

Abstract
Objective
To examine the effect of Ca2+ store depletion on the translocation of vanilloid transient receptor potential (TRPV) 4-C1 heteromeric channels to the plasma membrane.
Methods and Results
Vesicular trafficking is a key mechanism for controlling the surface expression of TRP channels in the plasma membrane, where they perform their function. TRP channels in vivo are often composed of heteromeric subunits. Experiments using total internal fluorescence reflection microscopy and biotin surface labeling show that Ca2+ store depletion enhanced TRPV4-C1 translocation into the plasma membrane in human embryonic kidney 293 cells that were coexpressed with TRPV4 and canonical transient receptor potential 1 (TRPC1). Fluorescent Ca2+ measurement and patch clamp studies demonstrated that Ca2+ store depletion enhanced 4α-PDD–stimulated Ca2+ influx and cation current. The translocation required stromal interacting molecule 1 (STIM1). TRPV4-C1 heteromeric channels were more favorably translocated to the plasma membrane than TRPC1 or TRPV4 homomeric channels. Similar results were obtained in native vascular endothelial cells.
Conclusion
Ca2+ store depletion stimulates the insertion of TRPV4-C1 heteromeric channels into the plasma membrane, resulting in an augmented Ca2+ influx in response to flow in the human embryonic kidney cell overexpression system and native endothelial cells.
Keywords: TRPV4-C1 heteromeric channel, depletion intracellular Ca2+ stores, endothelial cell, flow, translocation
An important means to regulate TRP channel function is to modulate the translocation of channel proteins to the plasma membrane.1 Hormones, growth factors, and agonists for Gq protein–coupled receptors, such as carbachol, can stimulate the translocation of several TRP channels, including TRPC3–6 and TRPV-1 to the plasma membrane.1 Multiple cellular factors, including soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE), Homer, and small G proteins, may participate in the translocation processes.1 Ca2+ store depletion may also stimulate the translocation of TRPC3 and TRPC6 to the plasma membrane.2,3 However, the exact molecular mechanism by which TRP channels insert into the plasma membrane is unknown. There is still a lack of a general model for TRP trafficking.1
TRP channels in vivo are often composed of heteromeric subunits. Heteromeric assembly usually occurs between the members within the same TRP subfamily, such as between TRPC1 and TRPC3.4 However, coassembly could also happen between the subunits from a different TRP subfamily, such as between TRPC1 and polycystic transient receptor potential 2 (TRPP2) (PKD2)5,6 and between TRPV4 and TRPP2.7 These heteromultimeric channels may display properties different from those of homomultimeric channels.8,9 To our knowledge, there is still no report about the vesicular translocation of heteromeric TRP channels. Recent findings indicate that TRPC1 can coassemble with TRPV4 to form heteromeric channels.10 In vascular tissues, the TRPV4-C1 heteromeric channels are the main channels responsible for flow-induced endothelial Ca2+ influx and subsequent vascular relaxation.10 In separate studies, we also found that Ca2+ store depletion potentiates flow-induced Ca2+ influx11 and subsequent vascular relaxation.12 In the present study, we tested the hypothesis that Ca2+ store depletion may enhance the translocation of TRPV4-C1 heteromeric channels to the plasma membrane. Our results show that Ca2+ store depletion triggers the translocation of TRPV4-C1 heteromers to the plasma membrane, resulting in an enhanced Ca2+ influx in response to flow. This scheme provides a mechanistic explanation for the potentiated Ca2+ influx to flow under the condition of Ca2+ store depletion in vascular endothelial cells.
Methods
Cell Culture, Clones, and Transfection
Human embryonic kidney (HEK) 293 cells and human umbilical vein endothelial cells (HUVECs) were cultured using standard protocol. The human TRPC1 gene (NM_003304) and the mouse TRPV4 gene (NM_022017) were cloned into pcDNA6 or pCAGGS vector for expression. Channel pore mutants TRPV4M680D and TRPC1Mut-pore were as described elsewhere.13,14 For total internal fluorescence reflection microscopy (TIRFM) experiments, TRPV4 was tagged with cyan fluorescent protein (CFP) at its C-terminus and TRPC1 was tagged with yellow fluorescent protein (YFP) at its N-terminus. Small interfering RNAs (siRNAs) and scramble control were from Ambion, Austin, Tex. HEK cells were transfected using Lipofectamine 2000. HUVECs were transfected by electroporation using Nucleofector II. Functional studies were performed 2 to 3 days after transfection.
TIRFM and Biotinylation of Cell Surface Proteins and Immunoblots
Briefly, the evanescent field fluorescence (EFF) intensity of single cells or vesicles was measured by TIRFM (Olympus, Tokyo, Japan); samples were excited by a 440-nm laser for enhanced cyan fluorescent protein (ECFP) and a 513-nm laser for enhanced yellow fluorescent protein (EYFP). The EFF values before thapsigargin (TG) (or tetrakis-(2-pyridylmethyl)ethylenediamine [TPEN], ATP, or bradykinin) treatment were normalized to 1. Most results were expressed as maximal change in EFF intensity (MaxΔEFF). A MaxΔEFF value of 1 represents an increase in EFF value by 1-fold (or 100%). Occasionally, raw EFF values were also presented.
Biotinylation of cell surface proteins was performed using a cell surface protein isolation kit based on the manufacturer’s instructions. Proteins were resolved by SDS-PAGE and analyzed by immunoblots using anti-TRPV4 (1:200), anti-TRPC1 (1:200), or anti-STIM1 (1:200) antibodies.
Double Immunolabeling, Fluorescence Resonance Energy Transfer, and [Ca2+]i Measurement
Double immunolabeling in HUVECs was performed by incubating the cells with a mixture of anti-TRPC1 plus anti-caveolin 1 or anti-TRPV4 plus anti-caveolin 1 antibodies, followed by fluorescence-labeled secondary antibodies. Fluorescence signals were detected by an FV1000 laser scanning confocal system.
For fluorescence resonance energy transfer (FRET), CFP (or YFP)-tagged TRPV4 and YFP (or CFP)-tagged TRPC1 were co-transfected into HEK cells. An inverted microscope equipped with 3-cube FRET filters and a charge-coupled device camera was used to measure the FRET ratio.
[Ca2+]i in cultured cells was measured as described elsewhere.10 In flow experiments, flow was initiated by pumping normal physiological saline solution into a specially-designed parallel plate flow chamber,10 in which the cells were adhered to the bottom. We used a flow rate with shear stress of approximately 5 dyne per square centimeter. If necessary, brefeldin A (BFA), 5 μmol/L, was introduced 30 minutes before the application of TG.
Whole Cell Patch Clamp
Whole cell current was measured with an EPC-9 patch clamp amplifier, as described elsewhere.14 Cells were pretreated with or without TG for 15 minutes. If necessary, BFA, 5 μmol/L, was introduced 30 minutes before the application of TG.
The t test was used for statistical comparison, with P<0.05 considered significant. For comparison of multiple groups, a 1-way ANOVA with a Newman-Keuls test was used.
For expanded experimental procedures, please see the supplemental information (available online at http://atvb.ahajournals.org).
Results
Effect of Ca2+ Store Depletion on Translocation of TRPV4-C1 Heteromeric Channels to the Plasma Membrane in the Overexpression System
TIRFM uses an evanescent wave to illuminate fluorophores within 250 nm of the plasma membrane.15 It is a valuable method for observing protein movements within the periplasmic space. In these experiments, HEK293 cells were first cotransfected with CFP-tagged TRPV4 and YFP-tagged TRPC1 to allow the formation of TRPV4-C1 heteromers.10 Treatment of the cells with TG, TPEN, or physiological agonists (ATP and bradykinin), each of which depletes intracellular Ca2+ stores, induced a time-dependent increase of TRPV4 and TRPC1 fluorescence in the periplasmic space (Figure 1A–D, supplemental Figure I, and supplemental Figure IIA [raw data]). The appearance of TRPV4 and TRPC1 fluorescence matched well temporally and spatially (Figure 1A and C, supplemental Figure I, and supplemental Figure IIA [raw data]), which is even more evident when single-vesicle fluorescence was examined (Figure 1C and supplemental Figure IC). A time-series plot shows that the total fluorescence of the cell surface peaked at approximately 2 minutes after TG treatment, then gradually and slowly decreased (Figure 1B and supplemental Figure IIA [raw data]). There were still significant fluorescence signals 15 minutes after TG treatment. BFA, a blocker of vesicular translocation, abrogated the fluorescence increase (Figure 1B and C). These data suggest that Ca2+ store depletion stimulates the translocation of TRPV4-C1–containing vesicles close to the plasma membrane. As a control, TG treatment had no effect on the fluorescence of yellow fluorescent protein tagged fluorescent plasma membrane marker (Pmem-YFP) (supplemental Figure IIIA and B), which is the fluorescent marker of the plasma membrane. This control excluded the possibility of plasma membrane movement in the axial direction during the experiments. There was no fluorescence signal in mock-transfected cells (supplemental Figure IIIC).
Figure 1.

Effect of Ca2+ store depletion on the translocation of TRPV4–C1 heteromeric channels to the plasma membrane, as measured by TIRFM. A through D, HEK cells were coexpressed with TRPV4-CFP plus TRPC1-YFP and were incubated with TG, 4 μmol/L. A and D, Time-series images of EFF for a representative single HEK cell (A) or a single vesicle (D). The bar indicates 5 μm in A and 1 μm in D. B, Time-series plot of EFF in representative cells for experiments similar to A. C, Summary data in experiments similar to A, showing the maximal change in EFF in response to TG or TPEN, 1 mmol/L. If necessary, BFA, 5 μmol/L, was introduced 30 minutes before TG application. E and F, HEK cells were expressed with either TRPV4-CFP or TRPC1-YFP alone. EFF images of a representative single cell (E) and summary data (F) before and after TG stimulation were shown. Controls in C and F were subjected to vehicle treatment (1% dimethyl sulfoxide). Data are given as the mean±SE (the number of experiments is labeled on top of the bars). #P<0.05 vs control, and *P<0.05 vs TG-treated cells without BFA.
More careful studies were performed on single vesicles. The fluorescence of single vesicles dissipated relatively faster than that of the whole cell surface. The average residency time of single vesicle fluorescence was 309±11 seconds (n=62 vesicles) (Figure 1D and supplemental Figure IC). The time-series images of single-vesicle fluorescence (Figure 1D and supplemental Figure IC) reflect the movement of a TRPV4-C1–containing vesicle toward the plasma membrane (shown as the gradual increase in vesicle fluorescence), followed by free diffusion of TRPV4-C1 proteins toward another area of the plasma membrane after vesicle fusion with the plasma membrane (shown as a decrease in vesicle fluorescence). Our confocal system could only resolve the structures that contain a cluster of fluorophores, such as TRPV4-C1–containing vesicles, but could not resolve a single fluorophore-containing molecule.
FRET was used to further confirm the interaction of TRPV4 and TRPC1 in the plasma membrane. Previously, we found the interaction of TRPV4 and TRPC1 by FRET at the whole cell level.10 Herein, we limited the FRET detection to the plasma membrane region only, and the results confirmed a physical interaction of CFP-tagged TRPV4 and YFP-tagged TRPC1 in the plasma membrane (supplemental Figure IV).
TIRFM detects the protein movement within the periplasmic space, but it does not necessarily define surface expression itself. Thus, cell surface biotinylation methods were used. TG treatment increased the amount of biotinylated TRPV4 and TRPC1 by 70±11% (n=4) and 64±14% (n=3), respectively, indicating an increase in cell surface TRPV4 and TRPC1 (Figure 2A and B). BFA abolished the stimulation effect of TG (Figure 2A and B). In HEK cells that were overexpressed with only 1 construct (TRPV4 or TRPC1 alone), TG treatment had little or no effect on the trafficking of these proteins, as determined by biotinylation (Figure 2C and D) and TIRFM (Figure 1E and F and supplemental Figure IIB–D), suggesting that TG enhances the translocation of TRPV4-C1 heteromeric channels more favorably than that of TRPV4 or TRPC1 homomeric channels. Supplemental Figure IID also shows that, under basal conditions without TG treatment, there was more surface fluorescence of TRPV4 and TRPC1 in cells that are coexpressed with TRPV4 plus TRPC1 when compared with the cells that were expressed with only 1 protein (either TRPV4 or TRPC1) (supplemental Figure IID).
Figure 2.
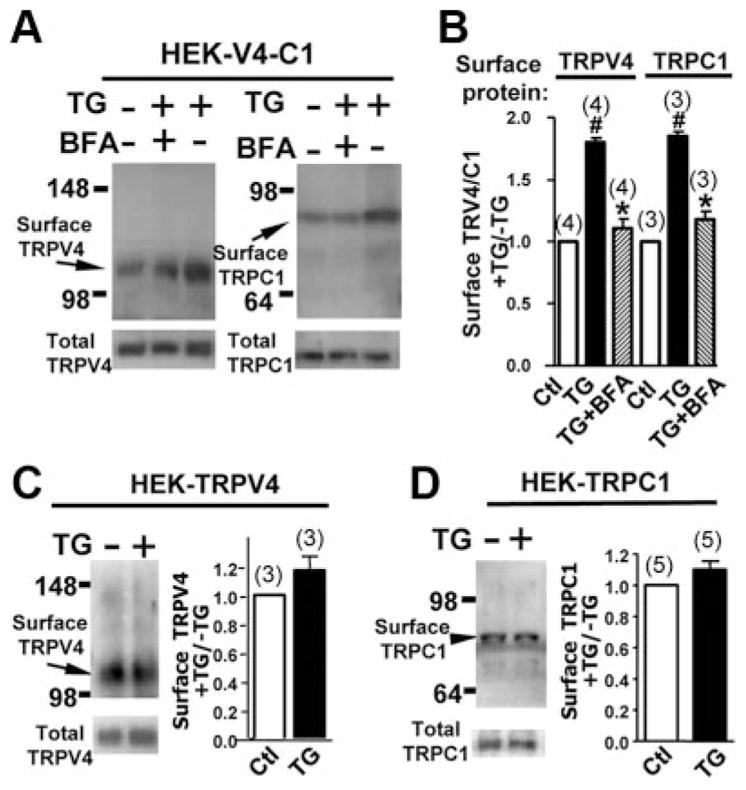
TG-induced translocation of TRPC1, TRPV4, and TRPV4–C1, as measured by cell surface biotinylation. A through D, HEK cells were transfected with TRPV4 plus TRPC1 (A and B), TRPV4 alone (C), or TRPC1 alone (D). The cells were then treated with or without TG, 4 μmol/L for 15 minutes, followed by a biotinylation assay. Representative images (A and left panel of C and D) and summary data (B and right panel of C and D) of cell surface biotinylation experiments were shown. If necessary, BFA, 5 μmol/L, was introduced 30 minutes before TG application. Data are given as the mean±SE (the number of experiments is labeled on top of the bars). #P<0.05 vs control (Ctl), and *P<0.05 vs the TG-treated cells without BFA.
The observed biotinylated TRPV4 and TRPC1 only reflected the overexpressed TRPV4 and TRPC1 but not the endogenous ones, because biotinylated signals were absent in nontransfected HEK cells (supplemental Figure V). Cell surface expression of endogenous TRPC1 and TRPV4 in nontransfected HEK cells, if any, was low and less than the detection level (supplemental Figure V). Two antibodies for immunoblots (anti-TRPC1 and anti-TRPV4) were previously shown to be highly specific to their respective targets.10,16,17
Functional Study of TRPV4-C1 Heteromeric Channels
4α-PDD is a synthetic phorbol ester that can activate TRPV4 homomeric channels18 and TRPV4-C1 heteromeric channels.10 However, the inactivation kinetics of the 4α-PDD–stimulated current differ greatly between TRPV4 homomeric and TRPV4-C1 heteromeric channels. For TRPV4 homomeric channels, the 4α-PDD–stimulated current is more transient and decays quickly,10 whereas for TRPV4-C1 heteromeric channels, this current is much more prolonged (Figure 3A).10 In whole cell recording, no apparent decay in current was observed within the duration of the experiments, which lasted for 6 to 10 minutes (Figure 3A). TG treatment, 4 αmol/L, for 15 minutes potentiated the 4α-PDD–stimulated cation current (Figure 3C); it also augmented the 4α-phorbol 12,13-didecanoate (4α-PDD)-stimulated Ca2+ influx (Figure 3E). These data match well with those from TIRFM (Figure 1C) and the cell surface biotinylation assay (Figure 2B), both of which show an increased surface expression of TRPV4-C1 heteromers 15 minutes after TG treatment. BFA abolished the potentiation effect of TG (Figure 3C–E).
Figure 3.
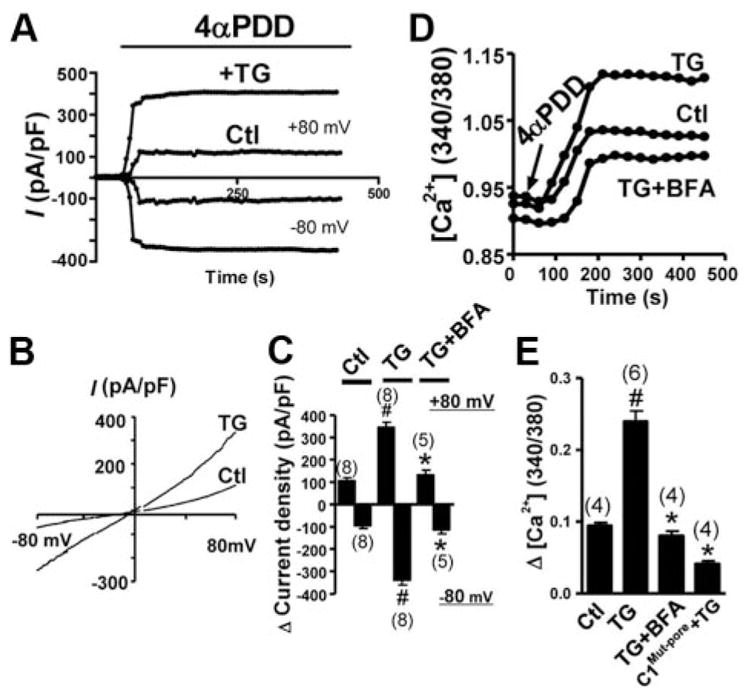
4α-PDD–stimulated cation current and [Ca2+]i increase. The HEK cells were cotransfected with TRPV4 and TRPC1. A through C, For cation currents, representative current traces were shown at ±80 mV (A), corresponding I–V curves (B), and summary data for the maximal change in current in response to 4α-PDD at ±80 mV (C). D and E, For [Ca2+]i measurement, representative traces (D) and summary data (E) for the maximal change in [Ca2+]i to 4α-PDD were shown. C1Multi-pore was used to replace TRPC1 in some experiments as labeled. TG, 4 μmol/L, was given for 15 minutes; 4α-PDD, 5 μmol/L; BFA, 5 μmol/L, was given 30 minutes before TG. Data are given as the mean±SE (the number of experiments is labeled on top of the bars). Ctl indicates control. #P<0.05 vs the cells without TG pretreatment, and *P<0.05 vs the bar labeled with # in the same panel.
Participation of STIM1
An STIM1-specific siRNA19 could effectively and selectively “knock down” the expression level of STIM1 proteins (supplemental Figure VI). TIRFM experiments show that this STIM1-siRNA was markedly reduced in TG-induced translocation of TRPV4-C1 heteromers in HEK cells that were coexpressed with TRPV4 and TRPC1 (Figure 4A and B). In functional studies, STIM1-siRNA reduced the 4α-phorbol 12,13-didecanoate (4α-PDD)-stimulated Ca2+ influx (Figure 4C). Immunoblots show that the anti-STIM1 antibody was highly specific to STIM1 (supplemental Figure VII).
Figure 4.
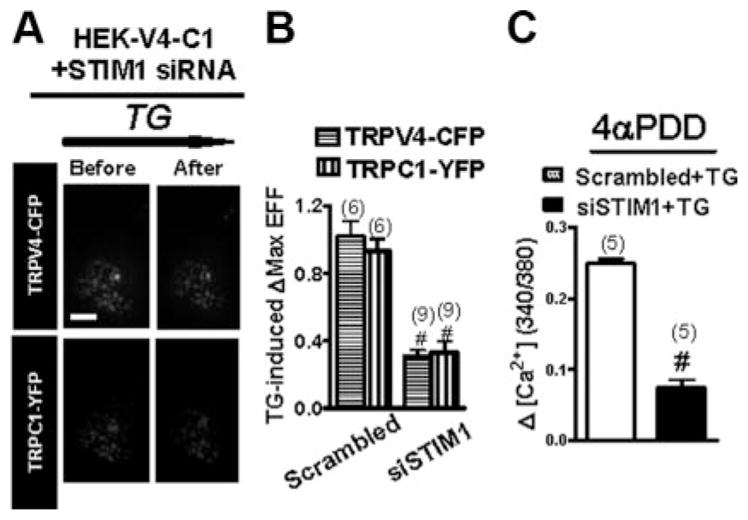
Involvement of STIM1 in the translocation of TRPV4–C1 heteromeric channels in response to Ca2+ store depletion. TRPV4–C1–coexpressing HEK cells were treated with STIM1-siRNA or scrambled siRNA, followed by TG treatment, 4 μmol/L. A, EFF images of a representative single cell in the presence of STIM1-siRNA before and after TG stimulation. B and C, Summary data showing that STIM1-siRNA diminished the TG-induced increase in EFF intensity (B) and 4α-PDD–stimulated Ca2+ influx (C). Data are given as the mean±SE (the number of experiments is labeled on top of the bars). #P<0.05 vs the scrambled siRNA.
Effect of TG on Translocation of TRPV4-C1 Heteromeric Channels in HUVECs
Next, we explored whether Ca2+ store depletion can facilitate the translocation of TRPV4-C1 heteromeric channels to the plasma membrane in HUVECs. Biotinylation experiments demonstrated that TG treatment, 4 μmol/L for 15 minutes, markedly increased the TRPV4 and TRPC1 proteins in the plasma membrane (Figure 5A); it had no effect on the total amount of cellular TRPV4 and TRPC1 proteins (Figure 5A). TRPC1-siRNA and TRPV4-siRNA were used, each of which was capable of “knock downing” the expression of its targeted genes in HUVECs (supplemental Figure VIII). Intriguingly, suppressing the TRPC1 protein level by TRPC1-siRNA reduced the TRPV4 translocation to the plasma membrane (Figure 5B). Similarly, TRPV4-siRNA reduced the TRPC1 translocation to the plasma membrane (Figure 5B). STIM1-siRNA reduced the translocation of both TRPV4 and TRPC1 (Figure 5B). These results in HUVECs are consistent with those obtained in TRPV4-C1– coexpressing HEK cells, supporting the notion that Ca2+ store depletion stimulates the translocation of TRPV4-C1 heteromeric channels to the plasma membrane and that TRPV4-C1 heteromeric channels are more favorably delivered to the plasma membrane than TRPV4 or TRPC1 homomeric channels.
Figure 5.

TG-induced translocation of TRPV4 –C1 heteromeric channels to the plasma membrane in HUVECs. A, Representative images (left and middle) and summary (right) of TG, 4 μmol/L–induced increases in cell surface TRPV4 (left) and TRPC1 (middle) proteins, as measured by a biotinylation assay. B, Similar to A, but showing the effect of TRPC1-siRNA, TRPV4-siRNA, or STIM1-siRNA. TG, 4 μmol/L, was given for 15 minutes. Data are given as the mean±SE (the number of experiments is labeled on top of the bars). The values in the absence of TG were normalized to 1. Ctl indicates control. #P<0.05 vs cells without TG, and *P<0.05 vs scrambled siRNA.
Role of TRPV4-C1 Heteromer Translocation in Flow-Induced Ca2+ Influx in HUVECs
An important function of TRPV4-C1 heteromeric channels is to mediate flow-induced Ca2+ influx in vascular endothelial cells.10 Previously, Ca2+ store depletion by TG potentiated the flow-induced Ca2+ influx in vascular endothelial cells.11 This was confirmed (Figure 6A). In addition, we found that this potentiated [Ca2+]i response to flow was abolished by BFA (Figure 6B), suggesting an involvement of vesicular trafficking. In the absence of TG treatment, BFA had no effect on flow-induced [Ca2+]i increase (supplemental Figure IX). The potentiated [Ca2+]i response to flow was also inhibited by TRPV4-siRNA, a pore mutant of TRPV4 (TRPV4M680D),14 TRPC1-siRNA, and a pore mutant of TRPC1 (TRPC1Multi-pore),13 suggesting an involvement of TRPV4-C1 heteromeric channels in the response (Figure 6B).
Figure 6.

Potentiation of flow-induced Ca2+ influx and 4α-PDD–stimulated [Ca2+]i increase by TG in HUVECs. A through D, Representative traces (A and C) and summary data (B and D) for the maximal change in [Ca2+]i in response to flow (A and B) or 4α-PDD (C and D) were shown. TG, 4 μmol/L, was given for 15 minutes; BFA, 5 μmol/L, was given for 30 minutes before TG. Data are given as the mean±SE (the number of experiments is labeled on top of the bars). #P<0.05 vs cells without TG, and *P<0.05 vs scrambled siRNA (for siRNA experiments) or wild type+TG (in BFA or C1Multi-pore experiments).
The possible caveolar localization of TRPV4-C1 heteromeric channels was explored. In double-labeling immuno-fluorescence experiments, we found strong overlapping of TRPC1 and TRPV4 fluorescence with that of caveolin-1 (supplemental Figure X), suggesting the localization of TRPC1 and TRPV4 in the caveolar compartment in HUVECs.
4α-PDD–stimulated Ca2+ influx and cation current were also studied in HUVECs (Figure 6C and D and supplemental Figure XI). TG treatment potentiated 4α-PDD–stimulated Ca2+ influx, the effect of which was abolished by BFA, TRPV4-siRNA, STIM1-siRNA, TRPC1-siRNA, and TRPC1Multi-pore (Figure 6C and D). In whole cell patch clamp recordings, TG potentiated the 4α-PDD–stimulated cation current, the effect of which was inhibited by BFA and TRPC1Multi-pore (supplemental Figure XI). These data support an involvement of TRPV4-C1 heteromeric channels and vesicular trafficking in the TG potentiation of 4α-PDD–stimulated Ca2+ and cation current responses.
Discussion
The major findings of this study are as follows: (1) TIRFM, cell surface biotinylation, and functional studies show that Ca2+ store depletion enhances the vesicular trafficking and insertion of TRPV4-C1 heteromeric channels into the plasma membrane in the HEK cell overexpression system, (2) STIM1 is important in controlling the vesicular trafficking and insertion of TRPV4-C1 heteromeric channels into the plasma membrane, (3) TRPV4 and TRPC1 are colocalized with caveolin-1 in the caveolar compartment of the plasma membrane in HUVECs, and (4) Ca2+ store depletion causes an increased delivery of TRPV4-C1 heteromeric channels to the plasma membrane in HUVECs, resulting in an augmented Ca2+ influx in endothelial cells in response to shear flow.
Regulated translocation of channel proteins to the plasma membrane is an important means to control the plasma membrane expression of TRP channels and their function.1 In the present study, we used the unique feature of our TIRFM system to simultaneously track the forward trafficking of CFP-tagged TRPV4 and YFP-tagged TRPC1 to the plasma membrane in the HEK cell overexpression system. The results show that Ca2+ store depletion by TG, TPEN, or physiological agonists (bradykinin and ATP) caused an enhanced translocation of TRPV4-C1 heteromeric channels to the plasma membrane. The increased TRPV4-C1 translocation is related to vesicular trafficking because BFA abolished it. These results were confirmed by cell surface biotinylation experiments and functional studies, in which 4α-PDD–stimulated cation current and Ca2+ influx were examined. The appearance of TRPV4 and TRPC1 fluorescence on the plasma membrane matched well temporally and spatially in TIRFM, suggesting that TRPC1 and TRPV4 are inserted into the plasma membrane as TRPV4-C1 heteromers.
Heteromultimerization of different TRP subunits could affect their translocation to the plasma membrane.8 TRPC4, TRPM7, and TRPP1 may facilitate the translocation of TRPC1, TRPM6, and TRPP2, respectively, to the plasma membrane.8,20,21 In the present study, TIRFM experiments show that there was more surface fluorescence of TRPV4 and TRPC1 in cells that are coexpressed with TRPV4 plus TRPC1 than those expressed with only 1 protein alone (either TRPV4 or TRPC1) (supplemental Figure IID). These data seem consistent with previous reports,8,20,21 suggesting that coexpression of heteromers facilitates their translocation to the plasma membrane. Interestingly, we found that Ca2+ store depletion preferentially stimulated the translocation of TRPV4-C1 heteromeric channels to the plasma membrane but had little or no effect on that of TRPV4 or TRPC1 homomeric channels. The reason for this preferential effect of TG is not clear. One speculation could be that TRPV4-C1 heteromers are preferentially packaged into vesicles; thus, their trafficking is more subjected to regulation by Ca2+ store depletion.
Substantial evidence indicates that STIM1 is the Ca2+ sensor in the endoplasmic reticulum and that it serves to sense Ca2+ store depletion.22 In the present study, we found that knocking down of STIM1 using STIM1-siRNA markedly suppressed the TG-stimulated delivery of TRPV4-C1 heteromers to the plasma membrane, suggesting an important role of STIM1 in TG-induced translocation of TRPV4-C1 heteromeric channels to the plasma membrane.
Recent studies have found that TRPV4-C1 heteromeric channels mediate flow-induced endothelial [Ca2+]i influx and subsequent vascular relaxation.10 In vascular endothelial cells, Ca2+ store depletion can potentiate flow-induced Ca2+ influx.11 Such an interaction between Ca2+ store filling status and flow-induced Ca2+ influx has profound physiological importance. Vascular endothelial cells are exposed to circulating blood that contains metabolites, hormones, growth factors, and cytokines, many of which can induce Ca2+ release from intracellular Ca2+ stores, resulting in Ca2+ store depletion. The depletion of Ca2+ stores would then augment flow-induced endothelial Ca2+ influx11 and subsequent vascular dilation.12 However, the mechanism of how Ca2+ store depletion could enhance flow-induced Ca2+ influx remains elusive. In the present study, a cell surface biotinylation assay shows that Ca2+ store depletion increases the cell surface expression of TRPV4-C1 heteromeric channels in HUVECs. In agreement, functional studies also show that Ca2+ store depletion increases 4α-PDD–stimulated Ca2+ influx and cation current in HUVECs. More important, after measuring the flow-induced [Ca2+]i increase, we found that TG potentiates the increase in HUVECs. This potentiation effect of TG was abolished by BFA, TRPV4-siRNA, TRPV1-siRNA, and TRPV4 and TRPC1 pore mutants, suggesting the involvement of vesicular trafficking and TRPV4-C1 heteromeric channels. Our working scheme is that Ca2+ store depletion causes an increased delivery of TRPV4-C1 heteromeric channels to the plasma membrane in HUVECs, resulting in an augmented Ca2+ influx in endothelial cells in response to shear flow. This scheme provides a mechanistic explanation for the potentiation effect of Ca2+ store depletion on flow-induced endothelial Ca2+ influx and vascular relaxation.11,12
Caveolae in endothelial cells have been implicated as plasma membrane microdomains that sense or transduce hemodynamic changes into biochemical signals that regulate vascular function.23 Previously, TRPV4 was reported to be colocalized with caveolin-1 in the caveolar compartment of the plasma membrane in HUVECs.24 In the present study, we found that both TRPV4 and TRPC1 are localized with caveolin-1 in the caveolar compartment. The functional implication of this colocalization in flow-induced Ca2+ influx in endothelial cells should be resolved.
In conclusion, we demonstrated that depletion of intracellular Ca2+ stores induced a rapid insertion of TRPV4-C1 heteromeric channels into the plasma membrane in TRPV4-C1–overexpressing HEK cells and native endothelial cells.
Supplementary Material
Acknowledgments
We thank Q. Xia, PhD, Department of Physiology, Zhejiang University, China, for assistance.
Sources of Funding
This study was supported by grants Chinese University of Hong Kong (CUHK)477307, CUHK477408, and CUHK479109 from Hong Kong Research Grants Council; the Focused Investment Scheme of CUHK; and the Li Ka Shing Institute of Health Sciences.
Footnotes
Disclosures
None.
References
- 1.Cayouette S, Boulay G. Intracellular trafficking of TRP channels. Cell Calcium. 2007;42:225–232. doi: 10.1016/j.ceca.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 2.Kim JY, Zeng W, Kiselyov K, Yuan JP, Dehoff MH, Mikoshiba K, Worley PF, Muallem S. Homer 1 mediates store- and inositol 1,4,5-trisphosphate receptor-dependent translocation and retrieval of TRPC3 to the plasma membrane. J Biol Chem. 2006;281:32540–32549. doi: 10.1074/jbc.M602496200. [DOI] [PubMed] [Google Scholar]
- 3.Cayouette S, Lussier MP, Mathieu EL, Bousquet SM, Boulay G. Exocytotic insertion of TRPC6 channel into the plasma membrane upon Gq protein-coupled receptor activation. J Biol Chem. 2004;279:7241–7246. doi: 10.1074/jbc.M312042200. [DOI] [PubMed] [Google Scholar]
- 4.Lepage PK, Boulay G. Molecular determinants of TRP channel assembly. Biochem Soc Trans. 2007;35:81–83. doi: 10.1042/BST0350081. [DOI] [PubMed] [Google Scholar]
- 5.Bai CX, Giamarchi A, Rodat-Despoix L, Padilla F, Downs T, Tsiokas L, Delmas P. Formation of a new receptor-operated channel by heteromeric assembly of TRPP2 and TRPC1 subunits. EMBO Rep. 2008;9:472–479. doi: 10.1038/embor.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsiokas L, Arnould T, Zhu C, Kim E, Walz G, Sukhatme VP. Specific association of the gene product of PKD2 with the TRPC1 channel. Proc Natl Acad Sci U S A. 1999;96:3934–3939. doi: 10.1073/pnas.96.7.3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kottgen M, Buchholz B, Garcia-Gonzalez MA, Kotsis F, Fu X, Doerken M, Boehlke C, Steffl D, Tauber R, Wegierski T, Nitschke R, Suzuki M, Kramer-Zucker A, Germino GG, Watnick T, Prenen J, Nilius B, Kuehn EW, Walz G. TRPP2 and TRPV4 form a polymodal sensory channel complex. J Cell Biol. 2008;182:437–447. doi: 10.1083/jcb.200805124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hofmann T, Schaefer M, Schultz G, Gudermann T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci U S A. 2002;99:7461–7466. doi: 10.1073/pnas.102596199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strubing C, Krapivinsky G, Krapivinsky L, Clapham DE. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron. 2001;29:645–655. doi: 10.1016/s0896-6273(01)00240-9. [DOI] [PubMed] [Google Scholar]
- 10.Ma X, Qiu S, Luo J, Ma Y, Ngai CY, Shen B, Wong CO, Huang Y, Yao X. Functional role of vanilloid transient receptor potential 4-canonical transient receptor potential 1 complex in flow-induced Ca2+ influx. Arterioscler Thromb Vasc Biol. 2010;30:851–858. doi: 10.1161/ATVBAHA.109.196584. [DOI] [PubMed] [Google Scholar]
- 11.Kwan HY, Leung PC, Huang Y, Yao X. Depletion of intracellular Ca2+ stores sensitizes the flow-induced Ca2+ influx in rat endothelial cells. Circ Res. 2003;92:286–292. doi: 10.1161/01.res.0000054625.24468.08. [DOI] [PubMed] [Google Scholar]
- 12.Liu C, Ngai CY, Huang Y, Ko WH, Wu M, He GW, Garland CJ, Dora KA, Yao X. Depletion of intracellular Ca2+ stores enhances flow-induced vascular dilatation in rat small mesenteric artery. Br J Pharmacol. 2006;147:506–515. doi: 10.1038/sj.bjp.0706639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu X, Singh BB, Ambudkar IS. TRPC1 is required for functional store-operated Ca2+ channels: role of acidic amino acid residues in the S5–S6 region. J Biol Chem. 2003;278:11337–11343. doi: 10.1074/jbc.M213271200. [DOI] [PubMed] [Google Scholar]
- 14.Voets T, Prenen J, Vriens J, Watanabe H, Janssens A, Wissenbach U, Bodding M, Droogmans G, Nilius B. Molecular determinants of permeation through the cation channel TRPV4. J Biol Chem. 2002;277:33704–33710. doi: 10.1074/jbc.M204828200. [DOI] [PubMed] [Google Scholar]
- 15.Steyer JA, Almers W. A real-time view of life within 100 nm of the plasma membrane. Nat Rev Mol Cell Biol. 2001;2:268–275. doi: 10.1038/35067069. [DOI] [PubMed] [Google Scholar]
- 16.Maroto R, Raso A, Wood TG, Kurosky A, Martinac B, Hamill OP. TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nat Cell Biol. 2005;7:179–185. doi: 10.1038/ncb1218. [DOI] [PubMed] [Google Scholar]
- 17.Yang XR, Lin MJ, McIntosh LS, Sham JS. Functional expression of transient receptor potential melastatin- and vanilloid-related channels in pulmonary arterial and aortic smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1267–L1276. doi: 10.1152/ajplung.00515.2005. [DOI] [PubMed] [Google Scholar]
- 18.Watanabe H, Davis JB, Smart D, Jerman JC, Smith GD, Hayes P, Vriens J, Cairns W, Wissenbach U, Prenen J, Flockerzi V, Droogmans G, Benham CD, Nilius B. Activation of TRPV4 channels (hVRL-2/mTRP12) by phorbol derivatives. J Biol Chem. 2002;277:13569–13577. doi: 10.1074/jbc.M200062200. [DOI] [PubMed] [Google Scholar]
- 19.Lefkimmiatis K, Srikanthan M, Maiellaro I, Moyer MP, Curci S, Hofer AM. Store-operated cyclic AMP signalling mediated by STIM1. Nat Cell Biol. 2009;11:433–442. doi: 10.1038/ncb1850. [DOI] [PubMed] [Google Scholar]
- 20.Hanaoka K, Qian F, Boletta A, Bhunia AK, Piontek K, Tsiokas L, Sukhatme VP, Guggino WB, Germino GG. Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature. 2000;408:990–994. doi: 10.1038/35050128. [DOI] [PubMed] [Google Scholar]
- 21.Chubanov V, Waldegger S, Schnitzler M, Vitzthum H, Sassen MC, Seyberth HW, Konrad M, Gudermann T. Disruption of TRPM6/TRPM7 complex formation by a mutation in the TRPM6 gene causes hypomagnesemia with secondary hypocalcemia. Proc Natl Acad Sci U S A. 2004;101:2894–2899. doi: 10.1073/pnas.0305252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cahalan MD. STIMulating store-operated Ca2+ entry. Nat Cell Biol. 2009;11:669–677. doi: 10.1038/ncb0609-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu J, Bergaya S, Murata T, Alp I, Bauer M, Lin M, Drab M, Kurzchalia T, Stan R, Sessa W. Direct evidence for the role of caveolin-1 and caveolae in mechanotransduction and remodeling of blood vessels. J Clin Invest. 2006;116:1284–1291. doi: 10.1172/JCI27100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saliez J, Bouzin C, Rath G, Ghisdal P, Desjardins F, Rezzani R, Rodella LF, Vriens J, Nilius B, Feron O, Balligand JL, Dessy C. Role of caveolar compartmentation in endothelium-derived hyperpolarizing factor-mediated relaxation: Ca2+ signals and gap junction function are regulated by caveolin in endothelial cells. Circulation. 2008;117:1065–1074. doi: 10.1161/CIRCULATIONAHA.107.731679. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.