

Abstract
Background
Limited information is available about the role of adipokines in the development and progression of acute pancreatitis (AP) in dogs.
Objectives
To determine whether the circulating concentrations of adipokines differed between healthy dogs and dogs with AP, and whether the circulating concentrations differed between AP survivors and AP nonsurvivors.
Animals
Twenty‐eight healthy dogs and 25 client‐owned dogs with AP.
Methods
Prospective observational cohort study of 25 client‐owned dogs with newly diagnosed AP and 28 otherwise healthy dogs with similar body condition scores. The serum concentrations of leptin, adiponectin, resistin, visfatin, interleukin (IL)‐1β, IL‐6, IL‐10, IL‐18, and tumor necrosis factor (TNF)‐α were measured.
Results
The serum concentrations of leptin (P = .0021), resistin (P = .0010), visfatin (P < .0001), IL‐1β (P < .0001), IL‐6 (P = .0002), IL‐10 (P < .0001), and IL‐18 (P < .0001) were significantly higher in dogs with AP than healthy dogs, whereas the adiponectin concentration (P = .0011) was significantly lower. There were significant differences in the serum concentrations of leptin (P = .028) and adiponectin (P = .046) in survivors and nonsurvivors. After the disappearance of clinical signs, the concentrations of resistin (P = .037) and IL‐1β (P = .027) decreased significantly, whereas the serum concentrations of leptin (P > .999), adiponectin (P = .11), visfatin (P = .83), IL‐6 (P = .82), IL‐10 (P = .82), IL‐18 (P = .56), and TNF‐α (P = .94) did not differ significantly.
Conclusion and Clinical Importance
This study showed that dysregulation of adipokines might be involved in the pathogenesis of AP. In addition, leptin and adiponectin are likely to be associated with mortality rate in AP.
Keywords: Adiponectin, Canine, Cytokine, Leptin, Resistin, Visfatin
Abbreviations
- AP
acute pancreatitis
- BCS
body condition score
- CI
confidence interval
- cPLI
canine pancreatic lipase immunoreactivity
- IL
interleukin
- NPO
nil per os
- TNF
tumor necrosis factor
Acute pancreatitis (AP) is defined as an acute inflammatory process of the pancreas with variable involvement of possible peripancreatic tissue or remote organ systems.1 It is widely accepted that pancreatic inflammation is relatively common, but its exact incidence in dogs is unknown because many dogs have subclinical or mild disease.2 However, AP can develop into a severe form, which is associated with local and systemic complications, and the reported mortality rate of AP is high, ranging from 27 to 58%.2, 3, 4 In human medicine, it is well known that obesity is a definitive risk factor for the development of local and systemic complications in AP and increases the likelihood of mortality rate with this disease.5, 6 In dogs, the risk of developing fatal AP is increased by an overweight body condition.7
Adipokines are biologically active substances derived from adipose tissue that act in an autocrine/paracrine or endocrine manner, which are important factors in the pathophysiology of obesity and its related conditions.8 Adipokines affect the regulation of energy metabolism, cardiovascular function, immune function, and inflammation,8 and they include prototypic adipokines such as leptin, adiponectin, resistin, and visfatin.9 In addition, a variety of inflammatory cytokines such as interleukin (IL)‐1, IL‐6, IL‐8, IL‐10, tumor necrosis factor (TNF)‐α, monocyte chemoattractant protein 1, and complement proteins are produced and released by adipose tissue, which may also be considered as adipokines.8, 9 In the context of AP, many inflammatory cytokines are activated after initial injury to the pancreatic acinar cells, which contribute to the inflammatory state.10 An experimental rodent study suggested that adipose tissue may become an important source of inflammatory mediators that participate in the progression of local abdominal damage to the systemic inflammatory response in the severe forms of the disease.11
Severe AP is characterized by lipase‐induced peripancreatic fat necrosis.12 Fat necrosis promotes high level infiltration of leukocytes into damaged areas of adipose tissue, and adipocytes, adipose tissue macrophages, and recruited immune cells become sources of inflammatory cytokines.11 In human medicine, it has been suggested that increases in the levels of circulating adipokines, especially resistin and visfatin, could be used as specific markers for peripancreatic fat necrosis.12, 13 However, little information is available about the circulating adipokine concentrations during the pathogenesis of AP in dogs. In our preliminary study, we measured the adipokine concentrations in dogs with AP,1 but the group sizes were small and it was difficult to reach any firm conclusions about the roles of adipokines during the development of AP in dogs. Therefore, the objective of this study was to examine whether there were differences in the circulating adipokine concentrations of dogs with AP and healthy dogs, as well as to determine whether the circulating adipokine concentrations differed between survivors and nonsurvivors among the dogs with AP.
Materials and Methods
Case Selection
Eighty‐two dogs with newly diagnosed AP were enrolled in this prospective, observational cohort study. Initially, dogs with another concurrent disease were not enrolled. Based on their body condition scores (BCS; 9‐point scale), 48 dogs with normal BCS (BCS = 5) were selected because we suspected that adipokines might be correlated with body fat mass in dogs.8 Among these 48 dogs, 2 dogs with recurrent bouts of AP and 5 dogs referred from other hospitals that suffered from AP with hospitalization for >3 days were excluded based on clinical data. Twelve intact bitches were also excluded. At follow‐up after AP treatment, 4 dogs were removed from the study because of hyperadrenocorticism, which has been reported to affect circulating adipokines concentrations.8, 14 Thus, 25 dogs with AP were included in this study. Twenty‐eight healthy, client‐owned dogs with the same BCS (BCS = 5) were included as controls. The healthy dogs were recruited from the same veterinary medical center when the dogs presented for health examinations and they had no histories of recent inflammatory disease. Informed consent was obtained from the owners and the University Ethics Committee approved all of the animal studies.
Diagnosis of Acute Pancreatitis
In the AP group, a diagnosis of AP was established only if all the abnormal findings were compatible with acute onset, ie, increased serum activity of amylase or lipase, morphologic evidence of pancreatitis obtained by ultrasonography,2 a positive SNAP cPL3 test, and increased Spec cPL3 concentrations.15, 16 Dogs exhibiting only some of the diagnosis criteria were not included in this study. The serum activities of amylase and lipase were measured with an auto‐analyzer4 and were considered to be elevated at >2,000 IU/L and >1,800 IU/L, respectively. Ultrasonographic findings suggestive of pancreatitis involvement in hypo/hyperechoic lesions, or mixed patterns, were recognized in the possibly enlarged and irregularly shaped pancreas.17, 18 In addition, alterations secondary to pancreatitis, such as hyperechoic mesentery, localized free abdominal fluid, thickened duodenal or gastric wall, spasmodic duodenum, irritated appearance of the adjacent intestines, and dilated common bile duct were also considered to be ultrasonographic evidence of AP.17, 18 The results of SNAP cPL tests were interpreted as abnormal only if the color of the sample spot was more intense than that of the reference spot.19 In the Spec cPL assays, concentrations >400 μg/L were considered to be consistent with pancreatitis.19
The dogs in the healthy group (Table 1) were considered to be healthy based on a physical examination, indirect measurement of their systolic blood pressure, examination of fecal specimens to determine the presence of parasites by a flotation technique, heartworm antigen testing, complete blood count analysis, serum biochemical analysis, urinalysis, adrenocorticotropic hormone response testing, and diagnostic imaging, including survey radiography and abdominal ultrasonography.
Table 1.
Characteristics of the dogs with acute pancreatitis (AP) and healthy dogs
| Dogs with AP (n = 25) | Healthy Dogs (n = 28) | |
|---|---|---|
| Sex | ||
| Female | 0 | 0 |
| Neutered female | 12 | 14 |
| Male | 5 | 8 |
| Neutered male | 8 | 6 |
| Body weight, kg (mean ± SEM; range) | 7.36 ± 1.45; 2.3–34 | 8.82 ± 1.44; 1.8–30 |
| Age, years (mean ± SEM; range) | 10.04 ± 0.81; 1–16 | 6.0 ± 0.77; 1–14 |
| Breed | ||
| Yorkshire Terrier | 6 | 3 |
| Miniature Schnauzer | 5 | 7 |
| Mixed breed | 3 | 3 |
| Shih Tzu | 3 | 3 |
| Miniature Poodle | 3 | 4 |
| Maltese | 2 | 2 |
| Pomeranian | 1 | 1 |
| Others | 2 | 5 |
Treatment and Grouping
Treatment was carried out as recommended in the literature at the time of the study.4, 20 The mainstay of the treatment was intravenous fluid therapy. The prescribed medication comprised pain killers, antiemetics, broad‐spectrum antibiotics, H2 receptor blockers, fresh plasma transfusion, and low‐molecular weight heparin, and nil per os (NPO) was maintained until vomiting stopped. Briefly, intravenous fluid therapy was initiated promptly upon hospitalization and dehydration was corrected using crystalloid fluid. Based on the assumption that abdominal pain could be present in all dogs with AP, butorphanol tartrate5 was administered (0.2 mg/kg IV q6h). Maropitant citrate6 was used as an antiemetic, which blocks centrally and peripherally mediated emesis (1 mg/kg SC q24h). Bacterial complications are rare in dogs with AP, but the dogs were treated with broad‐spectrum antibiotics if pyrexia, left shift neutropenia, or documented infection were present. Famotidine,7 an H2 receptor blocker, was also administered (0.5 mg/kg IV q12h). Dogs were maintained NPO if vomiting continued despite antiemetic treatment. If dogs had remained NPO and vomiting stopped, water was reintroduced slowly, which was followed by small amounts of a low‐fat diet on the next day.
Dogs with AP were categorized into “survivor” or “non‐survivor” groups. The survivor group included dogs that exhibited obvious improvements or even recovery at the end of hospitalization. The nonsurvivor group comprised dogs with a worsened clinical stage when they were discharged on the request of their owners and dogs that were euthanized or died, and the death was later verified by contacting the owner.
Assays
Blood samples were obtained from dogs with AP and control groups upon admission after fasting for ≥12 hours. Blood samples were also obtained from dogs with AP after their treatment at the time the clinical signs disappeared, again after fasting for ≥12 hours. For 15 dogs in the AP group, blood samples were only obtained upon their admission because of death (n = 13) or early discharge (n = 2). Serum was separated from clotted whole blood by centrifugation at 1,200 × g for 10 minutes within 1 hour of blood collection, and the sera were stored at −80°C until the assays. The following adipokines were analyzed: leptin, adiponectin, resistin, visfatin, IL‐1β, IL‐6, IL‐10, IL‐18, and TNF‐α.
The serum leptin concentrations were analyzed according to the manufacturer's protocol with a canine‐specific ELISA kit (Canine Leptin ELISA kit8 ), where the intra‐assay variability was 4% and the interassay variability was 6%, and the leptin assay sensitivity was 0.4 ng/mL. The serum adiponectin concentrations were analyzed with a canine‐specific ELISA kit (Canine Adiponectin ELISA kit8), where the intra‐ and interassay variabilities were <5% and <3%, respectively, and the assay sensitivity was 0.03 ng/mL. The serum resistin concentrations were analyzed with a canine‐specific ELISA kit (Canine Resistin ELISA kit9 ), according to the manufacturer's instructions, where the intra‐ and interassay variabilities were <5% and <7%, respectively. The serum visfatin concentrations were analyzed with a canine‐specific ELISA kit (Canine Visfatin ELISA kit9), where the intra‐ and interassay variabilities were 3% and 9%, respectively, and the assay sensitivity was 0.2 ng/mL. The serum IL‐1β concentrations were analyzed with a canine‐specific ELISA kit,10 where the intra‐ and interassay variabilities were <10% and <12%, respectively, and the assay sensitivity was 6.4 pg/mL. All samples, standards, and controls were assayed in duplicate. The optical density was determined at 450 nm with an automated microplate reader.11
The serum IL‐6, IL‐10, IL‐18, and TNF‐α concentrations were analyzed in duplicate with a Milliplex MAP Canine kit (Canine Cytokine/Chemokine MAGNETIC kit8). The intra‐ and interassay variabilities of all these assays were <5% and <15%, respectively, and the assay sensitivities for IL‐6, IL‐10, IL‐18, and TNF‐α were 3.7, 8.5, 5.8, and 6.1 pg/mL, respectively. The assays were quantified with a Luminex system.12
Statistical Analyses
All of the statistical analyses were carried out by a commercially available statistical program.13 Normality tests (D'Agostino‐Pearson omnibus test) were performed to determine whether the data were normally distributed. Mann‐Whitney U‐tests were used to compare the differences between the healthy and AP groups, and between the survivors and nonsurvivors in the AP group, and the data were expressed as medians (ranges). P‐values were calculated for two‐tailed tests, and the 95% confidence intervals (CIs) for the differences between medians were determined. When warranted, multiple comparisons were performed by the Holm‐Sidak method. Wilcoxon matched‐pairs signed rank tests were used to compare data for the AP group before and after treatment. The data were expressed as medians (ranges) and 95% CIs. P < .05 was considered significant.
Results
Circulating Adipokine Concentrations in Dogs with AP and Healthy Dogs
The median serum concentrations of leptin (95% CI for difference between medians = 0.63–5.63; P = .0021), resistin (95% CI for difference between medians = 7.0–27.8; P = .0010), and visfatin (95% CI for difference between medians = 0.10–0.14; P < .001) were significantly higher in the dogs with AP than in healthy dogs, whereas the median adiponectin concentration (95% CI for difference between medians = −6.96 to −1.42; P = .0011) was significantly lower in dogs with AP than in healthy dogs (Fig 1). The median serum concentrations of IL‐1β (95% CI for difference between medians = 44.20–95.90; P < .0001), IL‐6 (95% CI for difference between medians = 7.60–50.60; P < .0001), IL‐10 (95% CI for difference between medians = 10.30–25.80; P < .0001), and IL‐18 (95% CI for difference between medians = 15.50–37.40; P < .0001) were also significantly higher in dogs with AP than in healthy dogs, but the median TNF‐α concentration (95% CI for difference between medians = 0.0–1.88; P = .25) did not differ significantly between dogs with AP and healthy dogs (Fig 2).
Figure 1.
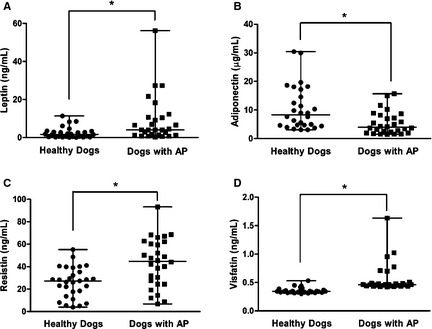
Comparison of the circulating concentrations of (A) leptin, (B) adiponectin, (C) resistin, and (D) visfatin in dogs with acute pancreatitis (n = 25) and healthy dogs (n = 28). The horizontal bars indicate the medians and ranges. *P < .05 (Mann‐Whitney U‐test).
Figure 2.

Comparison of the circulating concentrations of (A) interleukin (IL)‐1β, (B) IL‐6, (C) IL‐10, (D) IL‐18, and (E) tumor necrosis factor (TNF)‐α in dogs with acute pancreatitis (n = 25) and healthy dogs (n = 28). The horizontal bars indicate the medians and ranges. *P < .05 (Mann‐Whitney U‐test).
Differences Between Survivors and Nonsurvivors Among Dogs with AP
Compared with nonsurvivors, the median serum concentration of leptin (95% CI for difference between medians = −15.37 to −0.45; P = .028) was significantly lower in the dogs that survived AP, whereas the median adiponectin concentration (95% CI for difference between medians = 0.03–6.66; P = .046) was significantly higher (Fig 3). There were no significant differences in the median serum concentrations of other analytes between survivors and nonsurvivors within the AP group, including those of resistin (95% CI for difference between medians = −20.0 to 20.2; P = .76), visfatin (95% CI for difference between medians = −0.03 to 0.04; P = .86), IL‐1β (95% CI for difference between medians = −48.3 to 99.7; P = .80), IL‐6 (95% CI for difference between medians = −45.1 to 22.0; P = .74), IL‐10 (95% CI for difference between medians = −19.8 to 13.5; P = .66), IL‐18 (95% CI for difference between medians = −25.6 to 33.0; P = .40), and TNF‐α (95% CI for difference between medians = −1.88 to 5.1; P = .81) (Figs 3, 4).
Figure 3.
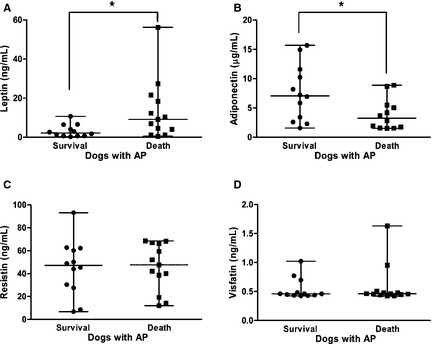
Differences in the circulating concentrations of (A) leptin, (B) adiponectin, (C) resistin, and (D) visfatin in the survivors (n = 12) and nonsurvivors (n = 13) among the dogs with acute pancreatitis. The horizontal bars indicate the medians and ranges. *P < .05 (Mann‐Whitney U‐test).
Figure 4.
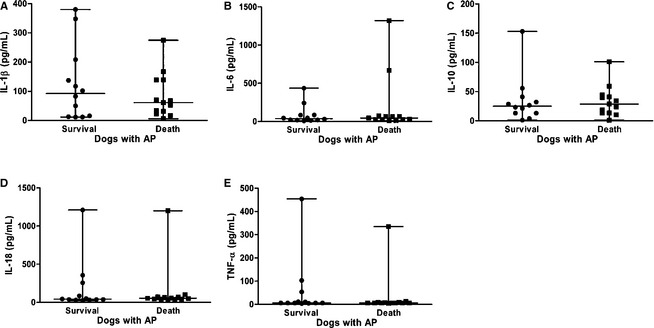
Differences in the circulating concentrations of (A) interleukin (IL)‐1β, (B) IL‐6, (C) IL‐10, (D) IL‐18, and (E) tumor necrosis factor (TNF)‐α in the survivors (n = 12) and nonsurvivors (n = 13) among the dogs with acute pancreatitis. The horizontal bars indicate the medians and ranges.
Comparison of the Survivors Before and After Treatment
In the dogs that survived, the median serum resistin concentration significantly decreased after treatment for AP (95% CI for difference between medians = −24.27 to −1.03; P = .037), but not the median concentrations of leptin (95% CI for difference between medians = −1.57 to 2.85; P > .999), adiponectin (95% CI for difference between medians = −0.05 to 3.67; P = .11), and visfatin (95% CI for difference between medians = −0.08 to 0.09; P = .83), compared with the concentrations before treatment (Fig 5). The median IL‐1β concentration (95% CI for difference between medians = −180.7 to −3.75; P = .027) decreased significantly, but not the median concentrations of IL‐6 (95% CI for difference between medians = −40.55 to 82.95; P = .82), IL‐10 (95% CI for difference between medians = −9.07 to 15.71; P = .82), IL‐18 (95% CI for difference between medians = −326.1 to 179.0; P = .56), and TNF‐α (95% CI for difference between medians = −11.37 to 24.60; P = .94) (Fig 6).
Figure 5.

Differences in the circulating concentrations of (A) leptin, (B) adiponectin, (C) resistin, and (D) visfatin before and after treatment in the dogs with acute pancreatitis (n = 10). The horizontal bars indicate the medians and ranges. *P < .05 (Wilcoxon matched‐pairs signed rank test).
Figure 6.
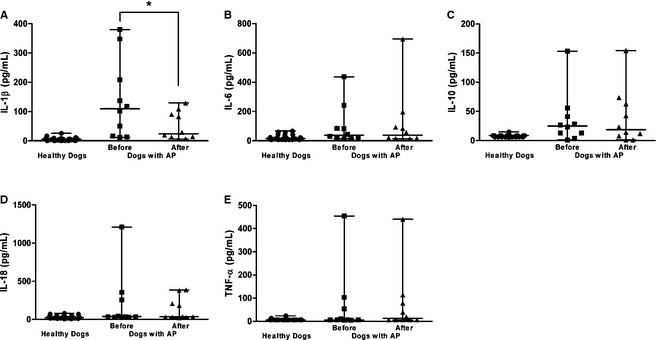
Differences in the circulating concentrations of (A) interleukin (IL)‐1β, (B) IL‐6, (C) IL‐10, (D) IL‐18, and (E) tumor necrosis factor (TNF)‐α before and after treatment in dogs with acute pancreatitis (n = 10). The horizontal bars indicate the medians and ranges. *P < .05 (Wilcoxon matched‐pairs signed rank test).
Discussion
Adipokines have been implicated in the development of AP, especially its severe form, in humans12, 21, 22, 23 and experimental animal models.11, 24, 25 Irrespective of the triggering factor, the disease progression typically follows a similar pathway after initial injury to the pancreatic acinar cells in humans, laboratory animals, and dogs.10 The results obtained in this study indicate that the circulating concentrations of serum adipokines differed between dogs with AP and healthy dogs, as well as between survivors and nonsurvivors. These results suggest a potential role for adipokines in the development and modulation of AP in dogs. This is the first study to determine the circulating concentrations of serum adipokines in dogs with AP.
This study showed that the serum leptin concentrations were upregulated in dogs with AP. The serum leptin concentrations of the AP survivors were also significantly lower compared with those of the AP nonsurvivors. Leptin is best known for its role in the regulation of food intake, body composition, and energy expenditure, where it acts mainly on the hypothalamus.9 Adipocytes are the main site of leptin synthesis and the main contributor to the serum leptin levels,8 but other tissue sites have been identified that can produce leptin. In dogs, leptin, leptin receptors, or both were recently detected in the stomach,26 mammary tissue,27 myocardium,28 corpus luteum,29 and even oral melanoma,30 but not in the pancreas, although it remains to be determined whether these findings were dependent on the experimental methods employed. However, it has been shown that leptin and its receptors are present in the pancreas in humans31 and laboratory animals,32 and it has been suggested that leptin may also play roles in the modulation of pancreatic function, as well as being involved in pancreatitis.32 Thus, several studies have evaluated the levels of leptin in AP, where high serum leptin levels were detected in AP groups compared with the control groups in humans32 and rats.25, 32, 33 These results were supported by other data that demonstrated the upregulation of the leptin mRNA and protein levels in the pancreas after the experimental induction of pancreatitis.32 Therefore, the inflamed pancreas might be a local source of leptin production in dogs. Further study on the transcriptional upregulation of leptin in the inflamed pancreas should help determine whether this is the case or not.
No clear data have associated the levels of leptin with the clinical severity of AP, although a small‐scale study reported that the leptin levels were elevated in severe AP compared with moderate or mild AP in humans,12 where it was suggested that peripancreatic fat cell necrosis might cause a massive release of adipokines in severe AP, which was positively associated with radiologic scores that described the amount of peripancreatic necrosis by computed tomography.12 However, adipose tissue does not accumulate contrast, which makes it difficult to evaluate its necrosis in computed tomography scans13 and criteria for peripancreatic necrosis have not been established in dogs. Thus, leptin might be a useful marker for adipose tissue necrosis, and high concentrations of leptin could facilitate the prediction of a severe disease course and the outcome of AP in dogs.
The circulating leptin concentrations were also increased in dogs with experimentally induced obesity34, 35 and in dogs with increased BCS.34, 36 Similar to humans and rodents, the main factor that affects the circulating leptin concentration in dogs appears to be the fat mass.8 It is already known that obesity is an independent risk factor for a poor outcome in severe AP,7 but the exact mechanism that underlies this relationship remains unknown. Therefore, further studies are needed to investigate the missing link in the casual relationship between leptin and severe AP.
This study also found that the circulating adiponectin concentrations were downregulated in dogs with AP. In addition, the serum adiponectin concentrations of survivors were elevated compared with those of nonsurvivors. These findings suggest that adiponectin might have a beneficial role in the modulation of the inflammatory response related to AP in dogs. The best‐characterized effects of adiponectin include the enhancement of insulin sensitivity,37 anti‐inflammatory properties,38 and inhibition of the development of atherosclerosis.39 Adiponectin is thought to be produced almost exclusively by mature adipocytes, and its gene expression is not observed in other tissues in dogs.8 Adipocytes are the most important source of adiponectin, but the serum adiponectin levels do not increase with the fat mass in the same way as the leptin levels.38 By contrast, an increase in the fat mass results in decreased circulating adiponectin, whereas weight loss results in increased adiponectin concentrations in humans and rodents.40 Similarly, adiponectin is inversely related to adiposity in dogs,41 although this inverse relationship is controversial.42 In human medicine, a previous study found that the serum adiponectin levels were inversely correlated with the body mass index and the severity of AP, which suggests that obesity increases the risk of severe AP markedly, possibly through the action of adiponectin.21 An experimental study also demonstrated that adiponectin plays a protective role in AP, where pancreatitis was induced in adiponectin‐knockout mice fed a high‐fat diet but not in wild‐type mice, and adenovirus‐mediated overexpression of adiponectin attenuated the severity of AP in adiponectin‐knockout mice.43 Another study also showed that the less obese mice among congenitally obese strains of mice had lower serum adiponectin concentrations but developed significantly more severe AP, thereby suggesting that the volume of adipose tissue itself is not solely responsible for the increased severity of pancreatitis in obesity and that other factors such as the adiponectin milieu are important modulators of the inflammatory response in AP.24 Thus, it was interesting to find that the serum concentrations of adiponectin were inversely related to the mortality because of AP in nonobese dogs in this study.
In this study, the circulating resistin concentrations were highly upregulated in dogs with AP, and the concentrations declined after treatment for AP. Resistin is a relatively recently described adipokine, which received its name based on the original observation that it induced insulin resistance in murine adipocytes.44 Resistin is expressed mainly by adipocytes in mice, whereas it appears to be produced primarily by macrophages in humans, although it is also expressed by adipocytes.38 In dogs, the expression of resistin has not yet been well demonstrated and little is known about its physiologic effects. In humans, it was suggested that resistin can be treated as a novel indicator of peripancreatic fat cell necrosis in patients with severe AP,12, 13, 22 which was based on the finding that the extent of fat cell necrosis causes a direct increase in passive adipokine release,12 with a similar pattern to leptin. In human medicine, it was also shown that the in vitro stimulation of macrophages with endotoxin or proinflammatory cytokines caused a marked increase in resistin production. Furthermore, the administration of endotoxin to human volunteers is associated with dramatically increased circulating resistin concentrations, which suggests that resistin may act as a critical mediator in inflammatory conditions.45 Therefore, changes in the circulating resistin concentration might be responsible for the modulation of inflammatory responses related to AP in dogs, although the ultimate source of resistin has not been identified in dogs.
No data are available on visfatin in dogs, which is a recently discovered adipokine that is produced mainly by visceral adipose tissue in humans and mice.46 It has been shown that visfatin binds and activates to the insulin receptor, thereby exerting insulin‐mimetic effects both in vitro and in vivo.47 In addition, visfatin corresponds to a protein that was identified previously as pre‐B‐cell colony‐enhancing factor, which is a cytokine that is significantly expressed in human acute lung injury and animal models of acute lung injury.47 In the context of AP, it was reported that the levels of visfatin were positively correlated with the clinical severity, clinical end points, such as death, and the need for intervention in human medicine.23 In this study, the serum visfatin concentrations were upregulated in dogs with AP, which suggests a potential role for visfatin in AP in dogs. However, further studies are required to fully understand the role of visfatin in dogs with AP, especially its potential associations with the clinical severity of canine AP.
In addition to macrophages, adipose tissues are the source of a variety of inflammatory cytokines, which are also regarded as adipokines.8, 9 It has been suggested that the systemic manifestations of AP are responsible for the majority of pancreatitis‐associated deaths,48 and several cytokines are involved in all aspects of the cascade that leads to systemic inflammatory response syndrome.49 In dogs, only 1 study has investigated the roles of cytokines during systemic complications, which showed that there were no differences in the total TNF‐α protein levels between groups with varying severity.50 Thus, this study analyzed proinflammatory cytokines, ie, IL‐1β, IL‐6, IL‐18, and TNF‐α, and an anti‐inflammatory cytokine, ie, IL‐10, to obtain further insights. However, there were no significant differences in the concentrations of all these cytokines between the survivors and nonsurvivors in the AP group. Only the serum IL‐1β concentration decreased significantly after the resolution of clinical signs, although we did not examine the source of this cytokine so it is not clear whether it was produced by adipose tissue. It is possible that IL‐1β is a potent facilitator of neutrophil migration, which is strongly associated with the development of systemic inflammation.51 In addition, the early activation of this cytokine has been demonstrated in experimental models of AP.52
This study was originally designed as a pilot study and it had several limitations. One limitation was the small number of dogs with AP, which constrains the reliability of the negative findings (such as the nonsignificant differences in the resistin, visfatin, and inflammatory cytokine(s) concentrations between survivors and nonsurvivors). The median concentrations were very close to each other, including those of some of the positive findings (such as the significant differences in the leptin and adiponectin concentrations) for survivors and nonsurvivors. Therefore, the clinical importance of those findings should be interpreted with caution. Another limitation was that the dogs with AP and the healthy dogs were not matched perfectly in terms of their sex and neuter status, which are factors that might affect the leptin concentrations.8 Future studies should avoid this limitation by matching the controls and cases rigorously and are required to clarify the relationship between the adipokine concentrations and the severity of AP in affected dogs.
In conclusion, this study suggests that the dysregulation of adipokines might be involved in the pathogenesis of AP. In particular, leptin and adiponectin are likely to be related to the risk of mortality because of AP in dogs. However, a cause‐and‐effect relationship between the biomarkers and the development of AP in dogs cannot be inferred because adipokine dysregulation is not specific to inflammatory pancreatic disease. Further studies are required to confirm these results using larger cohorts of dogs.
Acknowledgments
The authors thank all of the owners of the dogs included in this study. We also thank Dr Bo Kyoung Bae of IDEXX Korea for technical assistance and helpful discussions. This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), which was funded by the Ministry of Science, ICT, and Future Planning (2013R1A1A1011113).
Conflicts of Interest Declaration: The authors disclose no conflict of interest.
Footnotes
Lee IW, Chang DW, Yang MP, Kang JH. A pilot study to evaluate circulating levels of resistin in dogs with acute pancreatitis. J Vet Intern Med 2013;27:708 (abstract)
Alpha 5; Aloka Co., Tokyo, Japan
IDEXX Reference Laboratories, Seongnam‐si, South Korea
Hitachi 7020; Hitachi High‐Technologies Co., Tokyo, Japan
Butophan; Myungmoon Pharm. Co., Seoul, Korea
Cerenia; Pfizer, Pocé‐sur‐Cisse, France
Gaster Inj; Dong‐A ST, Seoul, Korea
Millipore Co., Billerica, MA
TSZ ELISA, Framingham, MA
Canine ELISA kit; USCN Life Sciences Co. Ltd, Wuhan, China
ELx 808; BioTek Instruments Inc., Winooski, VT
Luminex 200; Luminex Co., Billerica, MA
Prism 6; GraphPad Software Inc., La Jolla, CA
References
- 1. Ihse I, Lempinen M, Worning H. A clinically based classification system for acute pancreatitis. Summary of the ‘Atlanta Classification’. Scand J Gastroenterol 1994;29:95–96. [DOI] [PubMed] [Google Scholar]
- 2. Pápa K, Máthé A, Abonyi‐Tóth Z, et al. Occurrence, clinical features and outcome of canine pancreatitis (80 cases). Acta Vet Hung 2011;59:37–52. [DOI] [PubMed] [Google Scholar]
- 3. Cook AK, Breitschwerdt EB, Levine JF, et al. Risk factors associated with acute pancreatitis in dogs: 101 cases (1985–1990). J Am Vet Med Assoc 1993;203:673–679. [PubMed] [Google Scholar]
- 4. Mansfield C. Acute pancreatitis in dogs: Advances in understanding, diagnostics, and treatment. Top Companion Anim Med 2012;27:123–132. [DOI] [PubMed] [Google Scholar]
- 5. Martínez J, Johnson CD, Sánchez‐Payá J, et al. Obesity is a definitive risk factor of severity and mortality in acute pancreatitis: An updated meta‐analysis. Pancreatology 2006;6:206–209. [DOI] [PubMed] [Google Scholar]
- 6. Papachristou GI, Papachristou DJ, Avula H, et al. Obesity increases the severity of acute pancreatitis: Performance of APACHE‐O score and correlation with the inflammatory response. Pancreatology 2006;6:279–285. [DOI] [PubMed] [Google Scholar]
- 7. Hess RS, Kass PH, Shofer FS, et al. Evaluation of risk factors for fatal acute pancreatitis in dogs. J Am Vet Med Assoc 1999;214:46–51. [PubMed] [Google Scholar]
- 8. Radin MJ, Sharkey LC, Holycross BJ. Adipokines: A review of biological and analytical principles and an update in dogs, cats, and horses. Vet Clin Pathol 2009;38:136–156. [DOI] [PubMed] [Google Scholar]
- 9. Tilg H, Moschen AR. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol 2006;6:772–783. [DOI] [PubMed] [Google Scholar]
- 10. Mansfield C. Pathophysiology of acute pancreatitis: Potential application from experimental models and human medicine to dogs. J Vet Intern Med 2012;26:875–887. [DOI] [PubMed] [Google Scholar]
- 11. Franco‐Pons N, Gea‐Sorlí S, Closa D. Release of inflammatory mediators by adipose tissue during acute pancreatitis. J Pathol 2010;221:175–182. [DOI] [PubMed] [Google Scholar]
- 12. Schäffler A, Landfried K, Völk M, et al. Potential of adipocytokines in predicting peripancreatic necrosis and severity in acute pancreatitis: Pilot study. J Gastroenterol Hepatol 2007;22:326–334. [DOI] [PubMed] [Google Scholar]
- 13. Karpavicius A, Dambrauskas Z, Sileikis A, et al. Value of adipokines in predicting the severity of acute pancreatitis: Comprehensive review. World J Gastroenterol 2012;18:6620–6627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cho KD, Paek J, Kang JH, et al. Serum adipokine concentrations in dogs with naturally occurring pituitary‐dependent hyperadrenocorticism. J Vet Intern Med 2014;28:429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McCord K, Morley PS, Armstrong J, et al. A multi‐institutional study evaluating the diagnostic utility of the spec cPL™ and SNAP® cPL™ in clinical acute pancreatitis in 84 dogs. J Vet Intern Med 2012;26:888–896. [DOI] [PubMed] [Google Scholar]
- 16. Trivedi S, Marks SL, Kass PH, et al. Sensitivity and specificity of canine pancreas‐specific lipase (cPL) and other markers for pancreatitis in 70 dogs with and without histopathologic evidence of pancreatitis. J Vet Intern Med 2011;25:1241–1247. [DOI] [PubMed] [Google Scholar]
- 17. Hecht S, Henry G. Sonographic evaluation of the normal and abnormal pancreas. Clin Tech Small Anim Pract 2007;22:115–121. [DOI] [PubMed] [Google Scholar]
- 18. Penninck D. Pancreas In: Penninck D, d'Anjou M‐A, eds. Atlas of Small Animal Ultrasonography. Ames, IA; Wiley‐Blackwell; 2008;319–337. [Google Scholar]
- 19. Xenoulis PG, Steiner JM. Canine and feline pancreatic lipase immunoreactivity. Vet Clin Pathol 2012;41:312–324. [DOI] [PubMed] [Google Scholar]
- 20. Steiner JM. Canine pancreatic disease In: Ettinger SJ, Feldman EC, eds. Textbook of Veterinary Internal Medicine, 7th ed Philadelphia, PA; WB Saunders; 2010;1695–1704. [Google Scholar]
- 21. Sharma A, Muddana V, Lamb J, et al. Low serum adiponectin levels are associated with systemic organ failure in acute pancreatitis. Pancreas 2009;38:907–912. [DOI] [PubMed] [Google Scholar]
- 22. Schäffler A, Hamer O, Dickopf J, et al. Admission resistin levels predict peripancreatic necrosis and clinical severity in acute pancreatitis. Am J Gastroenterol 2010;105:2474–2484. [DOI] [PubMed] [Google Scholar]
- 23. Schäffler A, Hamer OW, Dickopf J, et al. Admission visfatin levels predict pancreatic and peripancreatic necrosis in acute pancreatitis and correlate with clinical severity. Am J Gastroenterol 2011;106:957–967. [DOI] [PubMed] [Google Scholar]
- 24. Zyromski NJ, Mathur A, Pitt HA, et al. A murine model of obesity implicates the adipokine milieu in the pathogenesis of severe acute pancreatitis. Am J Physiol Gastrointest Liver Physiol 2008;295:G552–G558. [DOI] [PubMed] [Google Scholar]
- 25. Kerem M, Bedirli A, Pasaoglu H, et al. Role of ghrelin and leptin in predicting the severity of acute pancreatitis. Dig Dis Sci 2007;52:950–955. [DOI] [PubMed] [Google Scholar]
- 26. Tsunoda Y, Yao H, Park J, Owyang C. Cholecystokinin synthesizes and secretes leptin in isolated canine gastric chief cells. Biochem Biophys Res Commun 2003;310:681–684. [DOI] [PubMed] [Google Scholar]
- 27. Ressel L, Finotello R, Innocenti VM, et al. Preliminary report on the expression of leptin and leptin receptor (ObR) in normal, hyperplastic and neoplastic canine mammary tissue. Res Vet Sci 2012;93:343–349. [DOI] [PubMed] [Google Scholar]
- 28. Fonfara S, Hetzel U, Tew SR, et al. Leptin expression in dogs with cardiac disease and congestive heart failure. J Vet Intern Med 2011;25:1017–1024. [DOI] [PubMed] [Google Scholar]
- 29. Balogh O, Kowalewski MP, Reichler IM. Leptin and leptin receptor gene expression in the canine corpus luteum during diestrus, pregnancy, and after aglepristone‐induced luteolysis. Reprod Domest Anim 2012;47(Suppl 6):40–42. [DOI] [PubMed] [Google Scholar]
- 30. Greene VR, Wilson H, Pfent C, et al. Expression of leptin and iNOS in oral melanomas in dogs. J Vet Intern Med 2013;27:1278–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Konturek PC, Konturek SJ, Brzozowski T, et al. Role of leptin in the stomach and the pancreas. J Physiol Paris 2001;95:345–354. [DOI] [PubMed] [Google Scholar]
- 32. Konturek PC, Jaworek J, Maniatoglou A, et al. Leptin modulates the inflammatory response in acute pancreatitis. Digestion 2002;65:149–160. [DOI] [PubMed] [Google Scholar]
- 33. Yavuz N, Unal E, Memisoglu K, et al. Plasma leptin levels in rats with pancreatitis. Tohoku J Exp Med 2004;204:243–248. [DOI] [PubMed] [Google Scholar]
- 34. Ishioka K, Soliman MM, Sagawa M, et al. Experimental and clinical studies on plasma leptin in obese dogs. J Vet Med Sci 2002;64:349–353. [DOI] [PubMed] [Google Scholar]
- 35. Sagawa MM, Nakadomo F, Honjoh T, et al. Correlation between plasma leptin concentration and body fat content in dogs. Am J Vet Res 2002;63:7–10. [DOI] [PubMed] [Google Scholar]
- 36. Ishioka K, Hosoya K, Kitagawa H, et al. Plasma leptin concentration in dogs: Effects of body condition score, age, gender and breeds. Res Vet Sci 2007;82:11–15. [DOI] [PubMed] [Google Scholar]
- 37. Yadav A, Kataria MA, Saini V, Yadav A. Role of leptin and adiponectin in insulin resistance. Clin Chim Acta 2013;417:80–84. [DOI] [PubMed] [Google Scholar]
- 38. Fantuzzi G. Adipose tissue, adipokines, and inflammation. J Allergy Clin Immunol 2005;115:911–919. [DOI] [PubMed] [Google Scholar]
- 39. Hopkins TA, Ouchi N, Shibata R, Walsh K. Adiponectin actions in the cardiovascular system. Cardiovasc Res 2007;74:11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hu E, Liang P, Spiegelman BM. AdipoQ is a novel adipose‐specific gene dysregulated in obesity. J Biol Chem 1996;271:10697–10703. [DOI] [PubMed] [Google Scholar]
- 41. Ishioka K, Omachi A, Sagawa M, et al. Canine adiponectin: cDNA structure, mRNA expression in adipose tissues and reduced plasma levels in obesity. Res Vet Sci 2006;80:127–132. [DOI] [PubMed] [Google Scholar]
- 42. Verkest KR, Rose FJ, Fleeman LM, et al. Adiposity and adiponectin in dogs: Investigation of causes of discrepant results between two studies. Domest Anim Endocrinol 2011;41:35–41. [DOI] [PubMed] [Google Scholar]
- 43. Araki H, Nishihara T, Matsuda M, et al. Adiponectin plays a protective role in caerulein‐induced acute pancreatitis in mice fed a high‐fat diet. Gut 2008;57:1431–1440. [DOI] [PubMed] [Google Scholar]
- 44. Steppan CM, Bailey ST, Bhat S, et al. The hormone resistin links obesity to diabetes. Nature 2001;409:307–312. [DOI] [PubMed] [Google Scholar]
- 45. Lehrke M, Reilly MP, Millington SC, et al. An inflammatory cascade leading to hyperresistinemia in humans. PLoS Med 2004;1:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fukuhara A, Matsuda M, Nishizawa M, et al. Visfatin: A protein secreted by visceral fat that mimics the effects of insulin. Science 2005;307:426–430. [DOI] [PubMed] [Google Scholar]
- 47. Ye SQ, Simon BA, Maloney JP, et al. Pre‐B‐cell colony‐enhancing factor as a potential novel biomarker in acute lung injury. Am J Respir Cirt Care Med 2005;171:361–370. [DOI] [PubMed] [Google Scholar]
- 48. Norman J. The role of cytokines in the pathogenesis of acute pancreatitis. Am J Surg 1998;175:76–83. [DOI] [PubMed] [Google Scholar]
- 49. Makhija R, Kingsnorth AN. Cytokine storm in acute pancreatitis. J Hepatobiliary Pancreat Surg 2002;9:401–410. [DOI] [PubMed] [Google Scholar]
- 50. Ruaux CG, Pennington HL, Worrall S, Atwell RB. Tumor necrosis factor‐alpha at presentation in 60 cases of spontaneous canine acute pancreatitis. Vet Immunol Immunopathol 1999;72:369–376. [DOI] [PubMed] [Google Scholar]
- 51. Dinarello CA. Biologic basis for interleukin‐1 in disease. Blood 1996;87:2095–2147. [PubMed] [Google Scholar]
- 52. Fink GW, Norman JG. Specific changes in the pancreatic expression of the interleukin 1 family of genes during experimental acute pancreatitis. Cytokine 1997;9:1023–1027. [DOI] [PubMed] [Google Scholar]