Data were analyzed with the Rosetta Resolver System software (Rosetta Biosoftware, USA). The criteria for identifying differentially expressed genes were: 1) an absolute log2 fold change ≥1; 2) a false discovery rate of <0.05; 3) an intensity difference of >1000 between two samples under comparison; 4) an individual intensity of >500. Genes showing significant differential expression were categorized into TB versus healthy controls, LTBI versus healthy controls, and LTBI versus TB. Our gene expression records can be found on the Gene Expression Omnibus under the accession number GSE62525.
Bioinformatics analysis
Differentially expressed genes were used as input for a series of bioinformatics analyses performed with the WEB-based GEne SeT AnaLysis Toolkit (WebGestalt) [53, 54]. WebGestalt is an open analytical platform that integrates gene ontology (GO) [55], KEGG [56], WikiPathway [57], protein interaction networks, microRNA binding sites and transcription factor targets [58], as well as cytogenetic band information, for a variety of enrichment analyses. The GO, KEGG, and protein interaction module tools were utilized to analyze the differentially expressed genes. Multiple testing bias was adjusted by a Benjamini-Hochberg threshold of p < 0.05. The enriched protein interaction network modules in each transcriptional profile were grouped according to their GO annotations or associated pathways. Experimentally confirmed interactions were cross-validated with the STRING database (v9.1) [19, 20] with a confidence level of 0.7 as the paratmeter setting. Visualization of the interaction networks was achieved using Cytoscape version 3.2.1 [59].
Real-time RT-PCR validation
The four differentially expressed genes selected for real-time RT-PCR validation were PTPRC, ASUN, NEMF, and DHX29. Total RNA from PBMC was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA) from 17 TB, 15 LTBI, and 15 healthy individuals. Each extracted RNA sample was reversely transcribed using the First Strand cDNA Synthesis kit (Roche, Boulder, CO, USA) according to the manufacturer’s instructions. Each cDNA sample was amplified with the FastStart Universal SYBR Green reagent (Roche, Mannheim, Germany) on the StepOnePlusTM Real-Time PCR system (Applied Biosystem, CA, USA). Briefly, the reaction conditions consisted of 10 ng of cDNA and 0.25 μM of primers in a final reaction volume of 20 μl in SYBR Green mixture. Each reaction was initiated with 10 min at 95 °C, followed by 40 cycles consisting of denaturation at 95 °C for 15 s, annealing at 60 °C for 1 min, and extension at 72 °C for 30 s. For each reaction, the β-actin gene was used as an endogenous control to normalize each sample. Primer sequences for each gene are listed in Additional file 13. The relative expression of each gene was compared using the 2-ΔΔ CT method and all experiments were run in triplicates and repeated three times. Differences in expression among TB, LTBI, and healthy controls were evaluated with one-way ANOVA followed by Tukey’s post hoc test in SPSS 18.0 (IBM Corporation, NY, USA). A p-value of <0.05 was regarded as statistically significant. A receiver operating characteristic (ROC) curve analysis was performed in the R statistical environment (3.1.1) to assess the specificity and sensitivity of each validated biomarker.
Construction of a diagnostic support model
We wished to test if the candidate biomarkers could be integrated in a diagnostic support system. As a proof of concept, we used the expression levels of PTPRC, ASUN, and DHX29 as features to build classification models based on the 57 volunteers who donated their blood samples for RT-PCR validation. Due to its low discriminating abiliy as evaluated by ROC analysis, NEMF was excluded. Experiments were conducted in LibSVM (version 3.12) [60] and WEKA, or Waikato Environment for Knowledge Analysis (version 3.6.5) [61]. Features were selected based on the previous ROC analysis results. The selected classifiers included the C4.5 decision tree algorithm [62], support vector machine (SVM) [63], Naïve Bayes [64, 65], and the random forest algorithm [66]. Models were built with single genes or a combination of the selected genes. Performance of each model was evaluated by 5-fold cross-validation.
Acknowledgements
This study was supported by the Ministry of Science and Technology of the Republic of China, Taiwan, under the contract number of MOST 101-2628-E-155-002-MY2, as well as by Taoyuan General Hospital, Taiwan, under the contract number of 10409.
Declarations
The publication cost of this manuscript is supported by the Ministry of Science and Technology of the Republic of China, Taiwan, under the contract number of MOST 101-2628-E-155-002-MY2. This article has been published as part of BMC Bioinformatics Volume 17 Supplement 1, 2016: Selected articles from the Fourteenth Asia Pacific Bioinformatics Conference (APBC 2016). The full contents of the supplements are available online at http://www.biomedcentral.com/bmcbioinformatics/supplements/17/S1.
Additional files
Average age and gender distribution of the participants (XLSX 11 kb)
List of differentially expressed genes among the TB, LTBI, and healthy individuals. (XLSX 19238 kb)
GO enrichment of genes differentially expressed in LTBI relative to healthy controls. (XLSX 111 kb)
GO enrichment of genes differentially expressed in TB relative to healthy controls. (XLSX 69 kb)
GO enrichment of genes differentially expressed in LTBI relative to TB. (XLSX 66 kb)
Pathway enrichment of genes differentially expressed in LTBI relative to healthy controls. (XLSX 19 kb)
Pathway enrichment of genes differentially expressed in TB relative to healthy controls. (XLSX 20 kb)
Pathway enrichment of genes differentially expressed in LTBI relative to TB. (XLSX 20 kb)
Enriched protein interaction modules associated with genes differentially expressed in LTBI relative to healthy controls. (XLSX 14 kb)
Enriched protein interaction modules associated with genes differentially expressed in TB relative to healthy controls. (XLSX 15 kb)
Enriched protein interaction modules associated with genes differentially expressed in LTBI relative to TB. (XLSX 20 kb)
Performance of diagnostic support models constructed using individual candidate biomarkers with various classifiers. (XLSX 11 kb)
RT-PCR Primer for the selected differentially expressed genes. (XLSX 11 kb)
Footnotes
From The Fourteenth Asia Pacific Bioinformatics Conference (APBC 2016) San Francisco, CA, USA. 11 - 13 January 2016.
Shih-Wei Lee and Lawrence Shih-Hsin Wu contributed equally to this work.
Competing interests
The authors have declared that no competing interests exist.
Authors’ contributions
JTYW, SWL, and LSHW conceived and designed the experiments. SWL diagnosed the patients and collected the samples. LSHW was responsible for RNA isolation and RT-PCR validation. GMH conducted the ROC and classification experiments. KYH and JTYW performed the bioinformatics analyses and JTYW wrote the manuscript. TYL assisted with the bioinformatics analyses. All authors read and approved the final manuscript.
Contributor Information
Shih-Wei Lee, Email: swl@mail.tygh.gov.tw.
Lawrence Shih-Hsin Wu, Email: lshwu@hotmail.com.
Guan-Mau Huang, Email: koko5696@gmail.com.
Kai-Yao Huang, Email: kaiyao.tw@gmail.com.
Tzong-Yi Lee, Email: francis@saturn.yzu.edu.tw.
Julia Tzu-Ya Weng, Email: julweng@saturn.yzu.edu.tw.
References
- 1.Kumar V, Cotran RS, Robbins SL. Robbins basic pathology. 8. New York: Saunders; 2007. [Google Scholar]
- 2.Dye C, Williams BG. The population dynamics and control of tuberculosis. Science. 2010;328(5980):856–61. doi: 10.1126/science.1185449. [DOI] [PubMed] [Google Scholar]
- 3.Skolnik RL. Global health 101. 2. Burlington, MA: Jones & Barlett Learning; 2012. [Google Scholar]
- 4.Lawn SD, Zumla AI. Tuberculosis. Lancet. 2011;378(9785):57–72. doi: 10.1016/S0140-6736(10)62173-3. [DOI] [PubMed] [Google Scholar]
- 5.McShane H. Tuberculosis vaccines: beyond bacille Calmette-Guerin. Philos Trans R Soc Lond Ser B Biol Sci. 2011;366(1579):2782–9. doi: 10.1098/rstb.2011.0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cole EC, Cook CE. Characterization of infectious aerosols in health care facilities: an aid to effective engineering controls and preventive strategies. Am J Infect Control. 1998;26(4):453–64. doi: 10.1016/S0196-6553(98)70046-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crowley L. An introduction to human disease: pathology and pathophysiology correlations. 9. Burlington, MA: Jones & Bartlett Learning; 2010. [Google Scholar]
- 8.Agarwal R, Malhotra P, Awasthi A, Kakkar N, Gupta D. Tuberculous dilated cardiomyopathy: an under-recognized entity? BMC Infect Dis. 2005;5:29. doi: 10.1186/1471-2334-5-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466(7309):973–7. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Constantoulakis P, Filiou E, Rovina N, Chras G, Hamhougia A, Karabela S, et al. In vivo expression of innate immunity markers in patients with Mycobacterium tuberculosis infection. BMC Infect Dis. 2010;10:243. doi: 10.1186/1471-2334-10-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacobsen M, Repsilber D, Gutschmidt A, Neher A, Feldmann K, Mollenkopf HJ, et al. Candidate biomarkers for discrimination between infection and disease caused by Mycobacterium tuberculosis. J Mol Med-Jmm. 2007;85(6):613–21. doi: 10.1007/s00109-007-0157-6. [DOI] [PubMed] [Google Scholar]
- 12.Sutherland JS, de Jong BC, Jeffries DJ, Adetifa IM, Ota MO. Production of TNF-alpha, IL-12(p40) and IL-17 can discriminate between active TB disease and latent infection in a West African cohort. Plos One. 2010;5(8) doi: 10.1371/journal.pone.0012365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chegou NN, Black GF, Kidd M, van Helden PD, Walzl G. Host markers in QuantiFERON supernatants differentiate active TB from latent TB infection: preliminary report. BMC pulmonary medicine. 2009;9:21. doi: 10.1186/1471-2466-9-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu B, Huang C, Kato-Maeda M, Hopewell PC, Daley CL, Krensky AM, et al. Messenger RNA expression of IL-8, FOXP3, and IL-12beta differentiates latent tuberculosis infection from disease. J Immunol. 2007;178(6):3688–94. doi: 10.4049/jimmunol.178.6.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pai M. Alternatives to the tuberculin skin test: interferon-gamma assays in the diagnosis of mycobacterium tuberculosis infection. Indian J Med Microbiol. 2005;23(3):151–8. doi: 10.4103/0255-0857.16585. [DOI] [PubMed] [Google Scholar]
- 16.Herrera V, Perry S, Parsonnet J, Banaei N. Clinical application and limitations of interferon-gamma release assays for the diagnosis of latent tuberculosis infection. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2011;52(8):1031–7. doi: 10.1093/cid/cir068. [DOI] [PubMed] [Google Scholar]
- 17.Bibova I, Linhartova I, Stanek O, Rusnakova V, Kubista M, Suchanek M, et al. Detection of immune cell response to M. tuberculosis-specific antigens by quantitative polymerase chain reaction. Diagn Microbiol Infect Dis. 2012;72(1):68–78. doi: 10.1016/j.diagmicrobio.2011.09.024. [DOI] [PubMed] [Google Scholar]
- 18.Lu C, Wu J, Wang H, Wang S, Diao N, Wang F, et al. Novel biomarkers distinguishing active tuberculosis from latent infection identified by gene expression profile of peripheral blood mononuclear cells. Plos One. 2011;6(8) doi: 10.1371/journal.pone.0024290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41(Database issue):D808–15. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39(Database issue):D561–8. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nemeth J, Rumetshofer R, Winkler HM, Burghuber OC, Muller C, Winkler S. Active tuberculosis is characterized by an antigen specific and strictly localized expansion of effector T cells at the site of infection. Eur J Immunol. 2012;42(11):2844–50. doi: 10.1002/eji.201242678. [DOI] [PubMed] [Google Scholar]
- 22.Verhagen LM, Zomer A, Maes M, Villalba JA, Del Nogal B, Eleveld M, et al. A predictive signature gene set for discriminating active from latent tuberculosis in Warao Amerindian children. BMC Genomics. 2013;14:74. doi: 10.1186/1471-2164-14-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wallis RS, Kim P, Cole S, Hanna D, Andrade BB, Maeurer M, Schito M, Zumla A. Tuberculosis biomarkers discovery: developments, needs, and challenges. Lancet Infect Dis. 2013;13(4):362–372. doi: 10.1016/S1473-3099(13)70034-3. [DOI] [PubMed] [Google Scholar]
- 24.Jo EK. Mycobacterial interaction with innate receptors: TLRs, C-type lectins, and NLRs. Curr Opin Infect Dis. 2008;21(3):279–86. doi: 10.1097/QCO.0b013e3282f88b5d. [DOI] [PubMed] [Google Scholar]
- 25.Chen M, Gan H, Remold HG. A mechanism of virulence: virulent Mycobacterium tuberculosis strain H37Rv, but not attenuated H37Ra, causes significant mitochondrial inner membrane disruption in macrophages leading to necrosis. J Immunol. 2006;176(6):3707–16. doi: 10.4049/jimmunol.176.6.3707. [DOI] [PubMed] [Google Scholar]
- 26.Grosset J. Mycobacterium tuberculosis in the extracellular compartment: an underestimated adversary. Antimicrob Agents Chemother. 2003;47(3):833–6. doi: 10.1128/AAC.47.3.833-836.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanchez D, Rojas M, Hernandez I, Radzioch D, Garcia LF, Barrera LF. Role of TLR2- and TLR4-mediated signaling in Mycobacterium tuberculosis-induced macrophage death. Cell Immunol. 2010;260(2):128–36. doi: 10.1016/j.cellimm.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 28.Palsson-McDermott EM, O’Neill LA. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology. 2004;113(2):153–62. doi: 10.1111/j.1365-2567.2004.01976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahmad S. Pathogenesis, immunology, and diagnosis of latent Mycobacterium tuberculosis infection. Clinical & developmental immunology. 2011;2011:814943. doi: 10.1155/2011/814943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shu CC, Wu MF, Hsu CL, Huang CT, Wang JY, Hsieh SL, et al. Apoptosis-associated biomarkers in tuberculosis: promising for diagnosis and prognosis prediction. BMC Infect Dis. 2013;13:45. doi: 10.1186/1471-2334-13-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen M, Divangahi M, Gan H, Shin DS, Hong S, Lee DM, et al. Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death. The Journal of experimental medicine. 2008;205(12):2791–801. doi: 10.1084/jem.20080767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arthur JS, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol. 2013;13(9):679–92. doi: 10.1038/nri3495. [DOI] [PubMed] [Google Scholar]
- 33.Mohr I, Sonenberg N. Host translation at the nexus of infection and immunity. Cell Host Microbe. 2012;12(4):470–83. doi: 10.1016/j.chom.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang SW, Pan WS, Lozano Beltran D, Oleyda Baldelomar L, Solano MA, Tuero I, et al. Gut hormones, appetite suppression and cachexia in patients with pulmonary TB. Plos One. 2013;8(1) doi: 10.1371/journal.pone.0054564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Daniel J, Maamar H, Deb C, Sirakova TD, Kolattukudy PE. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog. 2011;7(6) doi: 10.1371/journal.ppat.1002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Santucci N, D’Attilio L, Kovalevski L, Bozza V, Besedovsky H, del Rey A, et al. A multifaceted analysis of immune-endocrine-metabolic alterations in patients with pulmonary tuberculosis. Plos One. 2011;6(10) doi: 10.1371/journal.pone.0026363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yuksel I, Sencan M, Dokmetas HS, Dokmetas I, Ataseven H, Yonem O. The relation between serum leptin levels and body fat mass in patients with active lung tuberculosis. Endocr Res. 2003;29(3):257–64. doi: 10.1081/ERC-120025033. [DOI] [PubMed] [Google Scholar]
- 38.van Crevel R, Karyadi E, Netea MG, Verhoef H, Nelwan RH, West CE, et al. Decreased plasma leptin concentrations in tuberculosis patients are associated with wasting and inflammation. The Journal of clinical endocrinology and metabolism. 2002;87(2):758–63. doi: 10.1210/jcem.87.2.8228. [DOI] [PubMed] [Google Scholar]
- 39.Kim JH, Lee CT, Yoon HI, Song J, Shin WG, Lee JH. Relation of ghrelin, leptin and inflammatory markers to nutritional status in active pulmonary tuberculosis. Clin Nutr. 2010;29(4):512–8. doi: 10.1016/j.clnu.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 40.Marais BJ, Lonnroth K, Lawn SD, Migliori GB, Mwaba P, Glaziou P, et al. Tuberculosis comorbidity with communicable and non-communicable diseases: integrating health services and control efforts. Lancet Infect Dis. 2013;13(5):436–48. doi: 10.1016/S1473-3099(13)70015-X. [DOI] [PubMed] [Google Scholar]
- 41.Hermiston ML, Xu Z, Weiss A. CD45: a critical regulator of signaling thresholds in immune cells. Annu Rev Immunol. 2003;21:107–37. doi: 10.1146/annurev.immunol.21.120601.140946. [DOI] [PubMed] [Google Scholar]
- 42.Ordway D, Palanisamy G, Henao-Tamayo M, Smith EE, Shanley C, Orme IM, et al. The cellular immune response to Mycobacterium tuberculosis infection in the guinea pig. J Immunol. 2007;179(4):2532–41. doi: 10.4049/jimmunol.179.4.2532. [DOI] [PubMed] [Google Scholar]
- 43.Anderson MA, Jodoin JN, Lee E, Hales KG, Hays TS, Lee LA. Asunder is a critical regulator of dynein-dynactin localization during Drosophila spermatogenesis. Mol Biol Cell. 2009;20(11):2709–21. doi: 10.1091/mbc.E08-12-1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flynn JL, Ernst JD. Immune responses in tuberculosis. Curr Opin Immunol. 2000;12(4):432–6. doi: 10.1016/S0952-7915(00)00116-3. [DOI] [PubMed] [Google Scholar]
- 45.Bi XL, Jones T, Abbasi F, Lee H, Stultz B, Hursh DA, et al. Drosophila caliban, a nuclear export mediator, can function as a tumor suppressor in human lung cancer cells. Oncogene. 2005;24(56):8229–39. doi: 10.1038/sj.onc.1208962. [DOI] [PubMed] [Google Scholar]
- 46.Funasaka T, Tsuka E, Wong RW. Regulation of autophagy by nucleoporin Tpr. Scientific reports. 2012;2:878. doi: 10.1038/srep00878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci U S A. 2012;109(46):E3168–76. doi: 10.1073/pnas.1210500109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parsyan A, Shahbazian D, Martineau Y, Petroulakis E, Alain T, Larsson O, et al. The helicase protein DHX29 promotes translation initiation, cell proliferation, and tumorigenesis. Proc Natl Acad Sci U S A. 2009;106(52):22217–22. doi: 10.1073/pnas.0909773106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reiling N, Holscher C, Fehrenbach A, Kroger S, Kirschning CJ, Goyert S, et al. Cutting edge: Toll-like receptor (TLR)2- and TLR4-mediated pathogen recognition in resistance to airborne infection with Mycobacterium tuberculosis. J Immunol. 2002;169(7):3480–4. doi: 10.4049/jimmunol.169.7.3480. [DOI] [PubMed] [Google Scholar]
- 50.de Oliveira LR, Peresi E, Golim Mde A, Gatto M, Araujo Junior JP, da Costa EA, et al. Analysis of Toll-like receptors, iNOS and cytokine profiles in patients with pulmonary tuberculosis during anti-tuberculosis treatment. Plos One. 2014;9(2) doi: 10.1371/journal.pone.0088572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zarate-Blades CR, Silva CL, Passos GA. The impact of transcriptomics on the fight against tuberculosis: focus on biomarkers, BCG vaccination, and immunotherapy. Clinical & developmental immunology. 2011;2011:192630. doi: 10.1155/2011/192630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aranday Cortes E, Kaveh D, Nunez-Garcia J, Hogarth PJ, Vordermeier HM. Mycobacterium bovis-BCG vaccination induces specific pulmonary transcriptome biosignatures in mice. Plos One. 2010;5(6) doi: 10.1371/journal.pone.0011319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005;33(Web Server issue):W741–8. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res. 2013;41(Web Server issue):W77–83. doi: 10.1093/nar/gkt439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pico AR, Kelder T, van Iersel MP, Hanspers K, Conklin BR, Evelo C. WikiPathways: pathway editing for the people. PLoS Biol. 2008;6(7) doi: 10.1371/journal.pbio.0060184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27(12):1739–40. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chang C-C, Lin C-J. LIBSVM: A Library for Support Vector Machines. 2001. [Google Scholar]
- 61.Mark Hall EF, Holmes G, Pfahringer B, Reutemann P, Witten IH. SIGKDD Explorations. 2009. The WEKA data mining software: an update. [Google Scholar]
- 62.Quinlan JR. Improved use of continuous attributes in C4.5. J Artif Intell Res. 1996;4(1):77–90. [Google Scholar]
- 63.Corinna Cortes VV. Support-vector networks. Mach Learn. 1995;20(3):273–97. [Google Scholar]
- 64.Chen J, Huang H. Feature selection for text classification with Naïve Bayes. Expert Systems with Applications. 2009;36(3):5432–5. doi: 10.1016/j.eswa.2008.06.054. [DOI] [Google Scholar]
- 65.Guh R-S, Wu T-CJ, Weng S-P. Integrating genetic algorithm and decision tree learning for assistance in predicting in vitro fertilization outcomes. Expert Systems with Applications. 2011;38(4):4437–49. doi: 10.1016/j.eswa.2010.09.112. [DOI] [Google Scholar]
- 66.Breiman L. Random forests. 2001. [Google Scholar]