

Abstract
Binding of polo-like kinase 1 (Plk1) polo-box domains (PBDs) to phosphothreonine (pThr)/phosphoserine (pSer)-containing sequences is critical for the proper function of Plk1. Although high-affinity synthetic pThr-containing peptides provide starting points for developing PBD-directed inhibitors, to date the efficacy of such peptides in whole cell assays has been poor. This potentially reflects limited cell membrane permeability arising, in part, from the di-anionic nature of the phosphoryl group or its mimetics. In our current paper we report the unanticipated on-resin N(τ)-alkylation of histidine residues already bearing a N(π)- alkyl group. This resulted in cationic imidazolium-containing pThr peptides, several of which exhibit single-digit nanomolar PBD-binding affinities in extracellular assays and improved antimitotic efficacies in intact cells. We enhanced the cellular efficacies of these peptides further by applying bio-reversible pivaloyloxymethyl (POM) phosphoryl protection. New structural insights presented in our current study, including the potential utility of intramolecular charge masking, may be useful for the further development of PBD-binding peptides and peptide mimetics.
Keywords: Plk1, polo kinase, polo-box domain, crystal structure, cationic dialkyl histidine, intramolecular charge masking, phosphopeptides
INTRODUCTION
The polo-like family of serine/threonine protein kinases (Plk1- Plk5; collectively called Plks) plays pivotal roles in cell cycle regulation and cell proliferation.1–5 Plks1 – Plk3 contain N-terminal catalytic domains, as well as C-terminal polo-box domains (PBDs), which bind to “Ser- pSer/pThr”-containing motifs. These latter phospho-dependent protein-protein interactions (PPIs) mediate the sub-cellular localization required for proper Plk function.6–10 Plk1 has been recognized among Plk family members as a potential anticancer therapeutic target and significant effort has been directed at developing Plk1 kinase domain inhibitors.11–20 However, a potential drawback of kinase domain inhibitors is low specificity resulting from similarities among the ATP-binding clefts of Plks. Down regulation of Plk1 along with concomitant inhibition of the closely-related Plk2 or Plk3 is undesirable, due to the positive roles these latter kinases play in maintaining genomic stability.21, 22 The uniqueness of PBDs to the Plk family makes disruption of PBD-dependent protein-protein interactions (PPIs) an attractive alternative to kinase domain inhibitors.10, 23–29
In our efforts to develop highly potent Plk1 PBD-binding inhibitors, we recently reported the introduction of long chain alkyl phenyl groups onto the N(π) nitrogen (N3) of the His imidazole ring in the polo-box interacting protein 1 (PBIP1) – derived peptide “PLHSpT” (1) to form peptides such as 2a (Figure 1). This modification can impart up to 1,000-fold PBD-binding affinity enhancement through interaction of the N(π) His-adduct moiety within a cryptic hydrophobic binding pocket on the surface of the PBD (PDB accession code 3RQ7).30 Our subsequent work31–33 as well as the work of others,34, 35 has demonstrated that aryl functionality tethered from a variety of locations along the peptide chain can bind in the newly discovered hydrophobic channel. Among the peptides reported to date, the N(π)-alkyl-His residue in peptide 2a may be viewed as being “privileged” in its ability to access this new binding region, because of its close proximity to the key signature affinity residues, “Ser-pThr.” The compact nature of the “(alkyl)His-Ser-pThr” motif makes peptides such as 2a attractive starting points for inhibitor design. However, in spite of low-nanomolar PBD-binding affinities in in vitro assays, peptides related to 2a achieve effects in cell culture assays only at much higher concentrations.30 This low cellular efficacy could potentially result from poor cell membrane permeability, which may be attributable in part to the peptide phosphoryl di-anionic charge.
Figure 1.
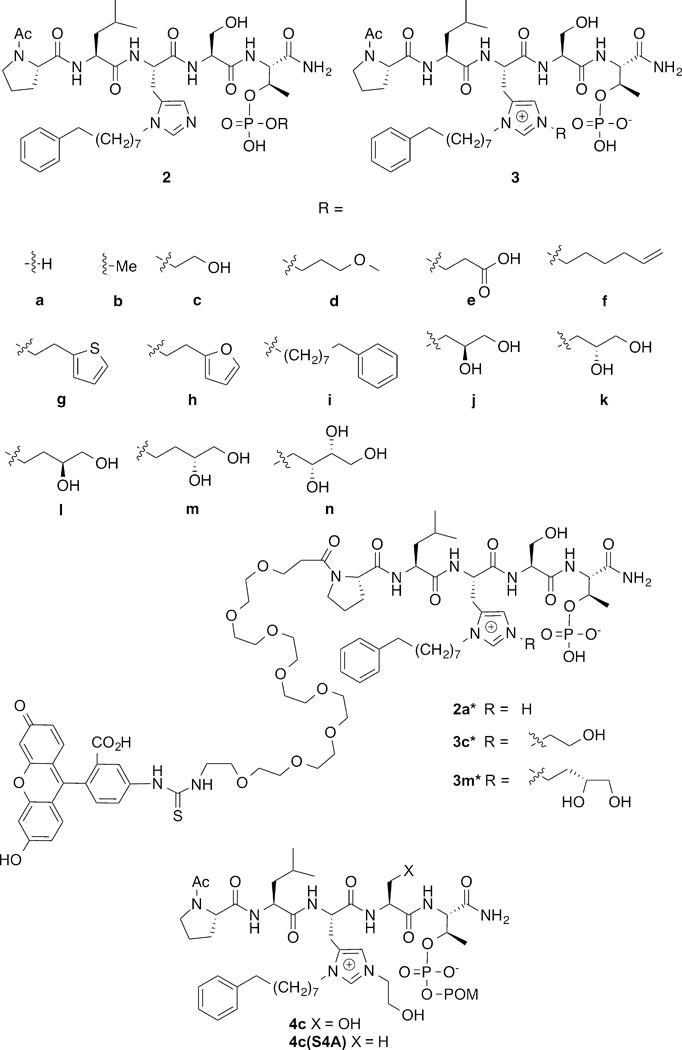
Structures of peptides discussed in the text.
Overcoming limitations imposed by poor cell membrane permeability of phosphoryl functionality is a general challenge to the development of inhibitors of phospho-dependent PPIs. Traditionally, enhancing cellular uptake by minimizing anionic charge has been approached either by using less charged phosphoryl mimetics or by blocking the acidic phosphoryl hydroxyls with bio-reversible prodrug moieties. In our current paper we report the unexpected on-resin Mitsunobu-catalyzed His N(τ)-alkylation of 2a to yield imidazolium-containing peptides (3, Figure 1) that exhibit enhanced cellular efficacies, potentially through intramolecular “charge masking.”36 As reported herein, we obtained further optimization of cellular efficacy by combining N(τ)-alkylation with bio-reversible phosphoryl prodrug protection.
RESULTS AND DISCUSSION
Unanticipated On-Resin Mitsunobu-Catalyzed His N(τ) Alkylation
Using 2a as a platform, we had previously examined replacing the pThr residue with mono-anionic non-phosphate-containing pThr mimetics. However, the resulting peptides exhibited significantly reduced Plk1 PBD-binding affinities.37 Therefore, our current work began as an attempt to revisit the issue of mono-anionic pThr esters within the context of the 2a platform. The objective was to prepare mono-phosphoryl esters of type 2b – 2n (Figure 1). In our earlier studies, when we subjected the resin-bound sequence, “Ac-Pro-Leu-His[N(τ)-Trt]-Ser(OtBu)-pThr[(OBn)(OH)]-amide resin” to Mitsunobu coupling using a series of alcohols, the major product resulted from esterification of a single pThr phosphoryl hydroxyl, as expected. However in general, these mono-esters displayed reduced PBD-affinities relative to 1.30 As we indicated in our prior report, along with the major pThr esterified peptides, we also identified minor byproducts arising from His N(π)-alkylation and some of these exhibited extremely high PBD-binding affinities. We subsequently confirmed that under Mitsunobu conditions, N(π)-alkylation of N(τ)-Trt-protected His residues can occur, but in very limited fashion.38
In our current work, we repeated the on-resin Mistunobu reactions as outlined above, except that instead of a His[N(τ)-Trt] residue we employed a His[N(π)-(CH2)8Ph] residue (designated as “His*”).38 Following acid-catalyzed resin cleavage and HPLC purification we obtained as the major products, peptides having molecular weights corresponding to the anticipated phosphoryl esters 2b – 2n. To unambiguously confirm the product structures, we subjected all members of the series to high-resolution electrospray ionization (ESI) LC-MS/MS analysis. As described below, to our surprise these analyses showed that rather than derivatizing a phosphoryl oxygen as had been anticipated, in most cases alkylation had occurred predominately at the His N(τ)-position. This yielded a series of isomeric products 3b – 3n (Figure 1).
Mass Spectral Characterization of His N(π)N(τ) Bis-alkylated Products
The mass spectral characterization of peptide 3m provides a model of the analytical approach that was applied to all members of the series. Electrospray ionization (ESI) of 3m generated both the protonated peptide (MH+) and the double-protonated peptide (M+2H)2+, which are the two ions that would also be expected for the isomeric phosphopeptide ester 2m. Structural differentiation of these two isomers required MS/MS analysis of these ions derived from the intact peptide (as illustrated schematically in Figure 2A). The primary MS/MS fragmentation observed for pSer- and pThr-containing peptides is loss of neutral phosphoric acid (H3PO4) or HPO3, with a much reduced fragmentation of the amide bond peptide backbone to generate “b” and “y” sequence-indicating ions.39 The abundance of these ions depends on the charge-state of the precursor ion, the structure of the peptide and the nature of the collision-induced dissociation (CID).40 For the phosphopeptide 3m, which contains a cationic, N(π),N(τ)- bis-alky-His residue and a free phosphate, the predominant CID-mediated neutral loss should be that of H3PO4 (98 da), resulting in a product ion at m/z 853.5182. For the corresponding phosphopeptide isomer 2m, which contains a N(π)-mono-alkylated His residue and an esterified pThr moiety, the expected neutral loss would be H2PO3(OR), where R = CH2CH2CH(OH)CH2OH, to result in an ion at m/z 765.4658 (Figure 2A). The “b3”and “b4” sequence-indicating ions would also be distinct, due to the difference in the nature of His alkylation.
Figure 2.
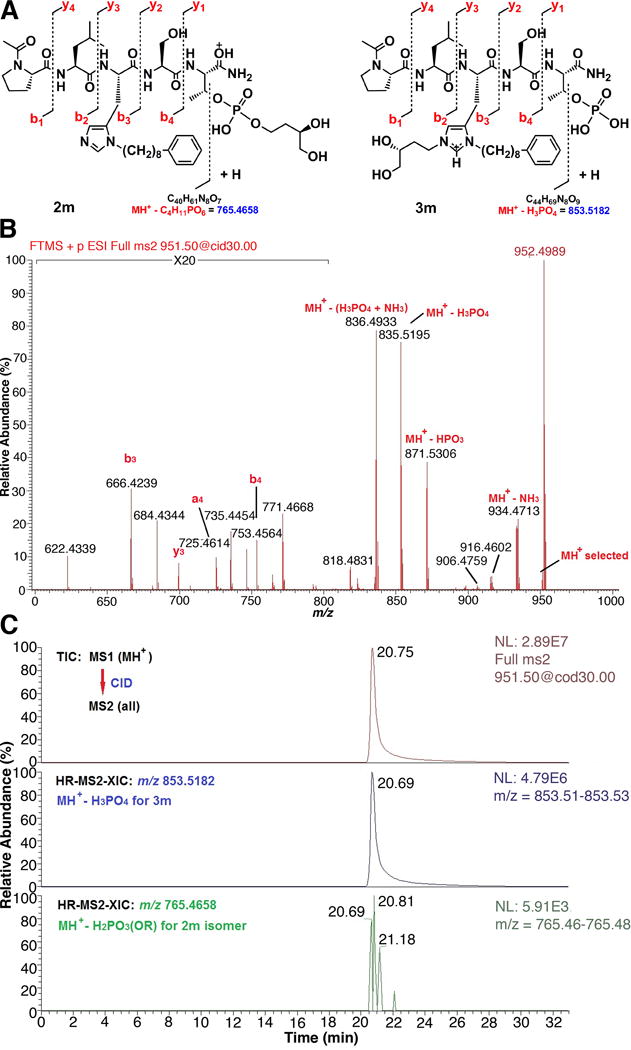
High resolution LC-MS/MS analysis of pThr-pentapeptide 3m. A) Structures and expected CID fragmentation of MH+ (m/z 951.4951) for the isomers 2m and 3m, respectively; B) Partial, high resolution CID MS/MS mass spectrum of the MH+ of 3m. The unequivocal presence of a free phosphate moiety is indicated by the product ions b3, a4, b4, y3, MH+-H3PO4, and MH+-HPO3. A more complete assignment of product ions and masses is available in the Supporting Information. C) LC-MS/MS estimation of the isomeric purity of 3m. Top trace – full total ion chromatogram (TIC) for CID MS/MS of MH+, middle trace – high-resolution (30 mDa window) MS/MS extracted ion chromatogram (HR-MS2-XIC) for MH+-H3PO4 of 3m (absolute peak height = 4.79 × 106 counts), bottom trace – HR-MS2-XIC for MH+-H2PO3(OR) of 2m where R = CH2CH2CH(OH)CH2OH (absolute peak height = 5.91 × 103 counts). Based on this analysis, pThr-pentapeptide 3m is predominantly the free phosphate (> 99%) and contains very little phosphopeptide ester 2m byproduct (approximately 0.1%).
A loss of both HPO3 and H3PO4, indicative of the free phosphate of compound 3m, was observed in both the CID (Figure 2B) and HCD MS/MS spectra derived from MH+. The expected fragment ion resulting from loss of H2PO3(OR), in the case of a mono-esterified pThr (e.g. 2m) was not detected. Furthermore, b3, b4 and y3 ions, which result from amide-bond cleavage and contain the bis-alkyl-His residue, were clearly observed in all MS/MS spectra of 3m. MSn (n = 2 & 3) analysis of the remaining members of the series 3b – 3n, including mono-esterified pThr peptides prepared by alternate synthetic procedures, confirmed the validity of the above observations. Because the isomeric N-alkylated and esterified peptides could not be completely separated by HPLC, a high-resolution MS/MS extracted ion chromatogram method was developed to define the ratio of isomers (Figure 2C). As discussed above, for each phosphopeptide isomer pair, a set of unique MS/MS product ions resulting from loss of a neutral phosphate-containing moiety is preferentially generated from MH+. For the bis-alkyl-His isomer, this is the product ion resulting from loss of H3PO4, e.g. m/z 853.5182 for 3m. For the phosphate ester isomer, the comparable product ion results from loss of the corresponding phosphoric acid ester or, in the case of 2m, m/z 765.4658. Because these are major product ions with expected similar abundances, the relative amounts of each can be estimated by extracting and comparing the absolute ion abundances when they are analyzed under similar conditions, i.e., LC/MS/MS. Certainty that these are the only ions extracted is increased by carrying out this procedure at a high resolution (20 mDa extraction window encompassing the targeted ion) that is matched to the resolution and mass accuracy of the MS/MS analysis (Figure 2C, middle and bottom traces). Here it can be seen that any 2m byproduct present in 3m is minimal since the diagnostic ion abundance is at the level of instrument noise. The peptides prepared by the on-resin Mitsunobu chemistry were primarily the N(π),N(τ)- bis-alky-His peptides (3). Only in the cases of 3d and 3f were more than 2% of the mono-esterified pThr isomers (2d and 2f) produced as byproducts and these were estimated at 15% and 17%, respectively.
Evaluation in Plk1 PBD ELISA Inhibition Assays
When we examined the peptide products (3b – 3n) in ELISA-based Plk1 PBD inhibition assays, we found that, in contrast to the loss of potent binding affinity observed with mono-anionic esters of 1,30 members of the currents series retained good PBD-binding affinity, which in some cases equaled or exceeded that of the parent peptide 2a (Table 1 and Figure S3).
Table 1.
Results of ELISA Plk1 PBD Competition Assaysa
| Entry | Compd. | IC50 (μM) |
|---|---|---|
| 1 | PLHST | NA |
| 2 | 1 | 9 |
| 3 | 2a | 0.003 |
| 4 | 2a†b | 0.003 |
| 5 | 3b | 0.005 |
| 6 | 3c | 0.001 |
| 7 | 3c(S/A) | 0.8 |
| 8 | 3d | 0.001 |
| 9 | 3e | 0.001 |
| 10 | 3f | 0.004 |
| 11 | 3g | 0.005 |
| 12 | 3h | 0.006 |
| 13 | 3i | 0.04 |
| 14 | 3j | 0.003 |
| 15 | 3k | 0.002 |
| 16 | 3l | 0.001 |
| 17 | 3m | 0.001 |
| 18 | 3m(S/A) | 0.15 |
| 19 | 3n | 0.015 |
Determined by ELISA competition assays as described. Binding curves are shown in Figures S3.
Given the similarities in inhibitory potencies among the series 3 in ELISA competition assays, we considered peptides showing marginally better IC50 values as potential candidates for cell-based studies (3c, 3d, 3e, 3l and 3m; IC50 = 0.001 μM, Table 1). Of these, 3e was omitted due to the presence of an anionic carboxylic acid group. Preliminary cellular evaluation of the remaining members of the series showed that the hydroxyl-containing analogs (3c, 3l and 3m) exhibited significantly higher potencies than the non-hydroxyl-containing analog (3d). This may potentially indicate a beneficial role for hydroxyl functionality in these assays. Interestingly, 3l and 3m showed identical potencies in spite of the diastereomeric hydroxyls in their N(τ)-alkyl functionalities. Based on the above, we selected peptides 3c and 3m for more in-depth evaluation.
To rule out promiscuous modes of binding,41, 42 the Ser to Ala mutants (S4A) of 3c and 3m were prepared, since the Ser residue provides key recognition features for canonical Plk1 PBD binding and replacement with an Ala residue is known to significantly reduce binding affinity.6 Consistent with non-promiscuous binding, our current study showed that the Ser to Ala substituted peptides exhibited more than two orders-of-magnitude reduced inhibitory potencies [3c(S4A) IC50 = 0.8 μM and 3m(S4A) IC50 = 0.15 μM] (Table 1 and Figure S3).
Plk1 PBD Co-crystal Studies
To unambiguously establish its mode of binding, we solved the X-ray co-crystal structure of 3m bound to the Plk1 PBD (Figure 3 and Figure S4). The Plk1 PBD interactions of the parent peptide 2a have been reported (PDB: 3RQ7).30. In our current work, we observed that both the protein backbone and the peptide ligand in the PBD•3m complex were essentially superimposable with the PBD•2a structure (Figure 3). Importantly, interactions of the His N(π)-(CH2)8Phe groups within a hydrophobic region identified in our earlier report, were essentially superimposable. Extra electron density in the region of the His N(τ)- position was consistent with a substituent at this position (Figure S4). The absence of more extended resolved density could indicate a degree of conformational flexibility for the substituent.
Figure 3.
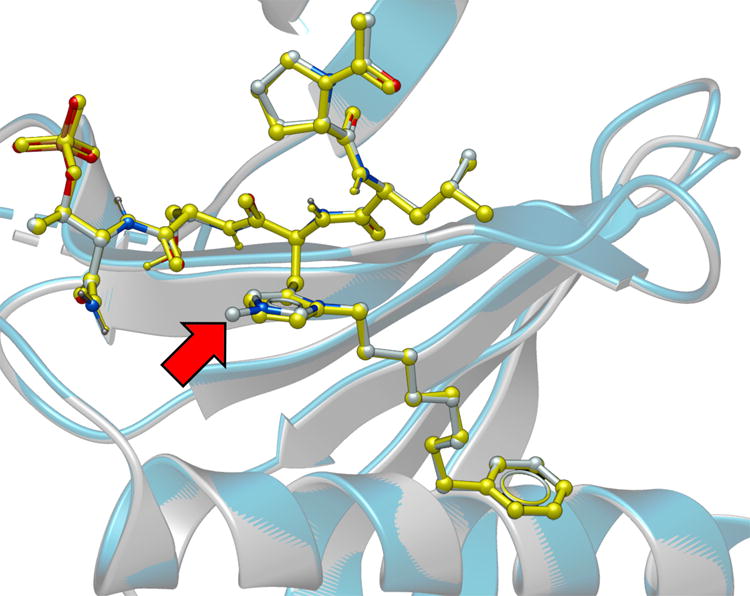
Plk1 PBD co-crystal structure of 3m superimposed on the co-crystal structure of PBD-bound 2a (PDB 3RQ7), highlighting the high degree of correspondence. The 3m structure is rendered with the protein backbone shown as grey ribbon and the ligand colored according to element (carbon = grey, nitrogen = blue and oxygen = red). The 2a structure is rendered with the protein backbone shown as blue ribbon and the ligand colored yellow. The red arrow indicates the location of the resolved portion of the N(τ) substituent. Most of the alkyldiol substituent is unresolved.
Determination of Plk1 PBD-Binding Specificities using Fluorescence Polarization
In order to evaluate Plk1 specificity, we introduced fluorescein isothiocyanate (FITC) groups into 2a, 3c and 3m via N-terminal PEG tethers (designated as 2a*, 3c* and 3m*) (Figure S5) and determined binding constants to PBDs from Plk1, 2 and 3 (designated as PBD1, PBD2 and PBD3, respectively) using fluorescence polarization techniques (Figure S6). We had previously shown for 2a, that such modification had little effect on binding affinity.30 In the current work, we found that 2a*, 3c* and 3m* bound to PBD1 with affinity constants (Kd values) of 12 nM, 2 nM and 3 nM, respectively (Table 2). Selectivities of 2a*, 3c* and 3m* for PBD1 relative to PBD2 were 33-fold (400 nM), 21-fold (43 nM) and 8-fold (24 nM), respectively, while the corresponding selectivities for PBD1 relative to PBD3 were 25-fold (306 nM), 120-fold (240 nM) and 20-fold (62 nM), respectively. In the absence of crystal structures for PBD2 and PBD3, the basis for selective binding to PBD1 relative to PBD2 and PBD3 are not obvious. However, alignment of the PBD sequences6, 9 provides insights into potential reasons for ligand selectivity. These include interactions of the N-terminal Pro-Leu residues with a region defined by Arg516 and Phe535 in PBD1 but which corresponds to Lys and Tyr residues, respectively, in PBD2 and PBD3.43
Table 2.
Determination of PBD Selectivity
| Kd (nM)a | ||||
|---|---|---|---|---|
| Entry | Compd.b | PBD1 | PBD2c | PBD3c |
| 1 | 2a* | 12 | 400 [33×] | 306 [25×] |
| 2 | 3c* | 2 | 43 [21] | 240 [120] |
| 3 | 3m* | 3 | 24 [8] | 62 [20] |
Determined by fluorscence polarization assays using FITC-labeled ligands.
Designation with an “*” indicates the presence of an N-terminal PEG-FITC group as described in Figure S5.
Values in brackets refer to the fold-change in binding affinity relative to PBD1. Binding curves are shown in Figure S6.
Evaluation of Bis-Alkyl-His Analogs in Cell-Based Studies
Next, we examined the effects of selected phosphopeptides in cultured HeLa cells. In these experiments, we employed 2a† as a reference. This is a previously reported variant of 2a, in which the pThr residue is replaced by a (2S,3R)-2-amino-3-methyl-4-phosphonobutyric amide residue44 and an N-terminal PEG group has been appended to increase water solubility.30 This analog represents the most potent peptide we have yet reported in cell-based assays.30 In our current work 2a† was able to induce partial mitotic block in asynchronously growing HeLa cells in a dose-dependent fashion, with the percentage of mitotically arrested cells at 24 h after treatment being more than doubled the percentage at 12 h after treatment. A maximal mitotic index of approximately 35% was observed after exposure for 24 h at the highest concentration used (600 μM) (Figure 4). Under these conditions, treatment with 3c achieved significant levels of mitotic block, with 200 μM and 400 μM concentrations resulting in mitotic indices of approximately 40% and 60%, respectively, after 24 h. Treatment with 3m resulted in similar, although slightly reduced, effects. In both cases, the corresponding S/A mutants, 3c(S4A) and 3m(S4A), respectively, gave significantly attenuated effects, supporting a Plk PBD-dependent mechanism of action (Figure 4).
Figure 4.
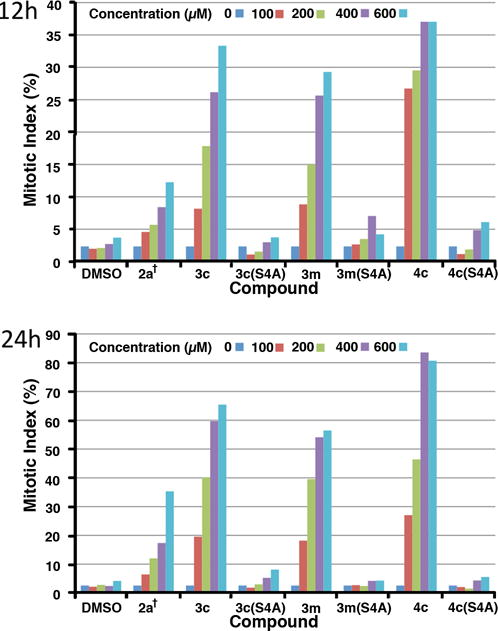
Percent of cultured HeLa cells in mitosis after treatment with the indicated concentrations of peptides for 12 h (upper) and 24 h (lower).
We have previously shown that treatment of cells with 2a† decreases cellular proliferation rate due to PBD inhibition-induced apoptotic cell death.30 In a similar fashion, the mitotic block observed in our current work is reflected in even stronger antiproliferative effects, which were measured by plotting relative cell numbers for each dose at 12, 24 and 48 h (Figure 5). An IC50 value of 320 μM for 2a† is consistent with the previously reported IC50 value of 380 μM.30 Approximate IC50 values of 85 μM and 90 μM obtained for 3c and 3m, respectively, represent marked improvements relative to 2a†, while the corresponding S/A mutants, 3c(S4A) and 3m(S4A), showed significantly reduced effects.
Figure 5.
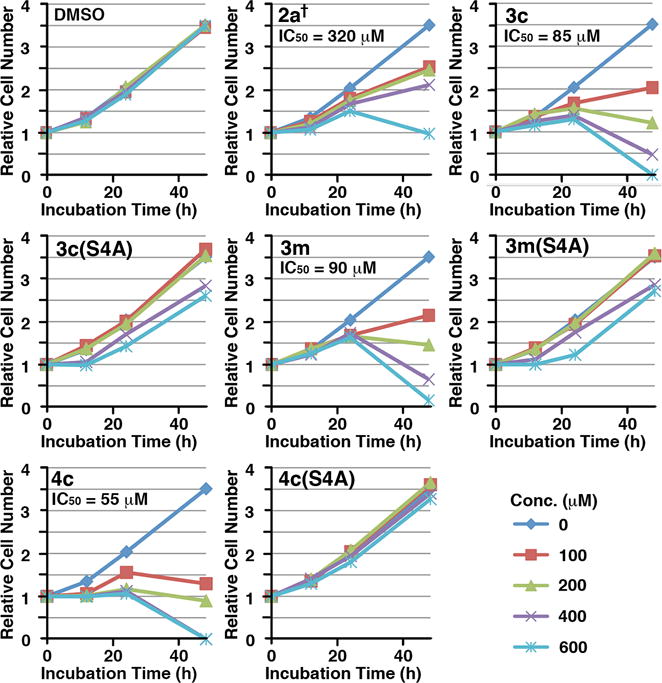
Dose-dependent inhibition for cell proliferation by the indicated peptides. Asynchronously growing HeLa cells were treated with various concentrations of the indicated compounds. The resulting cells were harvested at the 0, 12, 24 and 48 h time points and then quantified. The relative cell numbers for the indicated time points were determined in comparison to those of the respective 0 h time point sample. The IC50 values of cell proliferation inhibition for indicated compounds were calculated using the data points from the 48 h samples.
In ELISA PBD inhibition assays of the current study, 2a, 2a†, 3c and 3m all displayed similar IC50 values (Table 1). The lower IC50 value of 2a† relative to our previously reported value for this peptide (3 nM versus 30 nM)30 stems from variability in the ELISA assay (the ELISA O.D. 450 values vary depending on the length of developing time). The enhanced cellular activities shown by 3c and 3m relative to 2a† could be attributable to a number of factors. While 3c and 3m utilize phosphoryl esters and contain N-terminal acetyl groups, 2a† employs the phosphonic acid-based pThr mimetic, Pmab, and has an N-terminal PEG group. A component of the improved cellular efficacy of 3c and 3m may arise from improved cell membrane transit, potentially achieved as a consequence of neutralizing one phosphoryl anionic charge through intramolecular charge masking by the His imidazolium cation.36 We reasoned that neutralization of a second anionic charge could further enhance cellular activity.
Phosphoryl Prodrug Derivatization
A general strategy for increasing the bioavailability of phosphates is to block their acidic functionality with “prodrug” groups that can be removed enzymatically once the agent is within the cell.45, 46 The pivaloyloxymethyl (POM) moiety is a form of phosphoryl prodrug protection that has been widely employed in nucleotides.46 The POM group is removed enzymatically by esterase-mediated cleavage of the pivaloyloxymethyl ester linkage. This yields pivalic acid and an unstable hydroxymethyl ester group, which decomposes to formaldehyde with liberation of the free phosphoryl hydroxyl group. To our knowledge, with the exception of our recent synthetic methods paper,47 there have been no prior applications of POM phosphoryl protection of pThr or pSer-containing polypeptides within a biological context. Accordingly, we utilized our recently reported reagent, N-Fmoc-Thr[PO(OPOM)OH]-OH,47 along with our previously reported N-Fmoc-His*-OH38 to prepare resin-bound “Ac-Pro-Leu-His*-Ser-Thr[PO(OPOM)OH)]-amide resin. Mitsunobu alkylation followed by acid-catalyzed resin cleavage provided 4c as a variant of 3c bearing mono-POM protection of the phosphoryl group (see Supporting Information Figure S7).
Esterase Stability Assays
A successful prodrug strategy requires that the chosen derivative exhibit sufficient stability to allow delivery to the desired site of action, yet once there, that enzymatic activation occur within a biologically relevant time frame. Stability of esterase-labile prodrugs is often approximated using in vitro assays that employ readily available pig liver esterase (PLE). Since it was also important to examine the stability of the POM group within the more relevant contexts of cell culture media and intracellular milieu, we performed these experiments as well. We found that conversion of 4c to 3c occurred with a half-life of approximately 240 min in control PLE (Figure S9A). In culture media the half-life of 3c at a concentration of 1 μM was approximately 400 min (Figure S9B). In addition, at a more relevant concentration of 200 μM, conversion of 4c to 3c in culture media did not occur to any appreciable extent. In contrast, incubating 1 μM concentration of 4c with cell lysates showed that 50% conversion to 2c occurred in approximately 90 min (Figure S9C). These data indicate that in cell culture studies, 4c should persist in relatively unchanged form in the extracellular media, yet be rapidly converted to the active form 3c once inside the cell. Interestingly, since the in vitro ELISA–based PBD-inhibition assay utilizes cell lysates, conversion of 4c to 3c could occur during the course of a typical assay. Indeed, the inhibitory potency of 4c was found to increase from 0.02 μM to 0.002 μM by a 1.5 h pre-incubation prior to conducting the standard assay (Table 3 and Figure S8).
Table 3.
Pre-incubation Dependent Plk1 PBD Binding
| Entry | Compd. | IC50 (μM) | |
|---|---|---|---|
| Withouta | Withb | ||
| 1 | PLHSpT (1) | 16 | 2 |
| 2 | 2a | 0.002 | 0.001 |
| 3 | 3c | 0.002 | 0.001 |
| 6 | 3c(S4A) | 1.3 | 0.2 |
| 4 | 3m | 0.002 | 0.001 |
| 7 | 3m(S4A) | 0.3 | 0.06 |
| 5 | 4c | 0.02 | 0.002 |
| 8 | 4c(S4A) | 28 | 1 |
Cell lystates were incubated (1.5 h) without peptide prior to performing binding studies (1 h incubations)
Cell lysates were preincubated with peptide (1.5 h) prior to performing binding studies (1 h incubations). Binding curves are shown in Figure S8.
Evaluation of the Effects of Prodrug Protection in Cell-Based Studies
The effect of POM-protection in 4c was examined in asynchronously growing HeLa cells, as described above. These studies demonstrated that relative to parent 3c, peptide 4c showed an improved ability to induce mitotic block, reaching a maximum mitotic index of approximately 80% at 24 h at a concentration of 400 μM, as compared to approximately 60% for 3c under the same conditions and roughly 18% for 2a† (Figure 5). The potency of the S/A mutant 4c(S4A) was very similar to vehicle control, strongly supporting the PBD-dependence of mitotic block by 4c (Figure 5). The anti-proliferative potency of 4c (IC50 = 55 μM) was also improved relative to 3c (IC50 = 85 μM) (Figure 5).
MATERIALS AND METHODS
On-resin Synthesis of Peptides Containing N(π),N(τ)-alkyl His Residues (3b – 3m)
Fmoc-protected amino acids were purchased from Novabiochem. Phosphothreonine was incorporated using Fmoc-Thr[PO(OBzl)OH]-OH and Fmoc-His[N(π)-(CH2)8Ph)]-OH was prepared according to literature procedures.38 Peptides were synthesized on NovaSyn®TGR resin (Novabiochem, cat. no. 01-64-0060) or NovaSyn® TG Siber resin (Novabiochem, cat. no. 01-64-0092) using standard Fmoc-based solid-phase protocols in N-methyl-2-pyrrolidone (NMP). 1-O-Benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate (HBTU) (5.0 eq.), hydroxybenzotriazole (HOBT) (5.0 eq.) and N,N-diisopropylethylamine (DIPEA) (10.0 eq.) were used as coupling reagents. Amino terminal acetylation was achieved using 1-acetylimidazole (10 eq.) in DMF. The finished resins SI-1 or SI-2 (0.1 mmol) (Figure S1) were swelled in CH2Cl2 (15 min) and then treated with triphenylphosphine (262 mg, 1.0 mmol), diethyl azidodicarboxylate (DEAD) (0.46 mL, 40% solution in toluene, 1.0 mol) and alcohols b – n (Figure S1) (1.0 mmol) in dry CH2Cl2 at room temperature (4 h), then washed (CH2Cl2) and dried under vacuum (over night). Resins were then cleaved by treatment with trifluoroacetic acid (TFA): triispropylsilane (TIS) : H2O (95 : 2.5 : 2.5) (5 mL, 4 h). The resin was removed by filtrations and the filtrate was concentrated under vacuum, the residue was dissolved in 50% aqueous acetonitrile (CH3CN) (5 mL) and purified by reverse phase preparative HPLC using a Phenomenex C18 column (21 mm dia × 250 mm, cat. no: 00G-4436-P0) with a linear gradient from 20% aqueous CH3CN (0.1% TFA) to 90% CH3CN (0.1% TFA acid) over 30 min at a flow rate of 10.0 mL/minute. Lyophilization gave the products 3b – 3n as white powders (Figure S1). Peptide 2a was prepared as previously reported.38 Analytical data are provided in Table S1. Structure confirmation by MS/MS is described in the Supporting Information.
Synthesis of Fluorescein Isothiocyante (FITC)-Labeled Peptides (3*c and 3*m) for Plk Specificity Determination
The resin-bound peptide SI-3 (Figure S5) was synthesized by standard Fmoc solid-phase peptide-protocols as outlined above, and the amino-terminus was acylated with Fmoc-N-amido-dPEG®8-NHS ester (3.0 eq.) (Quanta Biodesign, Cat# 10995) by reacting with HBTU (5.0 eq.), HOBt (5.0 eq.) and DIPEA (10.0 eq.) at room temperature (4 h). On-resin Mitsunobu His-N(τ) alkylation was then performed as indicated above for the synthesis of peptides 3 (Figure S1) to form the resin-bound peptides SI-4 and SI-5 (Figure S5). Following Fmoc deprotection, the resins were treated with the fluorescein isothiocyanate (FITC) (open-form isomer I) (5.0 eq.) and DIPEA (10.0 eq.) in NMP (12 h). [Note: Performing installation of the FITC group prior to Mitsunobu esterification yielded complex products following resin cleavage.] Finished resins were washed sequentially with DMF, MeOH, CH2Cl2 and ether, dried under vacuum (1 h) and subjected to TFA-mediated cleavage as described above for the synthesis of peptides 3 and purified by HPLC to provide the desired peptides 3c* and 3m* as yellow powders. Analytical data are provided in Figure S5. Peptide 2a* was synthesized as previously described.30
Synthesis of the POM Prodrug-Protected Peptides 4c and 4c(S4A)
Synthesis of peptides 4c and 4c(S4A), which represent peptides 3c and 3c(S4A) having their one phosphoryl hydroxyl derivatized as a pivaloyloxymethyl (POM) ester, was achieved as outlined in Figure S7. Solid-phase peptide synthesis was conducted as indicated above for the synthesis of resin-bound peptides SI-1 and SI-2 (Figure S1) except that Fmoc-pThr[PO(OPOM)(OH)]-OH (SI-6)47 was used in place of Fmoc-Thr(PO(OBzl)OH)-OH to yield the resin-bound peptides SI-7 and SI-8 (Figure S7). On-resin Mitsunobu His-N(τ) alkylation was then performed as indicated above for the synthesis of peptides 3. The resulting resins were cleaved using TFA as indicated above and purified by HPLC to provide peptides 4c and 4c(S4A) as white powders. Analytical data are provided in Table S1.
Mass Spectrometry Analysis
Low resolution positive ion, electrospray ionization (ESI) mass spectra were obtained by LC/MS analysis on an Agilent LC/MSD single quadrupole system that was also equipped with an in-line diode-array UV detector. A narrow-bore (100 × 2.1 mm), small-particle (3.5-μm), Zorbax Rapid-Resolution reversed-phase C18 column coupled with a C18 guard column (12.5 × 2.1 mm) was eluted with a 5–90% gradient of CH3OH/H2O containing 0.1% CH3COOH at a flow rate of 300 μL/min to separate the components of crude reaction isolates, enzymatic and cellular incubation mixtures, and purified final products. In the case of the latter, both the total-ion chromatogram (TIC) and the UV-chromatogram at 220 nm were used to assess compound purity. Enzymatic and hydrolytic stability was monitored and quantified by generating the appropriate compound-indicating extracted ion chromatograms using the LC/MSD ChemStation software (version B.04.02 SP1).
High resolution LC/MS and LC/MS/MS analyses were conducted on a Thermo-Fisher LTQ-XL Orbitrap hybrid mass spectrometer system operated under Xcalibur (version 2.1.0 SP1) control for data acquisition and qualitative analysis. A narrow-bore (150 × 2 mm), small particle (3.0-μm) Imtakt, Cadenza CD-C18 reversed-phase column coupled with a guard column (5 × 2 mm) of the same stationary phase was eluted with a two-stage linear gradient of CH3CN/H2O containing 0.1% HCOOH at 250 μL/min. An initial shallow gradient of 2–50% CH3CN/H2O over 20 min was followed by a steeper gradient of 50–90% CH3CN/H2O in 10 min with a 3-min hold at the final conditions before LC reset and equilibration. Primary mass spectra (MS1) for accurate mass measurement of a molecular species ([M+H]+, [M+Na]+ or [M+2H]+2) were obtained at a resolution of 30,0000 (FWHM), while MS/MS (MS2) studies employing collision-induced dissociation (CID) or higher energy CID (HCD) were conducted at a resolution of 15,000. For these MS2 studies, [M+H]+ or [M+2H]+2 precursor ions were selected using a mass window that encompassed the isotopic profile of the ion of interest. For the compounds in question, CID and HCD were carried out at an optimized energy setting of 30 and 27.5, respectively, in order to give the highest intensity product ion mass spectrum. Product ion spectra containing multiply charged ions (i.e., those from [M+2H]+2 precursors) were deconvoluted to a single charge state using the Xtract software module of Xcalibur in order to facilitate structural assignment and analysis. MS2 high-resolution extracted ion chromatograms (HR-XICs) were generated post-analysis using a 20 or 30 mDa mass window centered on the observed accurate mass of the appropriate diagnostic ions. These HR-XICs were then used to locate and measure the relative amounts of isomeric dialkylhistidyl phosphopeptide 3 and esterified histidyl phosphopeptide 2 present in each sample. A summary of the high resolution MS1 and MS2 data for each compound can be found beginning in the Supporting Information on p S-6.
ELISA-based PBD-binding Inhibition Assays
A biotinylated p-T78 peptide was first diluted with 1X coating solution (KPL Inc., Gaithersburg, MD) to the final concentration of 0.3 μM, and then 100 μL of the resulting solution was immobilized onto a 96-well streptavidin-coated plate (Nalgene Nunc, Rochester, NY). The wells were washed once with PBS plus 0.05% Tween20 (PBST), and incubated with 200 μL of PBS plus 1% BSA (blocking buffer) for 1 h to prevent non-specific binding. Mitotic 293A lysates expressing HA-EGFP-Plk1 were prepared in TBSN buffer (~ 60 μg total lysates in 100 μL buffer), mixed with the indicated amount of the competitors (p-T78 peptide and its derivative compounds), provided immediately onto the biotinylated peptide-coated ELISA wells, and then incubated with constant rocking for 1 h at 25 °C. Following the incubation, ELISA plates were washed 4 times with PBST. To detect bound HA-EGFP-Plk1, the plates were probed for 2 h with 100 μL/well of anti-HA antibody at a concentration of 0.5 μg/mL in blocking buffer and then washed 5 times. The plates were further probed for 1 h with 100 μL/well of HRP-conjugated secondary antibody (GE Healthcare, Piscataway, NJ) at a 1:1,000 dilution in blocking buffer. The plates were washed 5 times with PBST and incubated with 100 μL/well of 3,3′,5,5′-tetramethylbenzidine (TMB) solution (Sigma, St. Louis, MO) as a substrate until a desired absorbance was reached. The reactions were stopped by the addition of 100 μL/well of stop solution (Cell Signaling Technology, Danvers, MA). The optical density (O.D.) was measured at 450 nm by using an ELISA plate reader (Molecular Devices, Sunnyvale, CA). Data are shown in Table 1 and Figure S3.
PBD Fluorescence Polarization Binding Assays for Plk1, Plk2 and Plk3
5-Carboxyfluorescein-labeled peptides 2a*, 3c* and 3m* were incubated, at a final concentration of 2 nM, with various concentrations of bacterially-expressed purified PBDs of Plk1, Plk2 and Plk3 in a binding buffer containing 10 mM Tris (pH 8.0), 1 mM EDTA, 50 mM NaCl, and 0.01% Nonidet P-40. Fluorescence polarization was analyzed 10 min after mixing of all components in a 384-well format using a Molecular Devices SpectraMax Paradigm Multi-Mode Microplate Detection Platform. All experiments were performed in triplicate. Obtained data were plotted using GraphPad Prism software version 6. IC50 values are provided in Table 2 and binding curves are shown in Figure S6.
Stability of Prodrug Protected Peptide 4c to Enzymatic Deprotection Pig Liver Esterase Hydrolysis of Peptide 4c
Following literature procedures,48 0.05M potassium phosphate buffer (pH 7.4) was placed in centrifuge tube and a solution of peptide 4c in MeOH was added to the buffer to achieve a concentration of 200 μM of 4c with MeOH being less than 1%. To a 1 mL aliquot of the above solution was added pig liver esterase (57.6 units) and the reaction mixture was incubated at 37 °C with gentle agitation. At various time points, aliquots of the reaction mixture (50 μL) were transferred to an Eppendorf tube containing MeCN (50 μL). Following filtration, the hydrolysis product was monitored by LC-MS. Data are shown in Figure S9.
Hydrolysis of Peptide 4c in Cell Culture Media
To an aliquot of culture medium consisting of Dulbecco’s modified eagle medium (DMEM) [high glucose, no glutamine, (cat. 11960, Life Technologies)]49 (500 mL); fetal bovine serum (FBS)(cat. SH30396, Thermo Scientific) (50 mL); Antimycotic (cat. 15240-112, Life Technologies), (5 mL), Glutamax (cat. 35050-079, Life Techonologies) (5 mL) and Hepes buffer (cat. 15630-130, Life Technologies), (12.5 mL) was added a solution of peptide 4c in MeOH to achieve a concentration of 1 μM of 4c with MeOH being less than 1%. The reaction mixture was then incubated at 37 °C with gentle shaking. At various time points, aliquots of the reaction mixture (50 μL) were transferred to an Eppendorf tube containing MeCN (50 μL). Following filtration, the hydrolysis product was monitored by LC-MS. Data are shown in Figure S9.
Hydrolysis of Peptide 4c by Total Cell Lysates
Mitotic 293A cells expressing HA-EGFP-Plk1 were lysed at 4 °C using lysis buffer consisting of 50 mM Tris-–HCl, pH 8.0; 120 mM NaCl; 5% nonyl phenoxypolyethoxylethanol (Tergitol-type NP-40); 5 mM ethylene glycol tetraacetic acid (EGTA); 1.5 mM ethylenediaminetetraacetic acid (EDTA); 4-nitrophenyl phosphate di(tris) salt [PNPP (cat. N3254, Aldrich)] (0.1 g /10 mL) and protease inhibitor [complete EDTA-free (cat. 11873580001, Roche]. The lysate was centrifuged (13,000 rpm, 15 min, 4 °C) and the resulting cell lysate supernatant was placed in 96-well plates. To the wells was added a solution of peptide 4c in DMSO so that the concentration of the peptide was 1 μM with DMSO being at a concentration of less than 1% and a total volume of 125 μL. The reaction mixtures were incubated at 37 °C with gentle shaking. At different time points, MeCN (125 μL) was added to each well to quench the reactions. Following filtration, the hydrolysis products were monitored by LC-MS. Data are shown in Figure S9.
Cell Culture, Analysis of Cellular Proliferation, Aberrant Mitotic Population and Indirect Immunofluorescence Microscopy
HeLa cervical carcinoma CCL2 and 293A cells were cultured as recommended by the American Type Culture Collection (Manassas, VA). To prepare mitotic 293A cells expressing HA-EGFP-Plk1, cells were infected with adenovirus expressing HA-EGFP-Plk1 and arrested with 200 ng/mL of nocodazole for 16 h. To analyze the effect of the indicated compounds in cultured cells, logarithmically growing HeLa cells were treated with various concentrations of the indicated compounds for 12, 24 and 48 h. Images of HeLa cells were acquired using a Zeiss Axiovert 100 M microscope (Carl Zeiss MicroImaging, Inc., Thornwood, NY). Effects of treatment on mitotic block and cellular proliferation are shown graphically in Figures 5 and 6.
X-Ray Crystallography
Protein Purification and Crystallization
Plk1 PBD protein (residues 371–603) was purified as previously described.28 Crystals were grown using the hanging drop vapor diffusion method. PBD protein at 12 mg/mL in 10 mM Tris pH 8, 0.5 M NaCl, 10 mM DTT, 1% (v/v) DMSO and 1 mM of peptide 3m was mixed with an equal volume of reservoir solution consisting of 13% (w/v) PEG 3350, 0.1 M HEPES pH 7.5 and 100 mM NaCl. Crystals began appearing overnight and reached maximum size over several days. Crystals grew in clusters that were manually broken up to obtain sufficiently single crystals suitable for data collection.
Data Collection and Structure Determination and Refinement
Crystals were cryo-protected in 26% (w/v) PEG 3350, 0.1 M HEPES pH 7.5, 150 mM NaCl, 6% (v/v) glycerol, 1 mM 3m, 1 % (v/v) DMSO and 10 mM DTT, and data were collected at 100 K on a Rigaku Raxis-IV image plate detector with a Rigaku RU-300 home X-ray source. The data were processed with the HKL50 and CCP451 software suites. The structure was solved by molecular replacement using AMoRe52 using chain A of structure 3FVH28 (RCSB accession code) as a search model, and refined using PHENIX53 with manual fitting in XtalView.54 Refinement statistics, a sigmaA weighted 2Fo – Fc electron density map and a SA omit Fo – Fc electron map contoured at 3σ and 1.45 Å are provided in Figure S4.
CONCLUSIONS
In spite of their potential therapeutic applications, inhibitors of phospho-dependent protein-protein interactions present particular challenges for the development of cell permeable binding antagonists, due to the critical roles traditionally played by the di-anionic phosphoryl moiety in ligand affinity and the impediments that these charged groups present to cell membrane transit. Our current work is significant in that it has resulted in the discovery of unanticipated N(π),N(τ)- bis-alky-His-containing peptides having low-nanomolar inhibitory potencies in in vitroPBD-binding assays. Importantly, the efficacies in cellular assays of some of these peptides are improved relative to the dianionic peptide 2a†. A key structural aspect of the improved cellular potencies may be related to reduction of overall net peptide charge through intramolecular charge masking of one phosphoryl anionic charge by the His imidazolium cations. We have also demonstrated that further improvement in cellular profiles can be achieved by utilizing POM phosphoryl protection. The strategy described herein represents a notable advance in the development of PBD-binding inhibitors that may also find use in the design and development of phosphate-containing inhibitors in other contexts.
Supplementary Material
Acknowledgments
This work was supported in part by the Intramural Research Program of the NIH, Center for Cancer Research, NCI-Frederick and the National Cancer Institute, National Institutes of Health (W.-J. Q., J.-E. P., C. C. L., J. A. K., K. S. L. and T. R. B.), An NCI Director’s Innovation Career Development Award (W.-J. Q.) and National Institutes of Health grants ES015339 and GM104047 (M. B. Y.). This work is based upon research conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines, which are supported by a grant from the National Institute of General Medical Sciences (P41 GM103403) from the National Institutes of Health. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Appreciation is expressed to Wei Dai, New York University School of Medicine, NY for reagents.
Footnotes
This is the peer reviewed version of the following article: Biopolymers (Pept. Sci.) 102, 444 – 455, 2014, which has been published in final form at DOI 10.1002/bip.22569. This article may be used for non-commercial purposes in accordance with Wiley Terms and Conditions for Self-Archiving.
SUPPORTING INFORMATION
Supplemental Information includes synthetic schemes and physical characterization of synthetic peptides, MS-MS data, binding curves, prodrug enzyme hydrolysis curves and X-ray crystallographic data. This supplemental data can be found with this article online at http://dx.doi.org/
The authors have no competing financial interests.
References
- 1.Barr FA, Sillje HHW, Nigg EA. Nat Rev Mol Cell Biol. 2004;5:429–441. doi: 10.1038/nrm1401. [DOI] [PubMed] [Google Scholar]
- 2.Dai W. Oncogene. 2005;24:214–216. doi: 10.1038/sj.onc.1208270. [DOI] [PubMed] [Google Scholar]
- 3.Lowery DM, Lim D, Yaffe MB. Oncogene. 2005;24:248–259. doi: 10.1038/sj.onc.1208280. [DOI] [PubMed] [Google Scholar]
- 4.van de Weerdt BCM, Medema RH. Cell Cycle. 2006;5:853–864. doi: 10.4161/cc.5.8.2692. [DOI] [PubMed] [Google Scholar]
- 5.Archambault V, Glover DM. Nat Rev Mol Cell Biol. 2009;10:265–275. doi: 10.1038/nrm2653. [DOI] [PubMed] [Google Scholar]
- 6.Elia AE, Rellos P, Haire LF, Chao JW, Ivins FJ, Hoepker K, Mohammad D, Cantley LC, Smerdon SJ, Yaffe MB. Cell. 2003;115:83–95. doi: 10.1016/s0092-8674(03)00725-6. [DOI] [PubMed] [Google Scholar]
- 7.Elia AEH, Cantley LC, Yaffe MB. Science. 2003;299:1228–1231. doi: 10.1126/science.1079079. [DOI] [PubMed] [Google Scholar]
- 8.Cheng K-Y, Lowe ED, Sinclair J, Nigg EA, Johnson LN. EMBO J. 2003;22:5757–5768. doi: 10.1093/emboj/cdg558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park J-E, Soung N-K, Johmura Y, Kang YH, Liao C, Lee KH, Park CH, Nicklaus MC, Lee KS. Cell Mol Life Sci. 2010;67:1957–1970. doi: 10.1007/s00018-010-0279-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee KS, Park J-E, Kang YH, Kim T-S, Bang JK. Mol Cells. 2014;37:286–294. doi: 10.14348/molcells.2014.0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goh KC, Wang H, Yu N, Zhou Y, Zheng Y, Lim Z, Sangthongpitag K, Fang L, Du M, Wang X, Jefferson AB, Rose J, Shamoon B, Reinhard C, Carte B, Entzeroth M, Ni B, Taylor ML, Stuenkel W. Drug Dev Res. 2004;62:349–361. [Google Scholar]
- 12.McInnes C, Mezna M, Fischer PM. Curr Top Med Chem. 2005;5:181–197. doi: 10.2174/1568026053507660. [DOI] [PubMed] [Google Scholar]
- 13.Gumireddy K, Reddy MVR, Cosenza SC, Nathan RB, Baker SJ, Papathi N, Jiang J, Holland J, Reddy EP. Cancer Cell. 2005;7:275–286. doi: 10.1016/j.ccr.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 14.Lansing TJ, McConnell RT, Duckett DR, Spehar GM, Knick VB, Hassler DF, Noro N, Furuta M, Emmitte KA, Gilmer TM, Mook RA, Cheung M. Mol Cancer Ther. 2007:450–459. doi: 10.1158/1535-7163.MCT-06-0543. [DOI] [PubMed] [Google Scholar]
- 15.Lenart P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, Rettig WJ, Kraut N, Peters JM. Curr Biol. 2007;17:304–315. doi: 10.1016/j.cub.2006.12.046. [DOI] [PubMed] [Google Scholar]
- 16.Lu LY, Yu X. Cell Div. 2009;4 doi: 10.1186/1747-1028-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reindl W, Yuan J, Kraemer A, Strebhardt K, Berg T. ChemBioChem. 2009;10:1145–1148. doi: 10.1002/cbic.200900059. [DOI] [PubMed] [Google Scholar]
- 18.Strebhardt K. Nat Rev Drug Discov. 2010;9:643–659. doi: 10.1038/nrd3184. [DOI] [PubMed] [Google Scholar]
- 19.McInnes C, Wyatt MD. Drug Discov Today. 2011;16:619–625. doi: 10.1016/j.drudis.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 20.Weiss L, Efferth T. Exp Hematol Oncol. 2012;1:38. doi: 10.1186/2162-3619-1-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burns TF, Fei P, Scata KA, Dicker DT, El-Deiry WS. Mol Cell Biol. 2003;23:5556–5571. doi: 10.1128/MCB.23.16.5556-5571.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie S, Xie B, Lee MY, Dai W. Oncogene. 2005;24:277–286. doi: 10.1038/sj.onc.1208218. [DOI] [PubMed] [Google Scholar]
- 23.Jang Y-J, Lin C-Y, Ma S, Erikson RL. Proc Nat Acad Sci USA. 2002;99:1984–1989. doi: 10.1073/pnas.042689299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee KS, Grenfell TZ, Yarm FR, Erikson RL. Proc Natl Acad Sci U S A. 1998;95:9301–9306. doi: 10.1073/pnas.95.16.9301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seong Y-S, Kamijo K, Lee J-S, Fernandez E, Kuriyama R, Miki T, Lee KS. J Biol Chem. 2002;277:32282–32293. doi: 10.1074/jbc.M202602200. [DOI] [PubMed] [Google Scholar]
- 26.Strebhardt K, Ullrich A. Nat Rev Cancer. 2006;6:321–330. doi: 10.1038/nrc1841. [DOI] [PubMed] [Google Scholar]
- 27.Reindl W, Yuan J, Kraemer A, Strebhardt K, Berg T. Chem Biol. 2008;15:459–466. doi: 10.1016/j.chembiol.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 28.Yun S-M, Moulaei T, Lim D, Bang JK, Park J-E, Shenoy SR, Liu F, Kang YH, Liao C, Soung N-K, Lee S, Yoon D-Y, Lim Y, Lee D-H, Otaka A, Appella E, McMahon JB, Nicklaus MC, Burke TR, Jr, Yaffe MB, Wlodawer A, Lee KS. Nat Struct Mol Biol. 2009;16:876–882. doi: 10.1038/nsmb.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watanabe N, Sekine T, Takagi M, Iwasaki J-i, Imamoto N, Kawasaki H, Osada H. J Biol Chem. 2009;284:2344–2353. doi: 10.1074/jbc.M805308200. [DOI] [PubMed] [Google Scholar]
- 30.Liu F, Park J-E, Qian W-J, Lim D, Graber M, Berg T, Yaffe MB, Lee KS, Burke TR., Jr Nat Chem Biol. 2011;7:595–601. doi: 10.1038/nchembio.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu F, Park J-E, Qian W-J, Lim D, Scharow A, Berg T, Yaffe MB, Lee KS, Burke TR. ACS Chem Biol. 2012;7:805–810. doi: 10.1021/cb200469a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu F, Park J-E, Qian W-J, Lim D, Scharow A, Berg T, Yaffe MB, Lee KS, Burke TR. ChemBioChem. 2012;13:1291–1296. doi: 10.1002/cbic.201200206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qian W-J, Park J-E, Lee KS, Burke TR., Jr Bioorg Med Chem Lett. 2012;22:7306–7308. doi: 10.1016/j.bmcl.2012.10.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sledz P, Stubbs CJ, Lang S, Yang Y-Q, McKenzie GJ, Venkitaraman AR, Hyvoenen M, Abell C. Angew Chem, Int Ed Engl. 2011;50:4003–4006. doi: 10.1002/anie.201008019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sledz P, Lang S, Stubbs CJ, Abell C. Angew Chem Int Ed Engl. 2012;124:7800–7803. doi: 10.1002/anie.201202660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Allentoff AJ, Mandiyan S, Liang H, Yuryev A, Vlattas I, Duelfer T, Sytwu I-I, Wennogle LP. Cell Biochem Biophys. 1999;31:129–140. doi: 10.1007/BF02738168. [DOI] [PubMed] [Google Scholar]
- 37.Qian W-J, Park J-E, Liu F, Lee KS, Burke TR., Jr Med Chem. 2013;21:3996–4003. doi: 10.1016/j.bmc.2012.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qian W, Liu F, Burke TR., Jr J Org Chem. 2011;76:8885–8890. doi: 10.1021/jo201599c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palumbo AM, Tepe JJ, Reid GE. J Proteome Res. 2008;7:771–779. doi: 10.1021/pr0705136. [DOI] [PubMed] [Google Scholar]
- 40.Boersema PJ, Mohammed S, Heck AJR. J Mass Spectrom. 2009;44:861–878. doi: 10.1002/jms.1599. [DOI] [PubMed] [Google Scholar]
- 41.McGovern SL. Compr Med Chem II. 2006;2:737–752. [Google Scholar]
- 42.Coan KED, Maltby DA, Burlingame AL, Shoichet BK. J Med Chem. 2009;52:2067–2075. doi: 10.1021/jm801605r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murugan RN, Ahn M, Lee WC, Kim H-Y, Song JH, Cheong C, Hwang E, Seo J-H, Shin SY, Choi SH, Park J-E, Bang JK. PLoS One. 2013;8:e80043. doi: 10.1371/journal.pone.0080043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu F, Park J-E, Lee KS, Burke TR. Tetrahedron. 2009;65:9673–9679. doi: 10.1016/j.tet.2009.09.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schultz C. Bioorg Med Chem. 2003;11:885–898. doi: 10.1016/s0968-0896(02)00552-7. [DOI] [PubMed] [Google Scholar]
- 46.Hecker SJ, Erion MD. J Med Chem. 2008;51:2328–2345. doi: 10.1021/jm701260b. [DOI] [PubMed] [Google Scholar]
- 47.Qian W-J, Lai CC, Kelley JA, Burke TR. Chem Biodiversity. 2014;11:784–791. doi: 10.1002/cbdv.201300202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Srivastva DN, Farquhar D. Bioorg Chem. 1984;12:118–129. [Google Scholar]
- 49.Dulbecco R, Freeman G. Virology. 1959;8:396–397. doi: 10.1016/0042-6822(59)90043-1. [DOI] [PubMed] [Google Scholar]
- 50.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. Acta Crystallogr D Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 51.The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 52.Navaza J. Acta Crystallogr D Biol Crystallogr. 2001;57:1367–1372. doi: 10.1107/s0907444901012422. [DOI] [PubMed] [Google Scholar]
- 53.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McRee DE. J Struct Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.