

Abstract
Background
Myocardial injury detected by cardiac troponin I and T (cTnI and cTnT) in cardiac disease is associated with increased risk of death in humans and dogs.
Hypothesis
Presence of myocardial injury predicts long‐term death in cats with hypertrophic cardiomyopathy (HCM), and ongoing myocardial injury reflects change in left ventricular wall thickness over time.
Animals
Thirty‐six cats with primary HCM.
Methods
Prospective cohort study. Cats with HCM were included consecutively and examined every 6 months. Echocardiography, ECG, blood pressure, and serum cTnI and cTnT were evaluated at each visit. Cox proportional hazards regression analysis was performed to evaluate prognostic potential of serum troponin concentrations at admission and subsequent examinations. Correlations were used to examine associations between troponin concentrations and cardiac hypertrophy.
Results
Troponin concentrations at admission were median [range] 0.14 [0.004–1.02] ng/mL for cTnI, and 13 [13–79.5] ng/L for cTnT. Both were prognostic for death (P = .032 and .026) as were the last available concentrations of each (P = .016 and .003). The final cTnT concentration was a significant predictor of death even when adjusting for the admission concentration (P = .043). In a model containing both markers, only cTnT remained significant (P = .043). Left ventricular free wall thickness at end‐diastole (LVFWd) at admission was correlated with cTnI at admission (r = 0.35, P = .035), however no significant correlations (r = 0.2–0.31, P = .074–.26) were found between changes in troponin concentrations and left ventricular thickness over time.
Conclusions and Clinical Importance
Myocardial injury is part of the pathophysiology leading to disease progression and death. Low sensitivities and specificities prevent outcome prediction in individual cats.
Keywords: Biomarker, Companion animals, Feline, Myocardial injury, Prognostic significance
Abbreviations
- AMI
acute myocardial infarction
- ATE
arterial thromboembolism
- cTnI
cardiac troponin I
- cTnT
cardiac troponin T
- HCM
hypertrophic cardiomyopathy
- IVSd
interventricular septal thickness at end‐diastole
- LVFWd
left ventricular free wall thickness at end‐diastole
- T4
thyroxine
Hypertrophic cardiomyopathy (HCM) is the most common cardiac disease in cats.1 Primary HCM is characterized by concentric hypertrophy of the left ventricle in the absence of predisposing factors such as aortic stenosis, systemic arterial hypertension, and hyperthyroidism.1, 2 The thickening and fibrosis of the left ventricle cause an impaired diastolic function which can ultimately lead to heart failure, arterial thromboembolism (ATE), and sudden cardiac death.1, 2
Cardiac troponins I and T (cTnI and cTnT) have long been recognized as sensitive and specific markers of cardiomyocyte injury that are most commonly used in the diagnosis of acute myocardial infarction (AMI) in humans.3 However, an association has also been found between disease severity and circulating cardiac troponin in patients with heart failure, first reported in 1997.4 The pathophysiology of myocardial injury in chronic heart diseases is most likely distinct from that of AMI. The troponin elevations seen in chronic heart diseases are lower than those of AMI,5 and patients experience chronically increased troponin concentrations from on going injury rather than from a single acute insult.6
Cardiac troponins have prognostic importance in humans with chronic heart diseases just as is the case with AMI.5, 7, 8, 9 Studies with specific focus on HCM have reported that patients with increased serum cardiac troponins have a higher risk of cardiovascular events.7, 10 Similarly, in dogs cTnI have been reported to be prognostic for survival in chronic heart diseases such as mitral valve disease and dilated cardiomyopathy.11, 12 It has been suggested, however, that the increase in cTnI in dogs that die because of cardiac disease occurs close to the time of death.12 Therefore, it has been speculated that admission measurement might not be a satisfactory prognostic indicator, and, accordingly, that longitudinal measurements may be important.
Cardiac troponin I concentrations are significantly higher in cats with HCM compared to healthy cats,6, 13 but the prognostic potential of cardiac troponins in these cats has not been examined. The aim of this study was to investigate cardiac troponins as prognostic indicators of death in HCM in a cohort of cats of breeds with a known or suspected familial HCM. Whether longitudinal measurements of cardiac troponins in the cats reflected the changes in left ventricular thickness over time was also examined.
Materials and Methods
Study Population
Purebred cats with HCM were recruited consecutively through the cardiology service at the Department of Veterinary Clinical and Animal Sciences, University of Copenhagen, Denmark, from February 2010 to October 2011 and followed every 6 months until October 2012. A clinical examination was carried out, and echocardiography, ECG, blood pressure, and serum cTnI and cTnT were evaluated at each visit. The diagnosis of HCM was established using a conventional echocardiographic protocol as previously described.14 Cats were eligible for inclusion in case of a positive diagnosis of HCM, based on an overall cardiac evaluation including measurements of maximal end‐diastolic thickness of the interventricular septum (IVSd) or left ventricular caudal free wall (LVFWd) >5.5 mm in multiple segments from 2‐dimensional right parasternal long‐axis and short‐axis views of the left ventricle, and in the absence of other cardiac or systemic diseases associated with left ventricular myocardial hypertrophy.14 Echocardiographic parameters were obtained from digitally stored images as an average of measurements from 3 consecutive heartbeats, except in the case of 1 visit for 2 cats for which only the on‐site measurements from the day of examination were available. All measurements were made by the same experienced observer (JK). Baseline systolic pressure was measured in a quiet room and was calculated as an average of 3–5 consecutive measurements.1 Cats with persistent systolic arterial blood pressure >160 mmHg, serum creatinine >170 μmol/L, or serum thyroxine (T4) >35 nmol/L were excluded from the study.14
Outcome registration ended in January 2013 at which point survival time was determined. Clinical outcome was defined as cardiac death, either sudden death or euthanasia because of ATE or progression of HCM (clinical signs of heart failure or progression identified on echocardiography in a cat with declining quality of life), or noncardiac death and was determined through telephone contact with the owners. Survival time was calculated from the date of enrollment to the date of death, or the end of the study, and was considered censored for cats still alive at the end of the study.
Healthy cats were recruited through the HCM screening program at the Department of Veterinary Clinical and Animal Sciences, University of Copenhagen, and included as a control group based on normal findings on clinical examination, echocardiography, ECG, blood pressure, hematological and biochemical profiles, and serum T4. Blood was obtained from these cats only once.
The study was approved by the ethical committee of the Department of Veterinary Clinical and Animal Sciences, University of Copenhagen, Denmark, and informed owner consent was obtained.
Blood Sampling and Analysis
For each cat, a routine hematological and biochemical analysis and a serum T4 analysis were performed at the time of study inclusion and at each follow‐up visit. Serum for measurement of cTnI and EDTA plasma for measurement of cTnT were obtained at each visit. Serum was collected into 4 mL gel separator tubes, allowed to clot for 15 minutes at room temperature, centrifuged at 2,500 ×g for 5 minutes, separated, and stored in cryovials at −80°C within 6 hours of blood collection. Plasma was collected in 2 mL EDTA tubes, centrifuged, and treated as above. Samples were stored for a maximum of 17 months until batch analysis. Cardiac troponin I and cTnT were analyzed using commercially available high‐sensitivity immunoassays.2 , 3 The cTnI assay has recently been validated for use in cats and dogs,15 and the cTnT assay has been used previously for assessment of myocardial injury in dogs.16 Measured concentrations of cTnI and cTnT below the limits of detection of the assays (0.003 ng/mL and 13 ng/L) were assigned the value of the detection limits.
A storage error occurred for 1 sample from each of 14 cats which were stored for between 1 and 3 months at −20°C before being transferred to −80°C. Therefore, a 4 month stability study was carried out as follows: Serum samples from 5 different cats with varying degrees of HCM severity were collected and divided into 5 samples. One was placed at −80°C and the others at −20°C. After 1 month, 2 months, 3 months, and 4 months, a sample was moved from −20°C to −80°C. At the end, cTnI was measured in each sample for examination of cTnI recovery at 1 month intervals from 0 to 4 months with acceptable recovery set to 80–120%.
Statistical Analysis
Data were assessed for normality using the D'Agostino–Pearson omnibus test. Logarithmic transformation was applied where this made a Gaussian distribution acceptable. A two‐tailed t‐test was used to compare Gaussian data, and the Mann–Whitney U‐test was used to compare non‐Gaussian data.
For survival analysis, Cox proportional hazards regression models17 were computed. Cats were censored if they were alive at the end of the study period or if they died of noncardiac causes. To investigate the effect of cTnI measured at admission and at follow‐up, a Cox proportional hazard regression analysis was made with 2 covariates: cTnI concentration at admission and the last available cTnI concentration obtained at the last study visit. The effect of each covariate was tested separately (thus omitting the other covariate from the model) as well as in a combined model (testing whether cTnI at follow‐up had a significant prognostic effect beyond what was explained by cTnI at admission). Tests were carried out as likelihood ratio tests using the standard chi‐squared approximation. A similar analysis was made for cTnT, except that this variable was dichotomized as below or above the detection limit. Furthermore, the effects of cTnI and cTnT (dichotomized) were tested in a similar model containing both of these variables. For this analysis, the last available cTnI and cTnT concentrations were used. For visual assessment, Kaplan–Meier curves were also created with cats divided into high and low cTnI and cTnT groups, based on the median concentrations of the study population.
Correlations between admission measurements of LVFWd and IVSd and concentrations of cardiac troponins were examined. To evaluate whether changes in left ventricular thickness over time was correlated with changes in troponin concentrations, the differences between admission and last visit echocardiographic measurements (ΔLVFWd and ΔIVSd) and troponin concentrations (ΔcTnI and ΔcTnT) were then calculated for each cat, and correlations were assessed graphically as well as by Pearson's or Spearman's correlation coefficient where appropriate.
Statistical significance was defined as P < .05. All statistical analyses were conducted using commercial statistical software (Normality, t‐test, Mann–Whitney U‐test, correlations, Kaplan–Meier curves4 and Cox analyses by use of the function coxph in the R package survival5).
Results
Study Population Characteristics
Thirty‐six cats with HCM were consecutively included in the study (Table 1). These were 6 female intact, 4 female spayed, 4 male intact, and 22 male castrated cats with an age span of 6 months to 14 years (mean 4.9 years). Nineteen cats were Maine Coons, 13 cats were British Shorthairs, 2 were Norwegian Forest Cats, and 2 were Exotic Shorthairs.
Table 1.
Demographic data of the study population: 36 purebred cats with HCM.
| Breed | Age | Sex | |||
|---|---|---|---|---|---|
| Years Mean (range) | Intact Male | Castrated Male | Intact Female | Spayed Female | |
| MC | 4.8 (0.5–9) | 2 | 11 | 3 | 3 |
| BS | 4.6 (1–14) | 1 | 10 | 2 | 0 |
| NFC | 6.5 (6–7) | 1 | 0 | 1 | 0 |
| ES | 5.5 (4–7) | 1 | 0 | 0 | 1 |
MC, Maine Coon; BS, British Shorthair; NFC, Norwegian Forest Cat; ES, Exotic Shorthair.
23 healthy cats were included and consisted of 12 female intact, 7 female spayed, 3 male intact, and 1 male castrated cat with an age span of 11 months to 12 years (mean 4.3 years). Nine cats were Maine Coons, 8 were British Shorthairs, and 6 were Norwegian Forest Cats.
Clinical Outcome
At the end of the study, 23 of 36 cats with HCM were alive. Of the cats that died, 10 (28% of all cats) died of cardiac disease (1 sudden death, 2 euthanized due to ATE, and 7 euthanized because of progression of HCM). Three cats were euthanized for noncardiac reasons (1 had a broken leg after being hit by a car, 1 was 16 years old and had severe arthrosis, and 1 exhibited behavioral changes that were not because of progression of HCM). As these 3 cats were not critically ill, did not have signs of progression of cardiac disease, and were euthanized simply by request of the owner for the above reasons, they were counted as survivors in the survival analysis and were censored on the day of euthanasia. For 2 nonsurvivors, measurements were available only from study admission as the cats died within the first 6 months of admission. In the survival analysis, these 2 cats were, therefore, included with their admission concentration as both the admission and the last available concentration. Overall, median follow‐up time was 734 days.
Cardiac Troponins
The cTnI concentrations at admission for cats with HCM were median [range] 0.14 [0.004–1.02] ng/mL. Twenty‐four of the 36 cats with HCM (67%), including 9 of 10 nonsurvivors (90%), had cTnI concentrations above the concentration range of the healthy control cats (0.012 [0.003–0.09] ng/mL) at the time of admission. Nonsurvivors (0.40 [0.05–1.02] ng/mL) had significantly higher cTnI concentrations than survivors (0.10 [0.004–0.93] ng/mL) (t‐test, P = .0093) and control cats (t‐test, P < .001), and survivors had significantly higher cTnI concentrations than control cats (t‐test, P < .001) (Fig 1A). The last available cTnI concentrations were 0.56 [0.09–1.92] ng/mL for nonsurvivors and 0.10 [0.004–1.03] ng/mL for survivors. The longitudinal data are represented in Figure 2.
Figure 1.
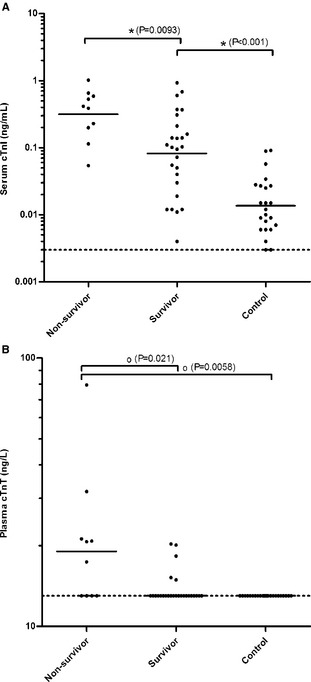
Serum cTnI (A) and plasma cTnT (B) concentrations of 36 cats with HCM (nonsurvivors and survivors) at admission and of healthy control cats. Geometric mean concentrations (A) for Gaussian and median concentrations (B) for non‐Gaussian data are shown as horizontal lines. Statistically significant differences between groups are symbolized with * (two‐tailed t‐test) and o (Mann–Whitney U‐test).
Figure 2.
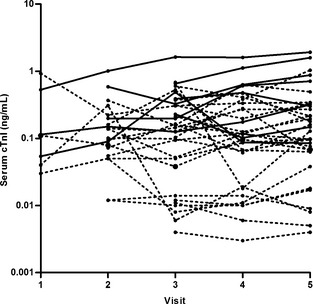
Longitudinal cTnI data of cats with HCM. Nonsurvivors (n = 8) are represented as solid black lines and survivors (n = 25) as dotted lines. Three cats (2 nonsurvivors and 1 survivor) are not represented as only admission concentrations were available for these cats. Visit 5 represents the last visit for each cat before death or the end of the study.
Cardiac troponin T concentrations at admission for cats with HCM were 13 [13–79.5] ng/L. Ten of the 36 cats (27.8%), including 6 of 10 nonsurvivors (60%), had cTnT concentrations above the detection limit of the assay (13 ng/L), whereas all healthy cats had cTnT concentrations below the detection limit. Nonsurvivors (19.1 [13–79.5] ng/L) had significantly higher cTnT concentrations than survivors (13 [13–20.3] ng/L) (Mann–Whitney, P = .021) and control cats (Mann–Whitney, P = .0058); however, no significant difference in cTnT concentrations was found between survivors and control cats (Mann–Whitney, P = .25) (Fig 1B). The last available cTnT concentrations were 27.2 [13–79.5] ng/L for nonsurvivors and 13 [13–27.8] ng/L for survivors.
Concentrations of both cTnI and cTnT at admission were prognostic for survival (P = .032 and .026) (visualized through Kaplan–Meier analyses, Fig 3) as were the last available concentrations of each marker (P = .016 and .003). For cats with cTnI concentrations ≥0.14 ng/mL (median concentration) 42% died compared to 16% of cats with lower concentrations corresponding to a sensitivity of 80% and a specificity of 61.5%. Similarly, 55% of cats with cTnT concentrations >13 ng/L (median concentration = assay detection limit) died compared to 19% of cats with undetectable cTnT corresponding to a sensitivity of 60% and a specificity of 84.6%. For cTnI, an increase in 1 ng/mL in the concentration measured at admission increased the hazard of death by a factor 9.1 (95% CI [1.4–61.4]). For the dichotomized cTnT, the hazard for cats with concentrations above the detection limit was 4.2 [1.2–15.1] times higher than for those below. The similar hazard ratios for the last available concentrations were 3.4 [1.3–8.7] for cTnI and 8.2 [1.7–39.0] for dichotomized cTnT. When knowing the cTnI concentrations at admission, the last available cTnI concentrations did not provide further prognostic information (P = .12), however the final cTnT concentrations were significant even when adjusting for the cTnT concentrations at admission (P = .043). In a model containing both cTnI and cTnT, only cTnT remained significant (P = .043).
Figure 3.

Kaplan–Meier survival curves comparing survival between cats with HCM with high and low serum concentrations according to median concentrations of cTnI (cutoff = 0.14 ng/mL) (A) and cTnT (cutoff = 13 ng/L) (B). Check marks indicate time of censoring for survivors.
LVFWd was found to be correlated with cTnI at admission (r = 0.35, Pearson, P = .035), but not with cTnT (r = 0.27, Spearman, P = .11). IVSd at admission was not correlated with either cTnI (r = 0.24, Pearson, P = .16) or cTnT (r = 0.09, Spearman, P = .61). No correlations were found between changes in left ventricular thickness measurements and changes in troponin concentrations over the course of the study (r = 0.20–0.31, Spearman, P = .074–.26).
In the stability study, 4 of 5 cats had cTnI concentrations in the range 0.087–0.18 ng/mL and were grouped (mean 0.12 ng/mL). The fifth cat had a cTnI concentration of 1.61 ng/mL and was examined separately. Recovery in both cases was found within an acceptable range from 0 to 3 months of storage at −20°C, but had decreased below acceptable limits (down to 72%) at 4 months for the low concentration group.
Discussion
This study documents a significant degree of chronic myocardial cell injury occurring in cats with HCM. Cardiac troponins measured at admission were significantly related to death in this cohort of cats as hypothesized, and a prognostically significant contribution of the longitudinal data was identified for cTnT, suggesting that myocardial injury is part of the pathophysiology that leads to disease progression and death. The change in cardiac troponin concentrations was not significantly correlated with the change in myocardial thickness, thus indicating that the chronically elevated and sometimes increasing troponin concentrations seen in these patients are not simply explained by changes in left ventricular thickness. Cardiac troponin I retained stability within acceptable limits for a maximum of 3 months at −20°C.
The presence of myocardial injury in HCM patients has previously been described in both humans and cats. The cause of injury is thought to be mild chronic ischemia, but its exact mechanism is still unknown. Histopathologically, the findings of HCM are cardiomyocyte hypertrophy and disarray (>5%), fibrosis, and abnormalities of the intramyocardial small vessels.18 Abnormal vessels can lead to cardiomyocyte necrosis and subsequent replacement fibrosis through an insufficient oxygen delivery.19, 20 In addition, presence of hypertrophy in the absence of adequate increases in myocardial capillary density may contribute to ischemia.21 As the disease is frequently genetic in origin,22, 23 it is also possible that cardiomyocyte abnormalities, determined by gene mutations, are directly contributing to the myocardial injury in HCM patients.24 A commonly observed effect of mutations is alterations in myocardial contractility,22, 25 resulting in an increased energy demand, and evidence also suggests that mutations predispose the heart to remodeling which could further contribute to cardiomyocyte injury.22 Other factors possibly involved in causing ongoing cell injury or death in HCM patients include activation of adrenergic, renin‐angiotensin‐aldosterone, or endothelin signaling pathways, inflammatory cytokines, degree of myocardial disarray, microthrombosis of coronary vessels, and oxidative and mechanical stress.5, 9, 26
Both cardiac troponin I and T were related to long‐term negative outcome in HCM cats in this study. Dogs with primary cardiac disease have long survival times associated with low cTnI concentrations,12 indicating absence of significant cardiomyocyte death. Supporting this finding, in human patients in heart failure an association exists between cardiac injury and death.5, 8, 9 Progressive cardiomyocyte loss is recognized as an important pathophysiological mechanism leading to cardiac dysfunction and heart failure.5 When looking specifically at cardiomyopathies, persistently high serum cardiac troponin concentrations have been reported in human patients with poor outcomes, also during stable periods without clinical signs.27 Therefore, an increase in cardiac troponins in such patients appears to reliably indicate subclinical ongoing myocyte injury. This is consistent with the findings of this study in which persistently higher troponin concentrations were found in nonsurvivors compared to survivors from the time of study inclusion till the end of the study. In humans with chronic heart failure, even small increases in troponin concentrations over time are predictive of cardiac events, whereas decreases over time reduce this risk.28, 29 Consequently, a rise in troponin concentration in the individual patient might indicate the need for more aggressive treatment, giving the troponins an important role in patient monitoring. However, in the overall human population with chronically and stably increased troponin concentrations in heart failure, longitudinal measurements do not add much discriminative power to baseline measurements.30 Our study indicates a similar situation in cats. Examining the contribution of the follow‐up data to the prognostic potential of the admission data revealed only a weakly significant contribution for cTnT and none for cTnI.
Whereas cTnI was the more sensitive marker in our study, revealing a higher percentage of cats with myocardial injury, cTnT appeared to be the superior prognostic marker. For both markers, however, the low sensitivities and specificities make them unsuitable for solitary prediction of outcome in individual cats, but they might supply supportive information to an echocardiographical evaluation with especially measurable cTnT concentrations indicating a poor prognosis. The superiority of cTnT could reflect the fact that increases in cTnT concentrations are generally found with more severe cardiomyocyte destruction, possibly because of the protein's larger size and to cTnT possibly having a closer structural binding to tropomyosin than cTnI.31 To the authors’ knowledge, this study is the first to report measurements of cTnT in cats with HCM.
Since a hypertrophy‐mediated mismatch between myocardial oxygen supply and demand is a suspected cause of chronic ischemia and myocardial injury, it might be expected that the degree of hypertrophy and myocardial injury were correlated. A correlation between maximum left ventricular wall thickness and cardiac troponins has been documented in humans10, 27 and weakly in cats,6, 13 and a weak association between LVFWd and cTnI at admission was also confirmed in this study. However, the degree of myocardial hypertrophy may change as the disease progresses, and this change in myocardial thickness (ΔLVFWd and ΔIVSd) was not found correlated with the concurrent changes in cardiac troponin concentrations (ΔcTnI and ΔcTnT) over the course of time in this study. Ongoing myocardial injury was, therefore, not simply explained by the degree of hypertrophy, and multiple factors are most likely involved in the pathogenesis of myocardial injury in HCM.
An important limitation of this study is a source of error inherent to veterinary survival studies in which nonsurvivors are defined to include animals that were euthanized. Some nonsurvivors of this study might, in fact, have survived longer, had treatment been continued. To minimize this error, cats that were euthanized were only counted as having suffered cardiac‐related death if euthanasia was due to progression of HCM (clinical signs of heart failure or progression identified on echocardiography in a cat with declining quality of life), or development of ATE. Thoracic radiographs were not available for the cats of this study, and, accordingly, confirming presence of heart failure was not possible, which is also a limitation of the study. However, clinical examination, echocardiography, and ECGs were sufficient to evaluate whether progression of HCM had occurred between visits. Thirdly, the sex distribution in HCM cats versus healthy controls is a limitation. Many male cats of the breeds included that went through the HCM screening program during the study were equivocal or were found to have false tendons on the echocardiographic examination and, therefore, were not eligible as controls of the study. As no difference in cardiac troponins between male and female cats has been reported to date, the resulting difference in the groups was accepted, whereas age‐matching of the groups was considered more important as higher cTnI concentrations in older cats have been documented.32 Finally, the storage error of a subset of the samples is a limitation which may have caused a minor decrease in troponin content. However, as the samples were stored for maximally 3 months and the stability study revealed acceptable recovery after 3 months of storage, this was not believed to impact the results.
In conclusion, cardiac troponins were significantly related to death in cats with HCM, suggesting that myocardial injury is part of the pathophysiology that leads to disease progression and death. Cardiac troponin I was highly sensitive in detecting myocardial injury in these cats while cTnT appeared to be the superior prognostic marker. Low sensitivities and specificities, however, prevent prediction of outcome by cardiac troponins in individual cats, thus troponins should be looked upon as a valuable adjunct to a thorough cardiovascular examination in HCM‐affected cats. Longitudinal measurements of cardiac troponins supplied only limited additional prognostic information when admission concentrations were available; however, a study including more cats would be valuable to fully appreciate the possible contribution of follow‐up data. Finally, whereas a correlation has been found between the thickness of the left ventricular free wall and cTnI concentration, the change in left ventricular thickness over time does not in itself explain changes in cardiac troponin concentrations, and a multifactorial cause of the chronic myocardial injury in cats with HCM is suspected.
Acknowledgments
The authors thank Professor Jens Peter Goetze, Department of Clinical Biochemistry, Rigshospitalet, University of Copenhagen, Denmark, for assistance with cardiac troponin T measurements and Veterinary Technician Michelle Dupont, Department of Veterinary Clinical and Animal Sciences, University of Copenhagen, Denmark, for assistance with blood sample collection.
Conflict of Interest Declaration: Authors disclose no conflict of interest.
This study was carried out at the Department of Veterinary Clinical and Animal Sciences, University of Copenhagen, Denmark, from 2010 to 2013.
Part of the study was funded through research grants from the Fund of Scientific Studies in Companion Animals, Denmark, and Agria & SKK's Research Fund for Companion Animals, Sweden.
Footnotes
PetMAP graphic System; Ramsey Chemical Inc, Tampa, FL
ADVIA Centaur CP TnI‐ultra; Siemens Healthcare Diagnostics Inc, Tarrytown, NY
Elecsys hs‐TnT; Roche Diagnostic Corporation, Indianapolis, IN
GraphPad Prism 5.02 for Windows; GraphPad Software, San Diego, CA
R 3.0.1 for Windows; R Foundation for Statistical Computing, Vienna, Austria
References
- 1. Abbott JA. Feline hypertrophic cardiomyopathy: An update. Vet Clin North Am Small Anim Pract 2010;40:685–700. [DOI] [PubMed] [Google Scholar]
- 2. Fox P, Liu S, Maron B. Echocardiographic assessment of spontaneously occurring feline hypertrophic cardiomyopathy ‐ an animal model of human disease. Circulation 1995;92:2645–2651. [DOI] [PubMed] [Google Scholar]
- 3. Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. J Am Coll Cardiol 2012;60:1581–1598. [DOI] [PubMed] [Google Scholar]
- 4. Missov E, Calzolari C, Pau B. Circulating cardiac troponin I in severe congestive heart failure. Circulation 1997;96:2953–2958. [DOI] [PubMed] [Google Scholar]
- 5. Horwich TB, Patel J, MacLellan WR, Fonarow GC. Cardiac troponin I is associated with impaired hemodynamics, progressive left ventricular dysfunction, and increased mortality rates in advanced heart failure. Circulation 2003;108:833–838. [DOI] [PubMed] [Google Scholar]
- 6. Herndon W, Kittleson M, Sanderson K, et al. Cardiac troponin I in feline hypertrophic cardiomyopathy. J Vet Intern Med 2002;16:558–564. [DOI] [PubMed] [Google Scholar]
- 7. Kubo T, Kitaoka H, Okawa M, et al. Combined measurements of cardiac troponin I and brain natriuretic peptide are useful for predicting adverse outcomes in hypertrophic cardiomyopathy. Circ J 2011;75:919–926. [DOI] [PubMed] [Google Scholar]
- 8. Latini R, Masson S, Anand IS, et al. Prognostic value of very low plasma concentrations of troponin T in patients with stable chronic heart failure. Circulation 2007;116:1242–1249. [DOI] [PubMed] [Google Scholar]
- 9. Peacock WF, De Marco T, Fonarow GC, et al. Cardiac troponin and outcome in acute heart failure. N Engl J Med 2008;358:2117–2126. [DOI] [PubMed] [Google Scholar]
- 10. Moreno V, Hernandez‐Romero D, Antonio Vilchez J, et al. Serum levels of high‐sensitivity troponin T: A novel marker for cardiac remodeling in hypertrophic cardiomyopathy. J Card Fail 2010;16:950–956. [DOI] [PubMed] [Google Scholar]
- 11. Fonfara S, Loureiro J, Swift S, et al. Cardiac troponin I as a marker for severity and prognosis of cardiac disease in dogs. Vet J 2010;184:334–339. [DOI] [PubMed] [Google Scholar]
- 12. Hezzell MJ, Boswood A, Chang Y, et al. The combined prognostic potential of serum high‐sensitivity cardiac troponin I and N‐terminal pro‐B‐type natriuretic peptide concentrations in dogs with degenerative mitral valve disease. J Vet Intern Med 2012;26:302–311. [DOI] [PubMed] [Google Scholar]
- 13. Connolly D, Cannata J, Boswood A, et al. Cardiac troponin I in cats with hypertrophic cardiomyopathy. J Feline Med Surg 2003;5:209–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Granstrom S, Godiksen MTN, Christiansen M, et al. Prevalence of hypertrophic cardiomyopathy in a cohort of British Shorthair cats in Denmark. J Vet Intern Med 2011;25:866–871. [DOI] [PubMed] [Google Scholar]
- 15. Langhorn R, Willesen J, Tarnow I, Kjelgaard‐Hansen M. Evaluation of a high sensitivity assay for measurement of canine and feline serum cardiac troponin I. Vet Clin Pathol 2013;42:490–498. doi:10.1111/vcp.12085. [DOI] [PubMed] [Google Scholar]
- 16. DeFrancesco TC, Atkins CE, Keene BW, et al. Prospective clinical evaluation of serum cardiac troponin T in dogs admitted to a veterinary teaching hospital. J Vet Intern Med 2002;16:553–557. [DOI] [PubMed] [Google Scholar]
- 17. Andersen PK, Gill RD. Cox regression model for counting processes ‐ a large sample study. Ann Stat 1982;10:1100–1120. [Google Scholar]
- 18. Liu SK, Roberts WC, Maron BJ. Comparison of morphologic findings in spontaneously occurring hypertrophic cardiomyopathy in humans, cats and dogs. Am J Cardiol 1993;72:944–951. [DOI] [PubMed] [Google Scholar]
- 19. Falk T, Ljungvall I, Zois NE, et al. Cardiac troponin‐I concentration, myocardial arteriosclerosis, and fibrosis in dogs with congestive heart failure because of myxomatous mitral valve disease. J Vet Intern Med 2013;27:500–506. [DOI] [PubMed] [Google Scholar]
- 20. Maron BJ, Wolfson JK, Epstein SE, Roberts WC. Intramural (small vessel) coronary artery disease in hypertrophic cardiomyopathy. J Am Coll Cardiol 1986;8:545–557. [DOI] [PubMed] [Google Scholar]
- 21. Krams R, Kofflard MJM, Duncker DJ, et al. Decreased coronary flow reserve in hypertrophic vardiomyopathy is related to remodeling of the coronary microcirculation. Circulation 1998;97:230–233. [DOI] [PubMed] [Google Scholar]
- 22. Gomes A, Potter J. Cellular and molecular aspects of familial hypertrophic cardiomyopathy caused by mutations in the cardiac troponin I gene. Mol Cell Biochem 2004;263:99–114. [DOI] [PubMed] [Google Scholar]
- 23. Meurs KM, Sanchez X, David RM, et al. A cardiac myosin binding protein C mutation in the Maine Coon cat with familial hypertrophic cardiomyopathy. Hum Mol Genet 2005;14:3587–3593. [DOI] [PubMed] [Google Scholar]
- 24. Cambronero F, Marin F, Roldan V, et al. Biomarkers of pathophysiology in hypertrophic cardiomyopathy: Implications for clinical management and prognosis. Eur Heart J 2009;30:139–151. [DOI] [PubMed] [Google Scholar]
- 25. Godiksen MTN, Kinnear C, Ravnsborg T, et al. Feline hypertrophic cardiomyopathy associated with the p. A31P Mutation in cMyBP‐C is caused by production of mutated cMyBP‐C with reduced binding to actin. Open J Vet Med 2013;3:95–103. [Google Scholar]
- 26. Cesta MF, Baty CJ, Keene BW, et al. Pathology of end‐stage remodeling in a family of cats with hypertrophic cardiomyopathy. Vet Pathol 2005;42:458–467. [DOI] [PubMed] [Google Scholar]
- 27. Sato Y, Taniguchi R, Nagai K, et al. Measurements of cardiac troponin T in patients with hypertrophic cardiomyopathy. Heart 2003;89:659–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miller WL, Hartman KA, Burritt MF, et al. Profiles of serial changes in cardiac troponin T concentrations and outcome in ambulatory patients with chronic heart failure. J Am Coll Cardiol 2009;54:1715–1721. [DOI] [PubMed] [Google Scholar]
- 29. Miller WL, Hartman KA, Burritt MF, et al. Serial biomarker measurements in ambulatory patients with chronic heart failure ‐ the importance of change over time. Circulation 2007;116:249–257. [DOI] [PubMed] [Google Scholar]
- 30. Masson S, Anand I, Favero C, et al. Serial measurement of cardiac troponin T using a highly sensitive assay in patients with chronic heart failure data from 2 large randomized clinical trials. Circulation 2012;125:280–288. [DOI] [PubMed] [Google Scholar]
- 31. Schober KE, Kirbach B, Oechtering G. Noninvasive assessment of myocardial cell injury in dogs with suspected cardiac contusion. J Vet Cardiol 1999;1:17–25. [DOI] [PubMed] [Google Scholar]
- 32. Serra M, Papakonstantinou S, Adamcova M, O'Brien PJ. Veterinary and toxicological applications for the detection of cardiac injury using cardiac troponin. Vet J 2010;185:50–57. [DOI] [PubMed] [Google Scholar]