

Abstract
Background
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited myocardial disease with high prevalence in the Boxer dog population. It is characterized by replacement of the myocardium with fatty or fibro‐fatty tissue. Several mechanisms for the development of ARVC have been suggested, including dysfunction of the canonical Wnt pathway, which is linked to many cellular functions, including growth and differentiation of adipocytes.
Hypothesis
Wnt pathway dysfunction is involved in the development of ARVC in the Boxer as evidenced by mislocalization of β‐catenin, an integral Wnt pathway modulator, and striatin, a known Wnt pathway component.
Animals
Five dogs without ARVC and 15 Boxers with ARVC were identified by 24‐hour Holter monitoring and histopathologic examination of the heart.
Methods
Right ventricular samples were collected and examined using confocal microscopy, Western blots, and quantitative (q) PCR.
Results
Confocal microscopy indicated that β‐catenin localized at sites of cell‐to‐cell apposition, and striatin localized in a diffuse intracellular pattern in hearts without ARVC. In hearts affected with ARVC, both β‐catenin and striatin were colocalized with the endoplasmic reticulum (ER) marker calreticulin. Western blots indentified a 50% increase in the amount of β‐catenin in ARVC samples. No change in β catenin mRNA was detected using qPCR.
Conclusions
Our data suggest that trafficking of Wnt pathway proteins from the ER to their proper location within the cell is inhibited in Boxers with ARVC. These results suggest that disturbances in the Wnt pathway may play a role in the development of ARVC in the Boxer.
Keywords: ARVC, Canine
Abbreviations
- ARVC‐Boxer
Boxer dog affected with ARVC
- ARVC
arrhythmogenic right ventricular cardiomyopathy
- ER
endoplasmic reticulum
- LV
left ventricle
- Non‐ARVC dog
a dog that is not a Boxer and not affected with ARVC
- qPCR
quantitative polymerase chain reaction
- RV
right ventricle
- LV
left ventricle
- Cp
crossing point, cycle number quantifying amount of RNA in sample values
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited disease clinically characterized by monomorphic ventricular tachycardia, heart failure, and sudden cardiac death.1, 2, 3 For many years, it has been documented that the structural changes observed in ARVC‐affected hearts of both humans and Boxer dogs are not restricted to the right ventricle (RV). Additionally, atrial arrhythmias have been documented in the atrial myocardium of the Boxer.1 The term arrhythmogenic cardiomyopathy currently is proposed as the term to describe this disease in humans,4 although the name change is not without controversy. In this study, we describe changes observed exclusively within the right ventricle of affected boxers, and as such, we have elected to use the term ARVC.
Clinical and histopathologic characteristics of ARVC are similar in both humans and Boxer dogs.1, 2 The histopathologic hallmark of ARVC is the fatty or fibro‐fatty replacement of right ventricular myocardium, originating in the region of the RV (the triangle of dysplasia5) and progressing from epicardium to endocardium. In its natural course, histopathologic manifestations encompass the entire right ventricular mass, and also may extend to the interventricular septum and left ventricle.1, 5 In the Boxer, histopathologic lesions of the left ventricle (LV) frequently are identified.1, 6, 7
The pathophysiologic mechanism of ARVC is poorly understood in humans and in the Boxer. In both, a loss of mechanical and electrical junctions at the intercalated disk has been documented.3, 6, 7 In humans, ARVC primarily is considered a disease of the desmosome, a mechanical junction within the intercalated disk that anchors to the intermediate filaments.3 Mutations in genes coding for many desmosomal proteins have been linked to humans with ARVC,3 and are thought to produce nonfunctional proteins, leading to loss of desmosomal integrity. Although loss of desmosomal integrity has been documented in the Boxer, mutations in the demosomal genes implicated in human ARVC have not been linked to cases of the disease.6, 8
A mutation in striatin, a scaffolding protein localizing to both the intermediate filaments and the intercalated disk,9 has been proposed as a cause of ARVC in the Boxer.10 However, the mechanism by which the loss of desmosomal or structural integrity leads to fibro‐fatty infiltration of the myocardium is not yet understood.
Although the pathophysiologic mechanism of ARVC progression remains unclear, it is thought to include molecular pathways involved in the formation of mechanical and electrical coupling, apoptosis, and migration and differentiation of epicardial‐derived cells.11 A decrease in Wnt signaling has been linked to increased adipo‐ and fibrogenesis, resulting in a transgenic mouse model of ARVC.12
The Wnt signaling pathway is conserved from mammals to Drosophila, and plays essential roles in the determination of cell fate, behavior, survival, and proliferation.13 Of particular relevance to understanding the phenotype of ARVC, Wnt signaling has been shown both to induce the epithelial‐to‐mesenchymal cell transition,14 and to regulate the proliferation of adipocytes.15 The particular pathways leading to fibroblast and adipocyte proliferation in the ARVC‐affected heart are unknown.
Through a complex signaling cascade, the Wnt protein is secreted from cells and leads to accumulation of β‐catenin within the cytosol. Cytosolic β‐catenin then translocates to the nucleus, where it binds to TCF/LEF transcription factors, leading to changes in cell proliferation and differentiation.13 In the developing heart, β‐catenin is necessary for the development and proliferation of cardiac progenitors, and Wnt signaling is required for cardiac myocyte formation.16 Although still an area of intense investigation, β‐catenin also appears to play a role in cardiac remodeling in the adult heart. For example, activation of the Wnt pathway and successful stabilization of cytoplasmic pools of β‐catenin have been linked to a decrease in apoptosis in the adult mouse heart.17 Another member of the catenin family, plakoglobin (γ‐catenin), is structurally and functionally similar to β‐catenin. Plakoglobin plays multiple roles within the cell: first, as a desmosomal protein, mutations in which have been linked to human ARVC,3 and second, as an antagonist of the Wnt/β‐catenin pathway that interacts and competes with β‐catenin in the nucleus.12 This interaction is associated with development of ARVC in mice.12 We previously identified mislocalization of plakoglobin in tissues collected from Boxers with ARVC,6 suggesting that Wnt signaling may play a role in the pathogenesis.
Our current studies stem from the hypothesis that dysfunction in the canonical Wnt/β‐catenin pathway is involved in the development of this disorder in the Boxer. In this study, we report mislocalization of β‐catenin and striatin, and an increase in the total amount of β‐catenin. These abnormalities in the Boxer may provide insight into the pathogenesis of ARVC in both humans and the Boxer dog.
Materials and Methods
Animals
Fifteen Boxers (10 males, 5 females; mean age, 8.1 years) affected with ARVC (ARVC‐Boxer) and 5 dogs without ARVC (Non‐ARVC dogs; 4 German Shepherds, 1 Beagle; 1 male, 4 females; mean age, 7.2 years) were studied. All dogs had been classified previously as being affected or not affected with ARVC based on extensive histopathologic examination (only the Beagle did not have histopathologic confirmation), and a complete clinical examination including 24‐hour Holter monitoring, when possible.6, 7 The histopathologic examinations were done by one of the investigators (PRF) without knowledge of the clinical presentation.
In humans, a premortem diagnosis of ARVC is based on a combination of major and minor criteria, which include structural, electrocardiographic, genetic, historical, and histologic characteristics as determined by an International Task Force in 2010.18 Postmortem diagnosis in humans is based primarily on the characteristic histopathologic lesions of cardiac tissue.19, 20, 21
A consensus of diagnostic criteria has not as yet been defined in dogs. Histopathologic diagnosis was considered to be the gold standard diagnostic technique in this study. Because of difficulty in obtaining Boxer tissues that were free of histopathologic lesions characteristic of ARVC, we additionally considered a combination of electrocardiographic, genetic, and historical characteristics, which mirror those proposed in human subjects, to confirm a diagnosis of ARVC. Other major criteria for diagnosis included presence of the striatin mutation (homozygous [n = 2], heterozygous [n = 4], negative [n = 7], not tested [n = 2]), history of sudden death (n = 3), evidence of cardiac dysfunction on echocardiogram (n = 5), or total ventricular ectopy (VE) of >10% in a 24‐hour period (n = 6). All Boxer dogs included in this study possessed, at minimum, 1 additional major criterion in addition to positive histopathology.
All samples were used for the confocal microscopy studies, whereas only a subset of these samples was available for additional experiments: Western blot (6 ARVC‐Boxer dogs, 5 Non‐ARVC dogs) and qPCR (6 ARVC‐Boxer dogs, 4 Non‐ARVC dogs). Available Boxer blood samples were submitted to Dr Kathryn Meurs to be screened for a mutation in the striatin gene. Dr Meurs and the authors were blinded to the mutation status of the samples until all additional experiments were completed.
The studies were performed in compliance with the NIH Guidelines for Care and Use of Laboratory Animals, and approved by IACUC at Cornell University. Informed consent was obtained from the owners of client‐owned dogs before inclusion in the study.
Tissue Sample Collection
All ARVC‐Boxers were client‐owned animals that died suddenly or were euthanized at the owners' request because of poor prognosis.6, 7 Non‐ARVC dogs were euthanized for nonrelated IACUC‐approved research, and hearts were donated for the purposes of our project. After euthanasia with sodium pentobarbital2 (86 mg/kg IV), hearts were collected via left thoracotomy from all dogs. Hearts were collected within 1 hour of death. Full thickness RV samples (2.5 cm in length and 2 cm in width) were fixed in 10% phosphate‐buffered formalin, embedded in paraffin and longitudinally sectioned into 4‐μm‐thick slices for confocal microscopy. When possible, additional RV samples were flash‐frozen in liquid nitrogen for Western blot and qPCR experiments. Samples from these dogs also were used in previous studies.6, 7
Confocal Microscopy
To determine the localization of β‐catenin, paraffin‐embedded tissue sections were stained with antibodies recognizing β‐catenin and calnexin, an endoplasmic reticulum (ER) marker. Protocols for immunolocalization of the relevant proteins followed those previously published.6, 20, 21 Briefly, sections were deparaffinized, and microwaved in citrate buffer for antigen retrieval. Sections then were blocked in 5% bovine serum albumin (BSA), 0.1% Triton buffer for 2 hours, and incubated overnight at 4°C with primary antibodies recognizing β‐catenin,3 calnexin,4 and striatin.4 After incubation, samples were rinsed in phosphate‐buffered saline (PBS) and treated with secondary antibodies for 45 minutes. Secondary antibodies used were Alexafluor 598 goat anti‐mouse, and Alexafluor 484 goat anti‐rabbit.5 Sections were examined using a Zeiss 510 Meta Confocal microscope equipped with a 63 × oil immersion lens and ZEN LE software.6
Western Blot and Quantitative (q) PCR
Western Blot
Western blotting was used to determine whether changes existed in the amounts of β‐catenin within the cardiac cells. The frozen RV tissue samples were homogenized in ice‐cold Laemmli buffer, and whole protein amounts measured by a Lowry protein assay.7 Fifteen micrograms of protein was loaded into each lane of an 8–16% tris‐glycine gradient gel.8 Proteins were separated by SDS‐PAGE, transferred onto nitrocellulose, blocked for 1 hour at room temperature with 5% nonfat milk in PBS‐Tween (0.5%), and probed overnight at 4º with a primary antibody recognizing β‐catenin.3 To ensure equal loading, blots were stripped with Restore Western Blot Stripping Buffer,9 and incubated with primary monoclonal antibody to α cardiac actin.10 All blots were visualized using chemiluminescence.9
Densitometry and Statistical Analysis
Densitometry was performed using Adobe Photoshop software.11 The densities of each band were measured, background subtracted, and adjusted for loading utilizing α cardiac actin as a control. Nonparametric statistical tests were used in all analyses. Data from 2 separate Western blots showed strong rank correlation between samples (Spearman's P = .86, P = .02). Data from each Western blot were normalized by computing Z‐scores and combined by averaging samples that were run independently in each Western blot. Box and whiskers plots were generated using R (v. 2.10.1).12 Boxes represent the 25th and 75th percentiles, and whiskers denote the points that lie within 1.5 times the interquartile range. The wide line shows the median. A 2‐sided Wilcoxon Rank Sum test was used to compare ARVC‐Boxer and Non‐ARVC canine samples.
Quantitative (q) PCR
To determine whether the increase in β‐catenin was the result of an increase in mRNA transcript levels, real‐time qPCR experiments were conducted. To isolate RNA, the frozen tissue samples were transferred to 2 mL microcentrifuge tubes containing 1 mL of Trizol Reagent.8 Samples were incubated at room temperature for 5 minutes, and then sonicated on ice for intervals of 20 seconds, until the tissue was broken down. Samples were centrifuged at 4o for 10 minutes at 7500 rpm to remove debris. The supernatant was transferred to a new tube, and 200 μL of chloroform was added; tubes then were vortexed for 15 seconds, and incubated for 3 minutes at room temperature. Tubes then were centrifuged for 15 minutes at 4o (12,000 × g). The aqueous phase was transferred to a new tube, and 500 μL ice‐cold isopropyl alcohol was added and the sample incubated for 10 minutes. Tubes were centrifuged for 10 minutes at 4o (12,000 × g). After centrifugation, the supernatant was discarded, and the pellet washed with 1 mL ice‐cold ethanol (75%), vortexed, and centrifuged at 4o for 5 minutes (7,500 × g). The supernatant was removed, and pellets air‐dried on ice for 10 minutes. Pellets were resuspended in RNase‐free H2O and incubated at 60o for 10 minutes. Final samples were stored at −80o.
Reverse transcription was performed using a PCR Express machine.n cDNA was made using the Superscript III First Strand Synthesis System,8 following instructions provided by the manufacturer. Briefly, for each reaction, 5 μL RNA, 1 μL random hexamer primers, 3 μL H2O, and 1 μL of dNTPs were incubated at 65o for 5 minutes, and then transferred to ice for 1 minute. A quantity of 10 μL of cDNA Synthesis Mix was added to each tube, consisting of 2 μL 10 × RT buffer, 4 μL of 25 mM MgCl2, 2 μL 0.1 M DTT, 1 μL RNase OUT, and 1 μL SuperScript III RT. Tubes were placed in a thermocycler and run through the following protocol: 25o for 10 minutes, 50o for 50 minutes, and 85o for 5 minutes. One microliter of RNAse H was added to each tube, and the samples were incubated at 37o for 20 minutes. Samples were stored at −20o.
Real‐time qPCR experiments were performed in a BioRad iCycler with an 18 μL/1 reaction volume containing 2 μL cDNA, 1 μL primers, 2 μL H2O, and 13 μL Platinum SYBR Green qPCR SuperMix‐UDG.8 Primers were designed for β‐catenin (forward: CCCTGAACTGACAAAACTGC; reverse: GGTCCACAGGAGTTTCTCG), Wnt (forward: GAGAGTGCAAGTGGCAGTTC; reverse: CAGGAGCGTGCCACTGAATG), TBP1 (internal control, forward: CCTGGAATCCCTATCTTCAG; reverse: CTGCTGCACTGCTGTCACAG), and β‐actin (internal control, forward: CATCACTATTGGCAACGAGC; reverse: CTTGCGGATGTCAACGTCAC).
Four controls were included for calibration and to ensure that all samples were within the linear rage of detection. A dilution series of a mixture of all DNA samples (1 : 1, 1 : 10, and 1 : 100) was included for calibration and to ensure that samples all were within the linear range of detection. Distilled deionized H2O was included as a negative control. A standard cycling program, based on SYBR Green qPCR Supermix8 instructions, was used. Samples were run for 50o for 2 minutes, 95o for 3 minutes, and then subjected to 55 cycles of 95o for 15 seconds followed by 60o for 60 seconds. Lastly, melting curves were calculated using the standard Roche 480 ramp (60o to 95o at 0.11°C per second).
Statistical Analysis
The “2nd Derivitive Max” setting in the Roche LightCycler 480 software (1.5.0)o was used to calculate Cp (crossing point). Results were imported into Microsoft Excel, and normalized using the “all DNA” dilution series. Concentrations were normalized for total RNA levels by dividing to a β‐Actin housekeeping control. As with the Western blot, R12 was used to generate boxplots and compute a 2‐sided Wilcoxon Rank Sum test to compare ARVC‐Boxer and Non‐ARVC canine samples.
Results
Histopathology: Presence of Fibro‐Fatty Infiltration and Myocarditis in ARVC Tissue
Histopathologic results from ARVC‐affected Boxers are shown in Figure 1. Panels A‐E show RV samples of myocardium from 5 different ARVC‐affected Boxers. Substantial fibro‐fatty infiltrates are noted in all samples. Panel F shows a higher magnification of a section taken from Panel E and shows the presence of lymphocytes, plasma cells, histiocytes, interstitial edema, and myocyte death because of myocarditis.
Figure 1.
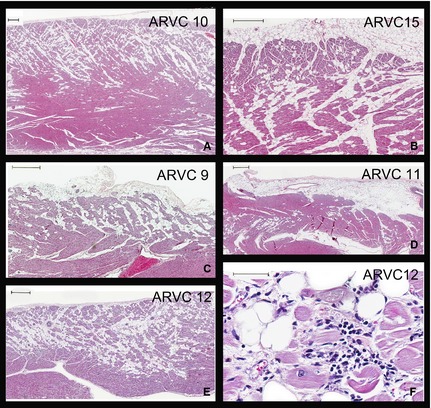
Representative photomicrographs of right ventricular sections from 5 Boxer dogs with ARVC (Panels A–E, Dog ID #s 10, 15, 9, 11, 12). All demonstrate classical patterns of fatty or fibro‐fatty myocardial replacement in a subepicardial to midmural distribution. Bars in upper left corners are 500 μm; Panel F. Higher magnification of a section taken from Panel E illustrates the additional presence of lymphocytes, plasma cells, histiocytes, interstitial edema, and myocyte death because of myocarditis; Bar in upper left corner is 50 μm. All sections were stained with hematoxylin and eosin.
Confocal Microscopy: Mislocalization of β‐Catenin and Striatin in Cardiac Cells
Confocal microscopy (Fig 2) indicated that in the Non‐ARVC canine RV sections, calnexin (green) localized to the area in and around the nucleus, corresponding to the ER (ER pattern). β‐catenin localized at sites of cell‐to‐cell apposition, corresponding to the intercalated disk. β‐catenin and calnexin were not found to colocalize in the Non‐ARVC sections. In the ARVC‐Boxer RV sections, calnexin still appeared to localize in an ER pattern. However, in contrast to the pattern observed in the Non‐ARVC canine sections, the localization appeared to spread out farther away from the nucleus on either end (Fig 2). Importantly, β‐catenin no longer localized to the intercalated disc; instead, β‐catenin colocalized with calnexin in an ER pattern around the nucleus.
Figure 2.
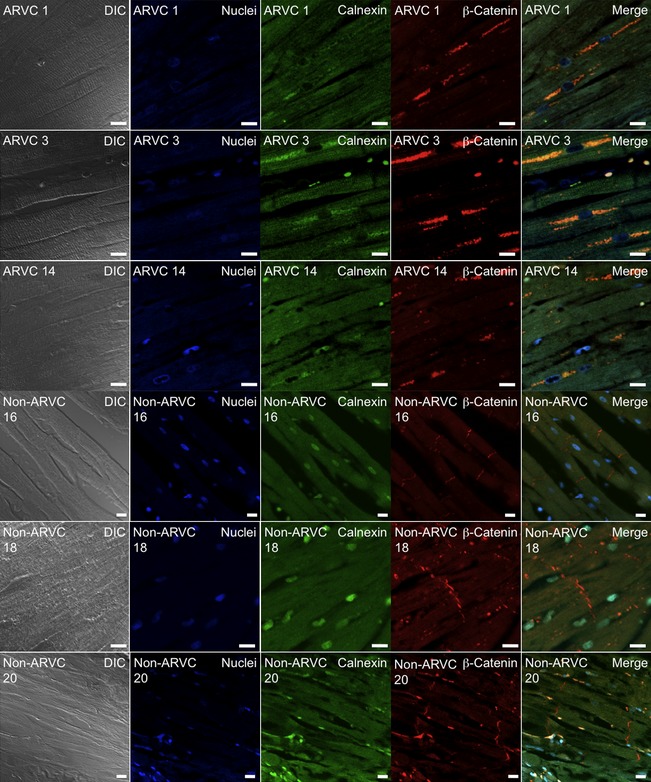
Colocalization of the endoplasmic reticulum (ER) marker calnexin (green, Column 3), and β‐catenin (red, Column 4) in right ventricular sections from Boxers affected with ARVC (Dog IDs #1, 3, 14) (Rows 1–3) and Non‐ARVC dogs (Dog IDs # 16, 18, 20) (Rows 4‐6). Columns 1–6 from left to right, Rows 1–6 from top to bottom. Column 1 shows Differential Image Contrast (DIC), whereas Column 6 shows merged images. In samples from Non‐ARVC dogs, calnexin localized to the area in and around the nucleus, corresponding to the ER, and β‐catenin localized at the intercalated disk. In the sections from Boxers with ARVC, β‐catenin and calnexin colocalized in an ER pattern (yellow) around the nucleus (blue).
Colocalization of striatin with β‐catenin was seen in the RV sections from both the Non‐ARVC dogs, and the ARVC‐Boxers. Within the Non‐ARVC canine samples, striatin localized in a “fish‐bone” pattern that corresponded to the intermediate filaments, as well as colocalizing with β‐catenin at the intercalated disk. In sharp contrast, striatin localized with β‐catenin in an ER pattern in the ARVC‐Boxers. (Fig 3)
Figure 3.
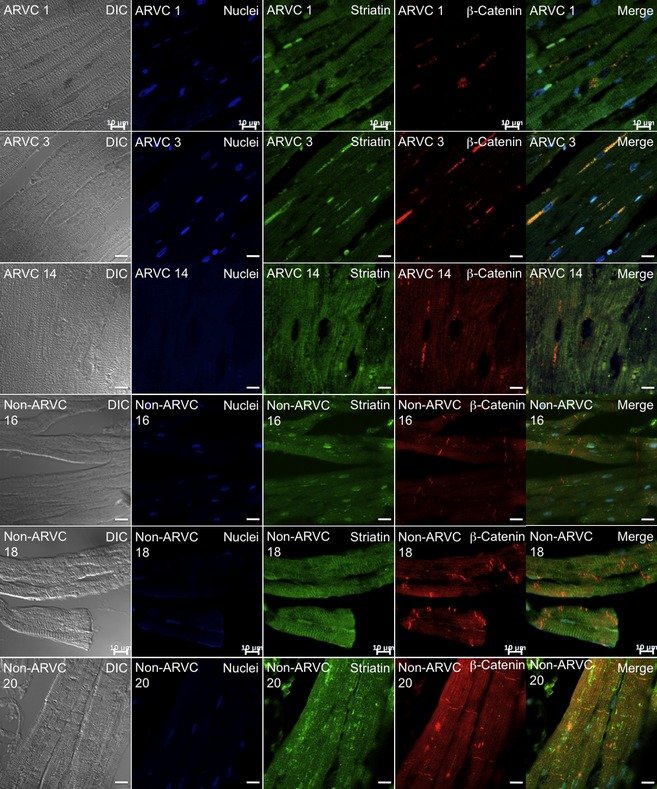
Colocalization of β‐catenin (red, Column 4) and striatin (green, Column 3) in RV sections from ARVC‐Boxers (Rows 1–3, Dog IDs #1, 3, 14) and Non‐ARVC dogs (Rows 4–6, Dog IDs #16, 18, 20). Columns 1–6 from left to right, Rows 1–6 from top to bottom. In the ARVC‐Boxer samples, β‐catenin and striatin colocalized in to the endoplasmic reticulum (an “ER‐pattern”) (yellow) surrounding the nucleus (blue). In the Non‐ARVC canine samples, β‐catenin localized at the intercalated disk and striatin localized at the intermediate filaments and intercalated disk.
Western Blot and qPCR: Increased β‐Catenin Protein Levels without Changes in mRNA
Figure 4A shows RV samples from the hearts of 4 Non‐ARVC dogs and 4 ARVC‐Boxers. Figure 4B shows a densitometry analysis of the Western blot, which shows a nearly 50% increase in the amounts of β‐catenin in the samples from ARVC‐Boxers compared to the Non‐ARVC dogs (P = .017). No changes in β‐catenin mRNA levels were detected in ARVC‐Boxer RV samples compared to Non‐ARVC canine RV samples (P = 1.0; Fig 4A). Additionally, we did not find evidence of changes in Wnt (Fig 5B) mRNA levels (P = .79).
Figure 4.
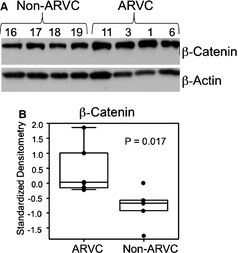
Western blot of right ventricular samples from 4 Non‐ARVC dogs and 4 ARVC‐Boxers dogs revealed an increase in total β‐catenin (A). β‐actin was used as a loading control. Densitometry analysis (B) revealed a statistically significant increase in the amount of β‐catenin by approximately 50% (P = .017).
Figure 5.
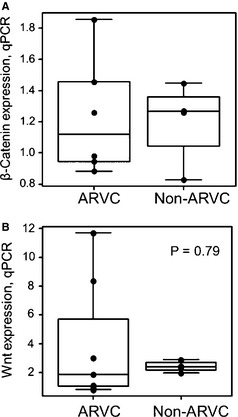
Quantitative PCR experiments for β‐catenin (A) and Wnt (B), using RNA from 6 ARVC and 4 Non‐ARVC RV samples. No significant changes in expression of β‐catenin or Wnt were observed.
Discussion
Our major findings include the mislocalization of β‐catenin and striatin to the ER in ARVC‐Boxers confirmed by histopathology. This mislocalization was observed in samples that were negative, heterozygous, and homozygous for the striatin mutation. Additionally, a 50% increase in β‐catenin protein levels was observed in samples from ARVC‐Boxers, compared to Non‐ARVC dogs, with no concurrent change in β‐catenin mRNA levels. We hypothesize that mislocalization of β‐catenin leads to decreased degradation of the protein, and decreased nuclear signaling via the canonical Wnt pathway. The Wnt pathway may play a role in the progression of fibro‐fatty infiltration in hearts from Boxers affected with ARVC. Involvement of the Wnt pathway in humans with ARVC also has been hypothesized.12
To explain our hypothesis that is based on the results reported herein, Figure 6 summarizes the normal Wnt pathway and how dysfunction of β‐catenin and striatin may disrupt Wnt signaling.13, 22 In its natural cycle, β‐catenin is produced in the nucleus, modified in the ER, and transported to the cell membrane, where it is an integral modulator of adherens junctions (Fig 6).23 β‐catenin then is phosphorylated and ubiquitinated in the cytoplasm by a group of proteins referred to as the “β‐catenin destruction complex”22 and degraded through the proteasomal pathway. In the presence of Wnt signaling, the β‐catenin destruction complex is inhibited, leading to cytosolic accumulation of β‐catenin. Accumulated cytosolic β‐catenin translocates to the nucleus, where it interacts with TCF/LEF transcription factors and is associated with many cellular functions, including cell proliferation and differentiation.22 Our finding of the mislocalization of β‐catenin to the ER suggests decreased trafficking of β‐catenin to the cell membrane and implies dysfunction of the canonical Wnt/β‐catenin pathway in cardiac tissue from Boxers with ARVC (Fig 6). Given the complex interactions of β‐catenin within various subcellular compartments, decreased trafficking has important implications for the association of β‐catenin with proteins at the plasma membrane, within the cytosol, and for its role in gene regulation within the nucleus.
Figure 6.
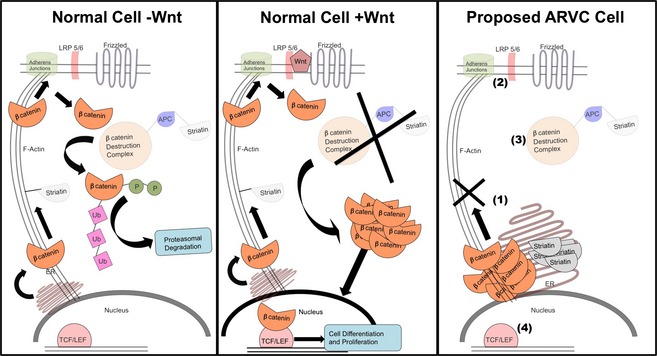
Left panel: In healthy cells, the absence of Wnt results in β‐catenin cycling from the nucleus to the adherens junctions and into the cytoplasm. Within the cytoplasm, interactions with the β‐catenin destruction complex (including APC and striatin) lead to phosphorylation (P), ubiquitination (Ub), and tagging β‐catenin for degradation through the proteasomal pathway. Middle panel: Activation of the Wnt pathway begins with the binding of Wnt to the membrane receptors, LRP5/6 and Frizzled. This initiates a cascade of events that ultimately inhibit the β‐catenin destruction complex. β‐catenin accumulates within the cytoplasm and is translocated to the nucleus. Here, β‐catenin binds to TCF/LEF transcription factors to initiate cellular pathways involved in differentiation and proliferation. Right panel: Our proposed dysfunction in ARVC‐affected cells from Boxers begins with accumulation of β‐catenin and striatin within the endoplasmic reticulum (ER). In this model, transport of β‐catenin to the membrane is compromised (1), resulting in a decreased number of adherens junctions (2), and decreased β‐catenin accumulating within the cytoplasm. Within the cytoplasm, less β‐catenin interacts with the β‐catenin destruction complex, and therefore decreased degradation of β‐catenin occurs (3). Finally, decreased translocation of β‐catenin into the nucleus results in decreased binding to TCF/LEF transcription factors and affects initiation of differentiation and proliferation pathways (4).
Previous studies have shown β‐catenin to be an integral component of the adherens junction, a mechanical junction that links apposing cardiomyocytes to the actin cytoskeleton.23 Decreased β‐catenin results in decreased assembly of adherens junctions24 and decreased cell adhesion.25, 26 During development, adherens junctions are the first macromolecular complexes to form at the intercalated disk.27, 28 In addition, failure of adherens junctions to form leads to failure of desmosomal and gap junction formation.29 These data are consistent with our previous studies, which identified decreased numbers of adherens junctions, desmosomes, and gap junctions at the intercalated disk in ARVC‐affected myocardium from Boxers.7 It seems plausible that decreased localization of β‐catenin at the plasma membrane may be associated with the loss of junctional complexes we previously reported.
Our results demonstrate an increase in β‐catenin protein levels, with no increase in β‐catenin mRNA or Wnt mRNA, and suggest decreased degradation of β‐catenin in RV samples from ARVC‐Boxers. We propose that ER mislocalization impedes the cycling of β‐catenin throughout the cell. This decreased trafficking of β‐catenin prevents or prolongs it from reaching its site of degradation within the cytosol (Fig 6).
The question remains as to whether mislocalization of β‐catenin at the ER results in a decreased cytosolic pool of β‐catenin available to translocate to the nucleus. Our current research does not quantify amounts of β‐catenin within the nuclei of ARVC‐Boxer versus Non‐ARVC canine samples. However, decreased amounts of β‐catenin binding to TCF/LEF transcription factors have been shown to result in an ARVC phenotype in transgenic mice.12 Additionally, stabilization and accumulation of β‐catenin within the cytosol have been linked to decreased apoptosis in adult cardiac tissue,17 and also are hypothesized to decrease adipogenesis in several cell types.15, 30 Thus, decreased trafficking of β‐catenin may lead to a reduction in the cytoplasmic pool, thereby, decreasing the translocation of β‐catenin to the nucleus. This disruption of the β‐catenin cycle may lead, directly or indirectly, to the increased apoptosis and adipogenesis observed in the development of ARVC in Boxers.
Confocal microscopy identified that striatin colocalized with β‐catenin in ARVC‐Boxer samples. It has been proposed that mutations in the 3′ UTR of striatin are linked to ARVC in Boxers, and result in disease caused by a change in the mRNA structure.10 Our results suggest that at least some striatin localizes to the ER in ARVC‐Boxer tissue, rather than forming the intermediate filament‐staining pattern observed in the Non‐ARVC canine samples. Striatin is thought to be associated with the canonical Wnt pathway (Fig 6), through its colocalization with the Wnt signaling regulator, adematous polyposis coli (APC).31 Additionally, striatin has been suggested to interact with F‐actin.31 Mutations in the striatin gene ultimately may result in its failure to traffic out of the ER (Fig 6). Interactions between β‐catenin and striatin are not documented, but it is possible that a direct or indirect reaction with striatin via F‐actin may aid in the trafficking of β‐catenin to the adherens junctions. Failure of striatin to be exported from the ER may have a deleterious effect on the subsequent trafficking of β‐catenin.
This study has several limitations. Previous sequencing reported by Meurs et al showed no increase in the LOD score in the locus containing β‐catenin,10 suggesting that a mutation in β‐catenin is unlikely. Therefore, the effects to β‐catenin observed in our studies are likely secondary to an as yet unknown process.
Localization of striatin in an ER pattern within ARVC‐affected cells was not previously reported.10 Although the reason for this discrepancy remains unclear, colocalization with β‐catenin, ER proteins, or both was not previously performed. We hypothesize that this localization pattern was not apparent with the use of different primary antibodies and colocalization techniques.
Because of our small sample size, we are unable to statistically predict the importance of striatin mutation status on the mislocalization of β‐catenin. Striatin mutation status was unknown at the time of data collection. In this study, no differences were noted among samples taken from striatin wild type, heterozygous, or homozygous samples. These data suggest that additional upstream events may be involved in the disruption of the Wnt pathway in cases of Boxer ARVC.
Next, we were restricted to analyzing cardiac tissue from affected dogs at the time of death or euthanasia. Importantly, this limited the number of available samples for use in our experiments. The use of cardiac tissue, rather than a cell culture model, also limits the type of experiments that we were able to conduct. For example, the use of siRNA techniques to silence β‐catenin, striatin or both in a cell culture model would be very useful in determining the individual importance of these genes in the development of the molecular abnormalities observed in cases of ARVC.
Finally, non‐Boxer control dogs were used in this study. Ideally, Boxers unaffected with ARVC would be the best controls for these studies, but it can be challenging to classify Boxers as not affected with ARVC. In our experience, some Boxers that test negative for the striatin mutation still demonstrate frequent characteristic left bundle branch block ventricular arrhythmias by 24‐hour Holter monitoring. Additionally, postmortem examinations of cardiac tissues from Boxers with normal cardiac examinations identify histopathologic lesions consistent with ARVC (Fig 1). Historically, postmortem histopathologic analysis of cardiac tissues has been considered the gold standard diagnostic test in both humans and in Boxer dogs. All of the Boxers examined in the course of this study had characteristic lesions of ARVC on histopathologic analysis, occasionally in the absence of antemortem signs of cardiovascular disease. To further ensure positive identification of affected animals, Boxers were required to meet at least 1 additional criterion in addition to ARVC‐positive histopathology. These additional criteria included presence of the striatin mutation, arrhythmias, history of sudden death, or evidence of cardiac dysfunction. All Boxers examined met a minimum of 2 criteria, and were considered affected with ARVC. Therefore, we chose to utilize nonBoxer, non‐ARVC dogs as the controls in these studies. Despite limitations, this research identifies abnormalities in the canonical Wnt pathway that may be relevant in Boxer dogs affected by ARVC.
Acknowledgments
The authors thank Dr Mark Roberson for sharing his enthusiasm, facilities, and thoughtful conversations. We also thank Dr Kathryn Meurs for the careful genetic screening of our samples. This research was supported by the Morris Animal Foundation grant #D09CA‐603.
Conflict of Interest Declaration: The authors disclose no conflict of interest.
Portions of this study were presented at the Morris Animal Foundation Conference, 2011, in Denver Colorado, and at the ACVIM Forum, 2011, in Denver, Colorado
Footnotes
Vila J, Oxford EM, Saelinger C, Moise NS, Fox PR, Pariaut R. Structural and molecular pathology of the atrium in boxer arrhythmogenic cardiomyopathy. J Vet Intern Med 2012;26:674 (Abstract)
Fatal‐Plus, Vortech Pharmaceutical, Dearborn, MI
BD Transduction Laboratories, San Jose, CA
Abcam, Cambridge, MA
Molecular Probes, Grand Island, NY
Carl Zeiss Microscopy, Thornwood, NY
BIO‐RAD, Hercules, CA
Invitrogen, Grand Island, NY
Thermo Fisher Scientific, Rockford, IL
Fitzgerald Industries International, Acton, MA
Adobe Systems Incorporated, San Jose, CA
References
- 1. Basso C, Fox PR, Meurs KM, et al. Arrhythmogenic right ventricular cardiomyopathy causing sudden cardiac death in boxer dogs: A new animal model of human disease. Circulation 2004;109:1180–1185. [DOI] [PubMed] [Google Scholar]
- 2. Sen‐Chowdhry S, Morgan RD, Chambers JC, et al. Arrhythmogenic cardiomyopathy: Etiology, diagnosis, and treatment. Annu Rev Med 2010;61:233–253. [DOI] [PubMed] [Google Scholar]
- 3. Sen‐Chowdhry S, Syrris P, McKenna WJ. Genetics of right ventricular cardiomyopathy. J Cardiovasc Electrophysiol 2005;16:927–935. [DOI] [PubMed] [Google Scholar]
- 4. Asimaki A, Saffitz JE. Gap junctions and arrhythmogenic cardiomyopathy. Heart Rhythm 2012;9:992–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Corrado D, Basso C, Thiene G, et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: A multicenter study. J Am Coll Cardiol 1997;30:1512–1520. [DOI] [PubMed] [Google Scholar]
- 6. Oxford EM, Everitt M, Coombs W, et al. Molecular composition of the intercalated disc in a spontaneous canine animal model of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Heart Rhythm 2007;4:1196–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Oxford EM, Danko CG, Kornreich BG, et al. Ultrastructural changes in cardiac myocytes from Boxer dogs with arrhythmogenic right ventricular cardiomyopathy. J Vet Cardiol 2011;13:101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Meurs KM, Ederer MM, Stern JA. Desmosomal gene evaluation in Boxers with arrhythmogenic right ventricular cardiomyopathy. Am J Vet Res 2007;68:1338–1341. [DOI] [PubMed] [Google Scholar]
- 9. Sanghamitra M, Talukder I, Singarapu N, et al. WD‐40 repeat protein SG2NA has multiple splice variants with tissue restricted and growth responsive properties. Gene 2008;420 48–56. [DOI] [PubMed] [Google Scholar]
- 10. Meurs KM, Mauceli E, Lahmers S, et al. Genome‐wide association identifies a deletion in the 3′ untranslated region of striatin in a canine model of arrhythmogenic right ventricular cardiomyopathy. Hum Genet 2010;128:315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Saffitz JE. Desmosome mutations in arrhythmogenic right ventricular cardiomyopathy: Important insight but only part of the picture. Circ Cardiovasc Genet 2009;2:415–417. [DOI] [PubMed] [Google Scholar]
- 12. Garcia‐Gras E, Lombardi R, Giocondo MJ, et al. Suppression of canonical Wnt/beta‐catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest 2006;116:2012–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. MacDonald BT, Tamai K, He X. Wnt/beta‐catenin signaling: Components, mechanisms, and diseases. Dev Cell 2009;17:9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heuberger J, Birchmeier W. Interplay of cadherin‐mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb Perspect Biol 2010;2:a002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shang YC, Zhang C, Wang SH, et al. Activated beta‐catenin induces myogenesis and inhibits adipogenesisin BM‐derived mesechymal stromal cells. Cytotherapy 2007;9:667–681. [DOI] [PubMed] [Google Scholar]
- 16. Kwon C, Arnold J, Hsiao EC, et al. Canonical Wnt signaling is a positive regulator of mammalian cardiac progenitors. Proc Natl Acad Sci USA 2007;104:10894–10899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zelarayan L, Gehrke C, Bergmann MW. Role of beta‐catenin in adult cardiac remodeling. Cell Cycle 2007;6:2120–2126. [DOI] [PubMed] [Google Scholar]
- 18. Marcus FI, McKenna WJ, Sherrill D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the task force criteria. Eur Heart J 2010;31:806–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Basso C, Calabrese F, Corrado D, Thiene G. Postmortem diagnosis in sudden cardiac death victims: Macroscopic, microscopic and molecular findings. Cardiovasc Res 2001;50:290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kaplan SR, Gard JJ, Carvajal‐Huerta L, et al. Structural and molecular pathology of the heart in Carvajal syndrome. Cardiovasc Pathol 2004;13:26–32. [DOI] [PubMed] [Google Scholar]
- 21. Kaplan SR, Gard JJ, Protonotarios N, et al. Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease). Heart Rhythm 2004;1:3–11. [DOI] [PubMed] [Google Scholar]
- 22. Cadigan KM. TCFs and Wnt/beta‐catenin signaling: More than one way to throw the switch. Curr Top Dev Biol 2012;98:1–34. [DOI] [PubMed] [Google Scholar]
- 23. Green KJ, Getsios S, Troyanovsky S, et al. Intercellular junction assembly, dynamics, and homeostasis. Cold Spring Harb Perspect Biol 2010;2:a000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Peifer M, Orsulic S, Sweeton D, et al. A role for the Drosophila segment polarity gene armadillo in cell adhesion and cytoskeletal integrity during oogenesis. Development 1993;118:1191–1207. [DOI] [PubMed] [Google Scholar]
- 25. Oyama T, Kanai Y, Ochiai A, et al. A truncated beta‐catenin disrupts the interaction between E‐cadherin and alpha‐catenin: A cause of loss of intercellular adhesiveness in human cancer cell lines. Cancer Res 1994;54:6282–6287. [PubMed] [Google Scholar]
- 26. Haegel H, Larue L, Ohsugi M, et al. Lack of beta‐catenin affects mouse development at gastrulation. Development 1995;121:3529–3537. [DOI] [PubMed] [Google Scholar]
- 27. Mathur M, Goodwin L, Cowin P. Interactions of the cytoplasmic domain of the desmosomal cadherin Dsg1 with plakoglobin. J Biol Chem 1994;269:14075–14080. [PubMed] [Google Scholar]
- 28. Vasioukhin V, Bauer C, Yin M, et al. Directed actin polymerization is the driving force for epithelial cell‐cell adhesion. Cell 2000;100:209–219. [DOI] [PubMed] [Google Scholar]
- 29. Kostetskii I, Li J, Xiong Y, et al. Induced deletion of the N‐cadherin gene in the heart leads to dissolution of the intercalated disc structure. Circ Res 2005;96:346–354. [DOI] [PubMed] [Google Scholar]
- 30. Taylor‐Jones JM, McGehee RE, Rando TA, et al. Activation of an adipogenic program in adult myoblasts with age. Mech Ageing Dev 2002;123:649–661. [DOI] [PubMed] [Google Scholar]
- 31. Breitman M, Zilberberg A, Caspi M, et al. The armadillo repeat domain of the APC tumor suppressor protein interacts with Striatin family members. Biochim Biophys Acta 2008;1783:1792–1802. [DOI] [PubMed] [Google Scholar]