

Abstract
Background
Since the biological function of c-Jun N-terminal kinase (JNK) in gastric cancer remains unclear, we investigated the clinical significance of JNK activation and its association with FOXO1 activation.
Methods
Immunohistochemical tissue array analysis of 483 human gastric cancer specimens was performed, and the results of the immunostaining were quantified. The correlation between JNK activation (nuclear staining for pJNK) and clinicopathological features, the proliferation index, prognosis or FOXO1 inactivation (cytoplasmic staining for pFOXO1) was analyzed. The SNU-638 gastric cancer cell line was used for in vitro analysis.
Results
Nuclear staining of pJNK was found in 38 % of the gastric carcinomas and was higher in the early stages of pTNM (P < 0.001). pJNK staining negatively correlated with lymphatic invasion (P = 0.034) and positively correlated with intestinal type by Lauren’s classification (P = 0.037), Ki-67-labeling index (P < 0.001), cyclin D1 (P = 0.045), cyclin E (P < 0.001) and pFOXO1 (P < 0.001). JNK activation correlated with a longer patients survival (P =0.008) and patients with a JNK-active and FOXO1-inactive tumor had a higher survival rate than the remainder of the population (P = 0.004). In vitro analysis showed that JNK inhibition by SP600125 in SNU-638 cells decreased cyclin D1 protein expression and increased FOXO1 activation. Further, JNK inhibition markedly suppressed colony formation, which was partially restored by FOXO1 shRNA expression.
Conclusions
Our results indicate that JNK activation may serve as a valuable prognostic factor in gastric cancer, and that it is implicated in gastric tumorigenesis, at least in part, through FOXO1 inhibition.
Keywords: JNK, Gastric cancer, Clinical significance, Proliferation, FOXO1
Background
Gastric cancer has been reported to be the fourth most common cancer and the second leading cause of cancer-related death worldwide [1]. More than 930,000 new cases are diagnosed and 700,000 deaths are attributed to gastric cancer annually [2]. Although gastric cancer develops through the accumulation of genetic alterations, such as oncogene (e.g. HER2, c-Met) activation and tumor suppressor gene (e.g. PTEN, p53) loss [3–6], the underlying molecular mechanisms of gastric carcinogenesis are largely unknown. Although anti-HER2 drugs such as trastuzumab and lapatinib are being used as molecularly targeted agents for gastric cancer patients with HER2 overexpression and patients responded clinically, acquired resistance to these anti-HER2 drugs has been observed in a subset of gastric cancer patients following chronic exposure [7]. Thus, better understanding of the molecular pathways involved in gastric cancer promotion will be helpful to improve gastric cancer therapies.
c-Jun N-terminal kinase (JNK) is a mitogen-activated protein kinase (MAPK), which regulates a wide range of cellular functions through both transcription-dependent and transcription-independent mechanisms [8]. In recent years, JNK has increasingly been recognized as an attractive molecular target for cancer treatment because of its broad roles in the regulation of cancer-associated cellular processes, including cell proliferation, differentiation and survival [8]. Since JNK acts as either a tumor promoter in breast cancer [9] and prostate cancer [10] or a tumor suppressor in skin cancer [11] and ovarian cancer [12], it is important to fully understand the role of JNK and the underlying molecular mechanisms in each tumor in order to validate the therapeutic potential of JNK.
With respect to gastric cancer, the biological significance of JNK has been controversial. Several in vitro studies have shown that JNK activation decreases gastric cancer cell survival [13–18]. In contrast, opposite finding have also been reported. JNK activation suppressed the apoptosis of gastric cancer cells in one study [19]. In addition, Shibata et al. [20] reported that JNK increased the development of gastric tumor in mice. Shibata et al. [20] also observed JNK activation in 40 % of 40 surgically resected human gastric carcinoma specimens. Thus, the role of JNK in gastric cancer remains elusive.
The subclass O of forkhead transcription factors (FOXO) consists of four members, FOXO1, FOXO3, FOXO4, and FOXO6. FOXO transcriptional activity is regulated by a complex array of post-translational modifications, including phosphorylation, acetylation, ubiquitination, and binding protein partners [21]. The two main post-translational modifications regulating FOXO phosphorylation are mediated by the AKT and JNK pathways [21]. Since FOXO proteins regulate diverse cellular functions, their dysregulations are considered to be potential targets of cancer therapy [22]. In gastric cancer, FOXO1 inactivation occurs and increases gastric cancer cell proliferation [23, 24] and angiogenesis [25]. Thus, restoration of FOXO1 activity may be a useful tool to suppress gastric tumor promotion.
JNK regulation of FOXO proteins is highly conserved across species [26], and a positive regulation of FOXOs by JNK has been shown in various human cancer cells, including colon cancer cells [27, 28], thyroid cancer cells [29], melanoma cells [30], and lung cancer cells [22, 31]. However, there has not been any study of that association in gastric cancer.
The present study was performed to evaluate the biological significance of JNK alone or in combination with FOXO1 in human gastric cancer regarding patient survival rate and tumor growth. Here, we evaluated the immunostaining for the active form of JNK phosphorylated at Thr183 and Tyr185 (pJNK) in 483 surgically resected human gastric carcinoma specimens and assessed its clinical significance. In cell culture experiments, SNU-638 gastric cancer cells were treated with a specific JNK inhibitor SP600125 to determine the direct role of JNK in the gastric cancer cell growth. In addition, the association between JNK and FOXO1 was examined in human gastric cancer specimens and gastric cancer cells.
Methods
Patients and tissue samples
Four hundred eighty-three surgically resected gastric carcinoma cases examined at the Department of Pathology, Seoul National University College of Medicine from 2 January to 29 December 2006 were analyzed. Gender, age, tumor size, tumor location, World Health Organization (WHO), Lauren’s classification, pathological tumor-node-metastasis (pTNM) stage and lymphatic invasion were evaluated by reviewing medical charts and pathological records [1], and examining the glass slides from each case. These information were summarized in Table 1. No patient received preoperative chemotherapy or radiotherapy. Patients’ clinical outcomes were followed from the date of surgery to either the date of death or 22 July 2013, resulting in the follow-up period ranged from 1 to 92 months. This protocol was reviewed and approved by the Institutional Review Board of Seoul National University (IRB No. 1309-087-522).
Table 1.
Clinicopathological profiles of 483 gastric cancer patients
| Clinicopathological features | Total cases (n = 483) | |
|---|---|---|
| Gender | Male | 337 |
| Female | 146 | |
| Age (years) | Mean ± SD | 58.3 ± 12.2 |
| Tumor size (cm) | Mean ± SD | 4.9 ± 3.1 |
| Tumor location | Low | 95 |
| Middle | 165 | |
| Upper | 222 | |
| Whole | 0 | |
| Remnant | 1 | |
| WHO | Papillary | 11 |
| WD | 65 | |
| MD | 157 | |
| PD | 182 | |
| Mucinous | 17 | |
| SRC | 47 | |
| Other | 4 | |
| Laurens’ classification | Intestinal | 236 |
| Diffuse | 193 | |
| Mixed | 54 | |
| Tumor stage | I | 271 |
| (TNM stage according | II | 91 |
| to AJCC system) | III | 62 |
| VI | 59 | |
| Lymphatic invasion | Absent | 273 |
| Present | 210 | |
| Follow-up (months) | Range (Mean) | 1-92 (69) |
SD standard deviation, WHO World Health Organization
Tissue array methods
Nine paraffin array blocks were prepared by Superbiochips Laboratories (Seoul, Korea), as previously described [32]. Briefly, core tissue biopsies (2 mm in diameter) were taken from individual paraffin-embedded gastric tumors (donor blocks) and arranged in a new recipient paraffin block (tissue array block) using a trephine apparatus. Each tissue array block was able to contain up to 60 cases, allowing nine array blocks to contain 483 cases. Each block contained an internal control consisting of non-neoplastic gastric mucosa from body, antrum and intestinal metaplasia. The staining results of the different intra-tumoral areas of gastric carcinomas in these tissue array blocks showed an excellent agreement [33] as described in the discussion section. A core was chosen from each case for analysis. We defined an adequate case as a tumor occupying more than 10 % of the core area.
Immunohistochemistry
Immunohistochemistry was performed using the streptavidin-peroxidase technique. Paraffin-embedded sections of 4 μm thicknesses were cut from each tissue array block and mounted on glass slides. Then, the tissue array slides were deparaffinized at 59 °C for 1 h followed by xylene treatment. After hydration in an ethanol series, antigen retrieval was performed by microwave method: slides immersed in 0.01 M citrate buffer (pH 6.0) were placed in microwave oven and microwaved for 15 min (700 W, medium, low, each for 5 min). After cooling in cold PBS, endogenous peroxidase activity was quenched by incubating the slides in 3 % hydrogen peroxide for 10 min. Nonspecific binding was blocked by treating sections with 5 % normal goat serum or 2 % normal horse serum (Vectastain ABC kit; Vector Laboratories, Burlingame, CA, USA) for 1 h. Sections were incubated with primary antibodies against phospho-JNKThr183/Tyr185 (1:50; rabbit polyclonal; Cell Signaling Technology, Beverly, MA, USA), phospho-FOXO1Ser256 (1:50; rabbit polyclonal; Cell Signaling Technology), Ki-67 (1:50; mouse monoclonal; DAKO, Glostrup, Denmark), cyclin D1 (1:500; rabbit polyclonal; Santa Cruz Biotechnology, Santa Cruz, CA, USA) or cyclin E (1:100; mouse monoclonal; Santa Cruz Biotechnology) at 4 °C overnight. After washing, sections were reacted with a secondary antibody, which is biotinylated goat anti-rabbit IgG (1:200, Vector Laboratories) or biotinylated horse anti-mouse IgG (1:100, Vector Laboratories) for 2 h at room temperature. Immunoreactions were visualized by incubation for 1 h at room temperature with streptavidin-horseradish peroxidase conjugate (Vector Laboratories), followed by a reaction with 0.025 % (w/v) 3,3-diaminobenzidine tetrahydrochloride (DAB) for 3 min and counterstaining with Meyer’s hematoxylin. Throughout the above analyses, controls were prepared by omitting the primary antibody. The expressions of proteins were assessed in a blinded fashion by two investigators (Y Kim and JS Pyo). For statistical analysis of immunostaining for proteins other than Ki-67, cases showing nuclear (pJNK, cyclin D1 and cyclin E) or cytoplasmic (pFOXO1) staining in 10 % or more of the tumor cells were considered to indicate positive staining. For Ki-67 staining, we evaluated 300 cells and counted the cells with nuclear staining for each specimen. The proliferation index was defined as follows: proliferation index (%) = 100 X Ki-67-positive cells/total cells.
Cell culture
A human gastric cancer cell line SNU-638 was obtained from the Korean Cell Line Bank (Seoul, Korea). Cells were cultured in RPMI1640 (Life Technologies, Grand Island, NY, USA) supplemented with 10 % fetal bovine serum (FBS), 2 mg/mL sodium bicarbonate, 100 U/mL penicillin, and 100 μg/mL streptomycin (Life Technologies) at 37 °C in a humidified 95 % air and 5 % CO2 atmosphere.
Treatment with a JNK inhibitor
To inhibit endogenous JNK activity, SNU-638 cells were treated with the indicated concentrations (5, 10, 20, and 30 μM) of a JNK inhibitor, SP600125 (Cell Signaling Technology), dissolved in dimethylsulfoxide (DMSO).
Western blotting
Cell lysates were prepared in 100–200 μl of 1 x sodium dodecyl sulfate (SDS) lysis buffer [125 mM Tris–HCl (pH 6.8), 4 % SDS, 0.004 % bromophenol blue, and 20 % glycerol]. Protein contents were measured using BCA Protein Assay Reagent (Pierce, Rockford, IL, USA). Equal amounts of proteins were loaded onto a 10 % discontinuous SDS/polyacrylamide gel and electrophoretically transferred to PVDF membranes (Millipore Corporation, Billerica, MA, USA) blocked with 5 % nonfat dry milk in phosphate-buffered saline (PBS)-Tween 20 (0.1 %, v/v) for 1 h. The membranes were then incubated at 4 °C overnight with or without 2 h incubation at room temperature with one of the following primary antibodies: anti-pJNK (1:1000, Cell Signaling Technology), anti-total JNK (1:1000, Cell Signaling Technology), anti-cyclin D1 (1:1000, Santa Cruz Biotechnology), anti-pFOXO1 (1:1000, Cell Signaling Technology), anti-total FOXO1 (1:1000, Cell Signaling Technology), and anti-β-actin (1:1000, Santa Cruz Biotechnology). Horse-radish peroxidase-conjugated anti-rabbit IgG (1:2000; Santa Cruz Biotechnology) or anti-mouse IgG (1:2500; Santa Cruz Biotechnology) was used as a secondary antibody. Enhanced chemiluminescence was used to detect the immunoreactive proteins. Equal protein loading was confirmed by β-actin.
Lentivirus-mediated short hairpin RNA (shRNA) silencing of FOXO1
FOXO1 shRNA lentiviral particles and non-targeting shRNA control particles were purchased from Sigma (St Louis, MO, USA). The sequence of the shRNA targeting FOXO1 used in the present study is the following: 5’-CCGGGCCTGTTATCAATCT-GCTAAACTCGAGTTTAGCAGATTGATAACAGGCTTTTTG-3’. The non-targeting shRNA control particles contain four base-pair mismatches within the short hairpin sequence to any known human or mouse gene. The viral infection was performed by incubating SNU-638 gastric cancer cells in the culture medium containing lentiviral particles for 12 h in the presence of 5 μg/mL Polybrene (Santa Cruz Biotechnology). Pooled puromycin (2 μg/mL)-resistant cells were harvested and stored for further analysis.
Luciferase reporter assay
Gastric cancer cells were seeded into 24-well plates at a density of 3 × 104 cells/well and were transiently co-transfected with 0.4 μg of forkhead responsive element (FHRE) -luciferase reporter plasmid (reporter construct in which a small region of the Fas ligand promoter containing the three FHREs, Addgene plasmid 1789) (Addgene Inco, Cambridge, MA, USA) and 0.4 μg of β-galactosidase vector, an internal control, using Lipofectamine Plus (Life Technologies). Twenty-four hours after transfection, assays for luciferase and β-galactosidase were carried out using a Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA). Luciferase activity was measured on an AutoLumat LB 9505c luminometer (Berthold Analytical Instruments, Nashua, Germany) and was normalized by β-galactosidase activity. Luciferase activity in control cells was arbitrarily set to 1.
Immunofluorescence staining
Cells were cultured on 4-well chamber slide (3 × 104 cells/chamber). After 24 h, cells were fixed with 4 % paraformaldehyde for 10 min and permeabilized with 0.5 % Triton X-100 for 5 min. After blocking with 5 % normal donkey serum for 5 min, cells were incubated overnight 4 °C with anti-pJNK (Cell Signaling Technology) and anti-FOXO1 (Cell Signaling Technology). Corresponding secondary antibody conjugated with Alexa Fluor-488 (green) or Alexa Fluor-555 (red) used for pJNK and FOXO1 respectively. For localization of nucleus, cells were counterstained with 4’,6-diamidino-2-phenylindole (DAPI) (Life Technologies) for 10 min. Immunofluorescence was detected under a fluorescence microscope (BX51; Olympus, Tokyo, Japan).
Colony formation assay
Gastric cancer cells (2.5 × 103 cells/well) were suspended in 0.3 % Bacto Agar (Sigma) in the RPMI1640 medium with 10 % FBS and overlaid onto a previously prepared 0.6 % Bacto Agar in 6-well culture plates (Thermo Scientific, Hudson, NH, USA). The agar plates were incubated with 1 mL of RPMI1640 medium, and medium was changed every 3 days. After 14 days of culture, surviving colonies were stained with 0.05 % crystal violet (Sigma) in 20 % methanol (Merck KGaA, Darmstadt, Germany). The number and size of colonies were determined using NIH Image Analysis software (version 1.46r; National Institutes of Health, Bethesda, MD, USA) as described previously [34]. For the inhibition of JNK activity, cells were cultured for 24 h in the media and treated with SP600125 (20 μM) for 14 days.
Statistical analysis
For tissue array analysis, statistical analyses were conducted using SPSS version 19.0 statistical software program (IBM, SPSS, Chicago, Ill., USA). To determine the significance of correlation between JNK activation and the clinicopathological factors or the other proteins, either the χ2 test or the Fisher’s exact test (two sided) was performed. Survival curves were estimated using the Kaplan-Meier product-limit method and the significance of differences between the survival curves was determined using the log-rank test. To determine whether JNK activation is an independent prognostic variable, multivariate survival analysis was performed using the Cox proportional hazard model. The relationship between JNK activation and the cell proliferation index was analyzed using the two-tailed Student’s t-test. For luciferase reporter assay and colony formation assay, data were analyzed using GraphPad Prism software for Windows Vista (version 4, San Diego, CA, USA) and the two-tailed Student’s t-test was used to determine the significances of the results. P values of < 0.05 were considered statistically significant for all statistical analyses.
Results
Immunohistochemical features
Figure 1 shows the representative findings of immunohistochemical tissue array analysis of 483 consecutive gastric carcinoma tissues. JNK activation was determined by immunostaining using an antibody against the active form of JNK (pJNK). In non-neoplastic gastric mucosa, pJNK (immuno)staining was observed throughout the nucleus of the proliferative zone of gastric glands (Fig. 1a). Nuclear pJNK staining was also detected in the areas of intestinal metaplasia with or without cytoplasmic staining (Fig. 1b). Gastric tumor cells showed pJNK staining mainly in the nucleus and/or in the cytoplasm (Fig. 1c), and nuclear staining was the criterion for pJNK [35, 36].
Fig. 1.
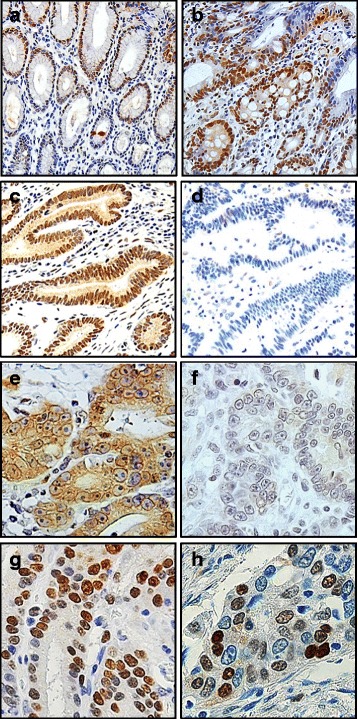
Representative immunohistochemical features of the gastric mucosa and cancer. Nuclear pJNK was expressed (a) in the proliferative zone of gastric mucosa and (b) in the areas of intestinal metaplasia. Gastric carcinomas displaying (c) positive or (d) negative expression of pJNK. Gastric carcinomas displaying (e) positive or (f) negative expression of pFOXO1. Gastric carcinoma cells showing nuclear expression of (g) Ki-67 or (h) cyclin D1. Original magnifications: ×100 (a-d) and × 400 (e-h)
For FOXO1, immunostaining was performed with an antibody against an inactivated form of FOXO1 phosphorylated at Ser 256 (pFOXO1). pFOXO1 staining was observed in the cytoplasm and, at times, in the nucleus of tumor cells (Fig. 1e). Tumor cells with cytoplasmic staining regardless of the nuclear staining were considered to exhibit FOXO1 inactivation [23]. For other proteins such as Ki-67 (Fig. 1g) and cyclin D1 (Fig. 1h), tumor cells with nuclear staining, regardless of cytoplasmic staining, were regarded as showing constitutive activation. For statistical analysis, immunohistochemical staining results were quantified as described in Methods.
JNK activation is associated with the clinicopathological factors in gastric cancer
In order to investigate the clinical significance of JNK activation in gastric cancer, the correlations between nuclear pJNK staining and the clinicopathologic features in 483 gastric cancer cases were analyzed (Table 2). Nuclear pJNK staining was found in 38 % of gastric cancer cases and was more likely to be found in the intestinal type by Lauren’s classification (P = 0.037). In addition, pJNK staining was more prevalent in the early pTNM stages (P < 0.001). Eighty-two percent of pJNK-positive tumors were in pTNM stages I and II, while 18 % were in stages III and IV. Moreover, pJNK staining inversely correlated with lymphatic invasion (P = 0.034). No association was found between JNK activation and age or gender.
Table 2.
Correlation between clinicopathological characteristics and pJNK expression in 483 gastric cancers
| pJNK | P | ||
|---|---|---|---|
| Positive (%) | Negative (%) | ||
| Total | 182 (38) | 301 (62) | |
| Age (years) | |||
| 0–39 | 13 (36) | 23 (64) | 0.432 |
| 40–65 | 103 (36) | 186 (64) | |
| 66–88 | 66 (42) | 92 (58) | |
| Gender | |||
| Male | 134 (40) | 203 (60) | 0.091 |
| Female | 48 (33) | 98 (67) | |
| Lauren’s classification | |||
| Intestinal | 104 (44) | 132 (56) | 0.037* |
| Diffuse | 62 (32) | 131 (68) | |
| Mixed | 16 (30) | 38 (70) | |
| pTNM stage | |||
| I | 127 (47) | 144 (53) | <0.001* |
| II | 22 (24) | 69 (76) | |
| III | 21 (34) | 41 (66) | |
| IV | 12 (20) | 47 (80) | |
| Lymphatic invasion | |||
| Absent | 113 (41) | 160 (59) | 0.034* |
| Present | 69 (33) | 141 (67) | |
pJNK phosphorylated c-Jun N-terminal kinase, TNM tumor-node metastasis
*Considered to be statistically significant (<0.05)
Patients with a pJNK-positive tumor showed a higher survival rate
To determine whether JNK activation is a significant prognostic factor for the survival of patients with gastric carcinoma, we used a log-rank test with Kaplan-Meier estimates. Of the 483 patients analyzed, those with nuclear pJNK staining (38 %) had a significantly higher survival rate than those with only cytoplasmic pJNK staining or negative staining (P = 0.008) (Fig. 2). Thus, JNK activation is a valuable biomarker of prognosis. However, multivariate Cox regression analysis including the pTNM stage revealed that pJNK staining was not an independent prognostic factor.
Fig. 2.
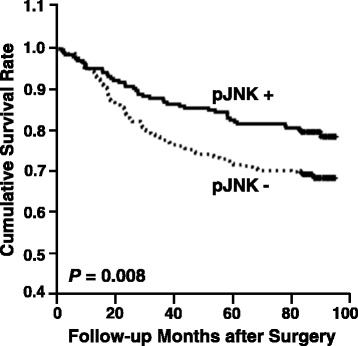
Kaplan-Meier survival curves for patient survival. Patients with a pJNK-positive tumor showed a more favorable prognosis than pJNK-negative carcinoma patients (P = 0.008)
JNK activation is positively associated with the tumor cell proliferation and growth
Tumor cell proliferation was evaluated by the immunohistochemical stainings for nuclear Ki-67 and cell cycle regulators, including cyclin D1 and cyclin E. Table 3 shows the results of the immunohistochemistry for Ki-67 performed on tissue array slides. pJNK staining positively correlated with the proliferation index (evaluated by Ki-67 labeling) (P < 0.001). Moreover, Table 4 shows positive correlations between pJNK and cyclin D1 (P = 0.045) or cyclin E (P < 0.001). Consistently, Western blotting of cultured SNU-638 cells showed that JNK inhibition by SP600125 treatment suppressed the protein expressions of pJNK and cyclin D1 in a dose-dependent manner (Fig. 3a). Taken together, our data from both human specimens and in vitro experiments suggested that JNK may promote gastric cancer cell proliferation through enhancement of cell cycle progression. Further, SP600125 treatment of gastric cancer cells suppressed colony formation of SNU-638 cells (Fig. 4a).
Table 3.
The expression of pJNK in relation to the proliferation index
| pJNK | P | ||
|---|---|---|---|
| Positive (n = 182) Mean ± SD (%) | Negative (n = 301) Mean ± SD (%) | ||
| Ki-67 (%) | 86.78 ± 51.34 | 54.65 ± 46.53 | <0.001* |
SD standard deviation
*Considered to be statistically significant (<0.05)
Table 4.
Correlation between the expression of pJNK and protumorigenic molecules in human gastric cancer
| pJNK | P | ||
|---|---|---|---|
| Positive (%) | Negative (%) | ||
| Cyclin D1 | |||
| Positive | 79 (42) | 107 (58) | 0.045* |
| Negative | 97 (34) | 186 (66) | |
| Cyclin E | |||
| Positive | 41 (58) | 30 (42) | <0.001* |
| Negative | 139 (34) | 266 (66) | |
*Considered to be statistically significant (<0.05)
Fig. 3.
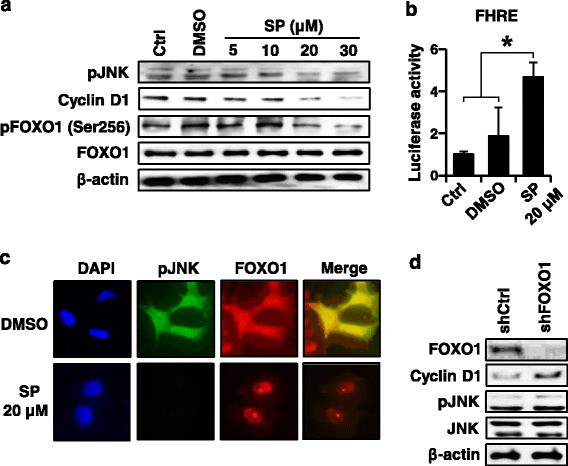
The effect of pharmacological inhibition of JNK on FOXO1 activation in cultured gastric cancer cells. (a-c). SNU-638 cells were cultured without treatment (Ctrl) or treated with DMSO vehicle control (DMSO) or SP600125 (SP) for 24 h. a Cells were treated with various concentrations of SP. Protein expressions were analyzed by Western blotting using specific antibodies against pJNKThr183/Tyr185 (pJNK), cyclin D1, pFOXO1Ser256, total FOXO1, and β-actin. b Cells were cultured in the presence or absence of SP (20 μM) and FOXO1 transcriptional activity was determined by luciferase reporter assay, which was normalized by β-galatosidase activity. Luciferase activity in Ctrl was arbitrarily set to 1 and the activities of other cells were adjusted accordingly. Each bar represents the mean ± standard deviation. * P < 0.05 compared with untreated control groups (Ctrl and DMSO). c The effect of JNK inhibition on the subcellular localization of FOXO1 was observed by double immunofluorescence staining for pJNK (green) and FOXO1 (red). 4’,6-diamidino-2-phenylindole (DAPI) staining was performed for nuclear localization (blue). d Cells were transfected with non-targeting shRNA (shCtrl) or FOXO1 shRNA (shFOXO1). The effects of FOXO1 silencing on the expressions of total FOXO1, cyclin D1, pJNK, and total JNK were evaluated by Western blotting
Fig. 4.
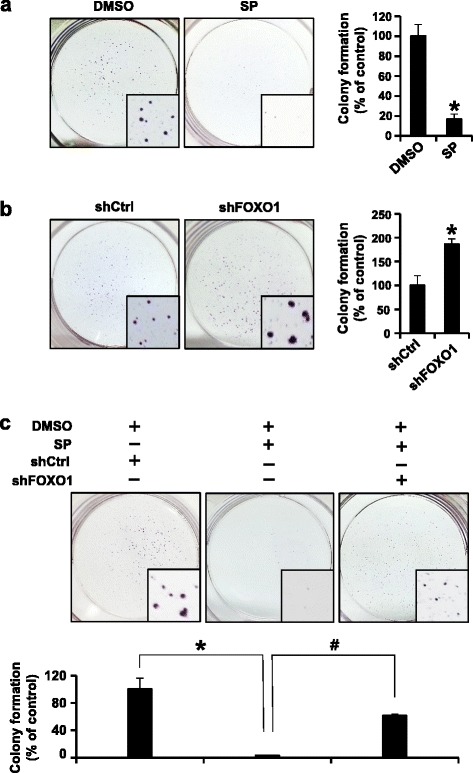
Role of JNK and FOXO1 in gastric tumor cell growth. SNU-638 cells were (a and c) cultured with or without 20 μM SP600125 (SP) for 14 days and/or (b and c) transfected with non-targeting shRNA (shCtrl) or FOXO1 shRNA (shFOXO1). Cell growth ability in vitro was evaluated by colony formation assay. Representative images of colonies are shown and the bar graphs represent the relative colony formation efficiencies. Percentage colony formations for control cells were arbitrarily set to 100 % and percentages for others were adjusted accordingly. a SP-treated cells showed a marked reduction in colony formation compared to DMSO control (* P = 0.013). b shFOXO1 transfection induced a significant enhancement in colony formation compared to shCtrl transfection (* P = 0.021). c SP treatment induced a marked suppression in colony formation compared to DMSO + shCtrl (* P = 0.01), and the combination with shFOXO1 transfection partially restored the colony formation compared to SP treatment (# P < 0.001). Each bar represents the mean ± standard deviation
Association between JNK and FOXO1 in gastric cancer specimens
To obtain a better understanding of the mechanism involved in the JNK-induced gastric tumor cell proliferation, we investigated the relationship between JNK and FOXO1. Immunohistochemical tissue array analysis showed that JNK activation was positively associated with FOXO1 inactivation in gastric cancer specimens, which indicates an inverse relationship between the activations of JNK and FOXO1 (P < 0.001) (Table 5). Consistently, FOXO1 inactivation, like JNK activation, was positively associated with the nuclear stainings for cyclin D1 (P = 0.003) and cyclin E (P < 0.001). In addition, FOXO1 staining in gastric cancer was not an independent prognostic factor.
Table 5.
Correlation between pFOXO1 expression and pJNK expression and protumorigenic molecules in gastric cancer
| pFOXO1 | P | ||
|---|---|---|---|
| Positive (%) | Negative (%) | ||
| pJNK | |||
| Positive | 169 (93) | 13 (7) | <0.001* |
| Negative | 206 (68) | 95 (32) | |
| Cyclin D1 | |||
| Positive | 157 (84) | 29 (16) | 0.003* |
| Negative | 208 (73) | 75 (27) | |
| Cyclin E | |||
| Positive | 68 (96) | 3 (4) | <0.001* |
| Negative | 301 (74) | 104 (26) | |
*Considered to be statistically significant (<0.05)
Pharmacological inhibition of JNK results in FOXO1 activation in gastric cancer cells
To further confirm the relationship between JNK and FOXO1, in vitro analysis was performed. First, SNU-638 gastric cancer cells were treated with a JNK inhibitor, SP600125. Western blotting (Fig. 3a) showed that SP600125 treatment of gastric cancer cells with a various concentrations resulted in a substantial decrease in the protein expression of the inactive form of FOXO1 (pFOXO1) in a dose dependent manner, whereas the protein expression of total FOXO1 was not changed. Consistently, luciferase reporter assay (Fig. 3b) showed that treatment of SNU-638 cells with SP600125 (20 μM) increased FOXO1 transcriptional activity compared to those without treatment (Ctrl and DMSO) with a statistical significance (P = 0.003 and P = 0.025, respectively).
Next, immunofluorescence stainings for pJNK and FOXO1 were performed to determine the effect of JNK inhibition on the subcellular localization of FOXO1 in SNU-638 cells (Fig. 3c). In the untreated DMSO control cells (upper row), immunofluorescence for pJNK (green) was shown both in the nucleus and the cytoplasm. After pharmacological inhibition of JNK with 20 μM SP600125 (lower row), gastric cancer cells showed negligible immunofluorescence for pJNK. On the other hand, immunofluorescence for FOXO1 was distributed both in the nucleus and the cytoplasm in control cells, whereas it was only accumulated in the nucleus of the SP600125-treated cells. Thus, it seems that JNK functions in the nucleus and that the translocation of FOXO1 from the nucleus to the cytoplasm was blocked by JNK inhibition. These findings are incompatible to previous reports in human cancer cells, which showed that JNK activation induced the nuclear translocation of FOXO1 proteins followed by an increase in FOXO transcriptional activity [22, 27–31].
In order to determine whether there is crosstalk between JNK and FOXO1, FOXO1 expression was silenced by transfection of FOXO1 shRNA into SNU-638 cells. Western blotting showed that FOXO1 silencing did not change protein expressions of pJNK and total JNK, whereas it increased cyclin D1 protein expression compared to control shRNA transfectants (Fig. 3d). Taken together, our data indicate that JNK acts as an inhibitory upstream molecule of FOXO1 in the FOXO1 pathway in gastric cancer cells, and that crosstalk between these molecules does not exist.
JNK induces gastric cancer cell growth through inhibition of FOXO1
The above results indicate that JNK activation in gastric cancer cells is associated with tumor cell proliferation and inhibition of FOXO1 activation. In order to determine the biological significance of the association between JNK and FOXO1 regarding tumorigenesis, colony formation assay was performed using SNU-638 cells. We found that SP600125 treatment markedly decreased colony formation (P = 0.013) (Fig. 4a), whereas FOXO1 silencing induced a significant increase in colony formation (P = 0.021) (Fig. 4b). In order to investigate whether JNK-induced colony formation is mediated by FOXO1 inactivation in gastric cancer cells, we used a combination of JNK inhibition and FOXO1 knockdown. In Fig. 4c, JNK inhibition markedly decreased colony formation (P = 0.01), and FOXO1 shRNA transfection combined with JNK inhibition substantially increased colony formation compared to JNK inhibition alone (P < 0.001). These results indicate that JNK activation, which induces gastric cancer cell growth, is mediated by FOXO1 inactivation. Thus, JNK/FOXO1 pathway seems to contribute to tumor growth of gastric cancer.
Combined status of pJNK-negative and pFOXO1-negative in relation to prognosis
We examined whether the combined status of pJNK and pFOXO1 is correlated with gastric cancer patient survival. Kaplan-Meier estimate and the log-rank test showed that patients with a pJNK-negative (JNK inactive) and pFOXO1-negative (FOXO1 active) tumor had significantly worse outcome than those with other combinations (P < 0.001) (Fig. 5a). Moreover, patients with a pJNK-positive (JNK active) and pFOXO1-postive (FOXO1 inactive) tumor had a higher survival rate than the remainder of the population (P = 0.004) (Fig. 5b).
Fig. 5.
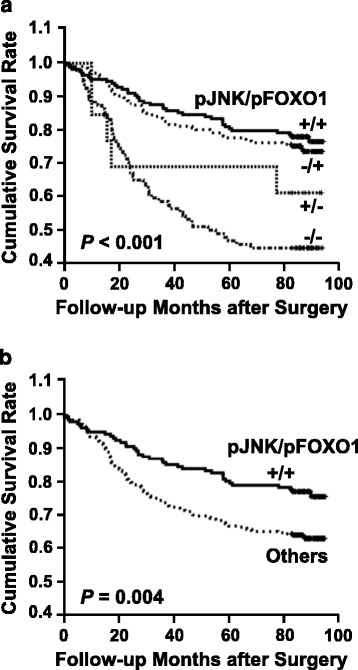
Kaplan-Meier survival curves for patient survival. a Patients with pJNK negative and pFOXO1-negative immunoreactivity showed the worst outcome (P < 0.001). b Patients with a pJNK-positive and pFOXO1-positive combination showed better outcome than the remainder of the population (P = 0.004)
Discussion
Although accumulating evidence supports that JNK activation is involved in cancer development and progression [37, 38], the biological significance of JNK in gastric cancer remains unclear. The present study showed that constitutive activation of JNK was associated with specific clinicopathological factors, including pTNM stages, lymphatic invasion, and a better prognosis. We believe that this is the first report regarding the clinical implications of JNK in human gastric cancer. Furthermore, we found that JNK negatively regulates FOXO1 activation in gastric cancer cells. This finding contrasts with the results of the previous studies [22, 27–31], which showed JNK-induced activation of FOXO proteins in human cancer cells.
In the present study, JNK activation (evaluated by pJNK staining) was mainly observed in the proliferative zone of the gastric gland and in the areas showing intestinal metaplasia, which is known to be a predictor of gastric neoplasia [39], in the non-neoplastic gastric mucosa. Since the intestinal metaplasia shows a higher proliferation index than the normal gastric mucosa [40], this staining pattern suggested a positive association between JNK activation and cell proliferation. Moreover, JNK activation in gastric cancer was positively correlated with the proliferation index (evaluated by Ki-67 labeling) and cell cycle-regulatory molecules such as cyclin D1 (P = 0.045) and cyclin E (P < 0.001), which are present more frequently in early-stage gastric carcinomas [41]. In cell culture, treatment of gastric cancer cells with a JNK inhibitor, SP600125, decreased cyclin D1 protein expression and colony formation. Taken together, these findings indicate growth-promoting activity of JNK in gastric cancer cells, which is a very important process in the gastric cancer promotion.
Previously, Shibata et al. [20] reported that JNK activation was found in 40 % of gastric cancer cases and that there was no association between JNK activation and Lauren’s classification. However, our results showed that JNK activation was found in 38 % of 483 gastric carcinoma cases and was more likely to be found in the intestinal type as determined by Lauren’s classification (P = 0.037). We speculate that these discrepancies between the results of Shibata et al. and ours may, at least in part, come from differences in the numbers of tumor cases analyzed (n = 40 versus n = 483 in the present study). Moreover, we found that JNK activation in gastric cancer was more prominent in early-stage pTNM tumors than in late-stage pTNM tumors (P < 0.001), and was negatively associated with lymphatic invasion (P = 0.034). Thus, it seems that JNK activation, at least in part, is required for cell proliferation and growth in early stage gastric carcinomas, which do not usually show lymphatic invasion.
Although previous studies suggested that FOXO1 inactivation may contribute to the development of gastric cancer [23–25], information on the molecular mechanisms underlying FOXO1 activation in gastric cancer is limited. Regarding the association between FOXO1 and JNK, there has been only one study by Ju et al. [22], which showed that JNK induced FOXO1 activation in lung cancer cells. In the present study, immunohistochemical tissue array analysis of gastric cancer specimens showed that JNK activation was positively correlated with FOXO1 inactivation. This relationship was further confirmed by in vitro analyses using a JNK inhibitor, SP600126. JNK inhibition in SNU-638 cells increased FOXO1 activation and suppressed the nuclear export of FOXO1, which suggested that FOXO1 is a nuclear substrate of JNK in gastric cancer cells. However, crosstalk between JNK and FOXO1 was not observed, because FOXO1 silencing did not affect the protein expression and activity of JNK. Thus, it seems that JNK is an upstream regulatory molecule of FOXO1, which increases the nuclear export and subsequent inactivation of FOXO1 (Fig. 6). These findings contrast with a previous report by Ju et al. [22]. Thus, we speculate that the relationship between these two molecules could be cell type-specific.
Fig. 6.
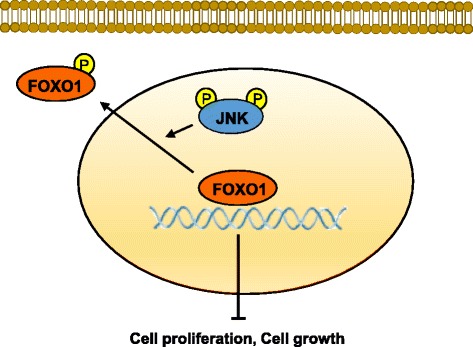
Schematic diagram showing the potential mechanism of cancer cell proliferation and growth regulated by the JNK/FOXO1 pathway in gastric cancer. As depicted, JNK activation induces cancer cell proliferation and growth by suppressing the transcriptional activity of FOXO1. When JNK is activated by phosphorylation, FOXO1 is phosphorylated, leading to its nuclear export and inactivation
Additionally, we found that colony formation of gastric cancer cells was decreased by pharmacological inhibition of JNK, whereas it was increased by FOXO1 shRNA transfection. Further, the combination of JNK inhibition and FOXO1 silencing partially restored the colony forming capability of gastric cancer cells compared to JNK inhibition alone. These results indicate that JNK activation, at least in part, induces gastric cancer cell growth through the inhibition of FOXO1. Since Altan et al. [42] reported that FOXO1 downregulation in a subset of gastric cancer cell lines inhibited 5-fluorouracil sensitivity, a JNK inhibitor may be used to restore the chemosensitivity to 5-fluorouracil through FOXO1 activation in the treatment of those gastric cancer patients.
Our results in the present study showed that JNK activation in gastric cancer specimens was positively correlated with early pTNM stages and with a better outcome. Similarly, FOXO1 inactivation was previously shown to be an early event and was positively correlated with better prognosis in gastric cancer [23]. Since gastric cancer specimens showed a positive correlation between JNK activation and FOXO1 inactivation in the present study, the combined status of JNK activation and FOXO1 inactivation was assessed in relation to survival. We found that patients with a tumor showing JNK activation and FOXO1 inactivation had a higher survival rate than those with other combinations (P = 0.004).
Tissue array method was used in the present study to evaluate the clinical significance of JNK activation and its association with FOXO1 inactivation. The potential limitations of tissue array method are mainly associated with the acquisition of information from only a tiny area in each tumor. In order to address the influence of tumor heterogeneity and to evaluate the ability of the tissue array method to yield information on the prognostic value of biomarkers, multiple replicate tissue array blocks had been constructed by several researchers [33, 43, 44]. In all of these studies, the data from each replica array showed an excellent agreement, i.e. they were almost identical within the statistical level of significance, and the prognostic associations of the markers were always as good as or better, when measured from the tissue array slides, than the analysis of individual large sections. Thus, it has been suggested that the effects of intra-tumoral heterogeneity were averaged out in such large scale analysis. Indeed, the tissue array method is used for population-level research to find molecular targets with therapeutic significance, but not for making clinical diagnoses of individual cases. In addition, core sampling from different tumor blocks from the same patient, perhaps including metastatic sites, are being used to compensate these limitations [45].
Conclusions
Our results indicate that JNK activation, alone or along with FOXO1 inactivation, is a candidate prognostic marker for the early gastric carcinoma and positively associated with pro-proliferation genes and FOXO1 inactivation. Since JNK activation increased gastric cancer cell growth through inhibition of FOXO1, combination of targeting JNK/FOXO1 pathway may further reduce gastric cancer cell growth. In-vivo experiments using this association as a potential target for anti-cancer therapy are needed.
Abbreviations
DMSO, dimethylsulfoxide; FBS, fetal bovine serum; FHRE, forkhead responsive element; FOXO, subclass O of forkhead transcription factors; JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; PBS, phosphate-buffered saline; pFOXO1, inactive form of FOXO1 phosphorylated at Ser256; pJNK, active form of JNK phosphorylated at Thr183 and Tyr185; pTNM, pathological tumor-node-metastasis; SDS, sodium dodecyl sulfate; shRNA, short hairpin RNA
Acknowledgements
We thank Superbiochips (Seoul, Korea) for the preparation of tissue array slides.
Funding
This work was supported by grant NO 04-2014-0300 from the SNUH Research Fund and the Education and Research Encouragement Fund of Seoul National University Hospital (2016).
Availability of data and materials
All the data supporting our findings are contained within the present manuscript.
Authors’ contributions
BLL has made substantial contributions to conception, design and drafting the manuscript and revising it critically for important intellectual content. YC and JP have made substantial contributions to acquisition of data, and analysis and interpretation of data, as well as have been involved in drafting the manuscript. YC, YSK, DAY, YK and JSP have made substantial contributions to acquisition of data, and analysis of data. BGJ, MAK and WHK have made substantial contributions to analysis and interpretation of data. All authors have given final approval of the version to be published.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
This study was approved by the Institutional Review Board of Seoul National University (IRB No. 1309-087-522) and also it was exempted from patient’s consent because of the following reasons: Most of the patients already died or are not visiting hospital anymore. This study will not cause hazard or exposure of personal information and will be used only for academic purposes.
References
- 1.Edge SB, Byrd DR, Campton CC, Fritz AG, Greene FL, Trotti A. AJCC Cancer Staging Manual. 7. New York: Springer; 2009. pp. 117–26. [Google Scholar]
- 2.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 3.Ko YS, Cho SJ, Park J, Kim Y, Choi YJ, Pyo JS, et al. Loss of FOXO1 promotes gastric tumor growth and metastasis through upregulation of human epidermal growth factor receptor 2/neu expression. Br J Cancer. 2015;113:1186–96. doi: 10.1038/bjc.2015.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee HE, Kim MA, Lee HS, Jung EJ, Yang HK, Lee BL, et al. MET in gastric carcinomas: comparison between protein expression and gene copy number and impact on clinical outcome. Br J Cancer. 2002;107:325–33. doi: 10.1038/bjc.2012.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kang YH, Lee HS, Kim WH. Promoter methylation and silencing of PTEN in gastric carcinoma. Lab Invest. 2002;82:285–91. doi: 10.1038/labinvest.3780422. [DOI] [PubMed] [Google Scholar]
- 6.Tamura G. Alterations of tumor suppressor and tumor-related genes in the development and progression of gastric cancer. World J Gastroenterol. 2006;12:192–8. doi: 10.3748/wjg.v12.i2.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim HP, Han SW, Song SH, Jeong EG, Lee MY, Hwang D, et al. Testican-1-mediated epithelial-mesenchymal transition signaling confers acquired resistance to lapatinib in HER2-positive gastric cancer. Oncogene. 2014;33:3334–41. doi: 10.1038/onc.2013.285. [DOI] [PubMed] [Google Scholar]
- 8.Bubici C, Papa S. JNK signaling in cancer: in need of new, smarter therapeutic targets. Br J Pharmacol. 2014;171:24–37. doi: 10.1111/bph.12432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mingo-Sion AM, Marietta PM, Koller E, Wolf DM, Van Den Berg CL. Inhibition of JNK reduces G2/M transit independent of p53, leading to endoreduplication, decreased proliferation, and apoptosis in breast cancer cells. Oncogene. 2004;23:596–604. doi: 10.1038/sj.onc.1207147. [DOI] [PubMed] [Google Scholar]
- 10.Yang YM, Bost F, Charbono W, Dean N, McKay R, Rhim JS, et al. C-Jun NH(2)-terminal kinase mediates proliferation and tumor growth of human prostate carcinoma. Clin Cancer Res. 2003;9:391–401. [PubMed] [Google Scholar]
- 11.She QB, Chen N, Bode AM, Flavell RA, Dong Z. Deficiency of c-Jun-NH(2)-terminal kinase-1 in mice enhances skin tumor development by 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 2002;62:1343–8. [PubMed] [Google Scholar]
- 12.Ono R, Matsuoka J, Yamatsuji T, Naomoto Y, Tanaka N, Matsui H, et al. M-RIP, a novel target of JNK signaling and a requirement for human cancer cell invasion. Int J Mol Med. 2008;22:199–203. [PubMed] [Google Scholar]
- 13.Chen Y, Wu Q, Song SY, Su WJ. Activation of JNK by TPA promotes apoptosis via PKC pathway in gastric cancer cells. World J Gastroenterol. 2002;8:1014–8. doi: 10.3748/wjg.v8.i6.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu K, Zhao Y, Li GC, Yu WP. c-Jun N-terminal kinase is required for vitamin E succinate-induced apoptosis in human gastric cancer cells. World J Gastroenterol. 2004;10:1110–4. doi: 10.3748/wjg.v10.i8.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao Y, Zhao X, Yang B, Neuzil J, Wu K. α-Tocopheryl succinate-induced apoptosis in human gastric cancer cells is modulated by ERK1/2 and c-Jun N-terminal kinase in a biphasic manner. Cancer Lett. 2007;247:345–52. doi: 10.1016/j.canlet.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 16.Kannaiyan R, Manu KA, Chen L, Li F, Rajendran P, Subramaniam A, et al. Celastrol inhibits tumor cell proliferation and promotes apoptosis through the activation of c-Jun N-terminal kinase and suppression of PI3 K/Akt signaling pathways. Apoptosis. 2011;16:1028–41. doi: 10.1007/s10495-011-0629-6. [DOI] [PubMed] [Google Scholar]
- 17.Xiao F, Liu B, Zhu QX. c-Jun N-terminal kinase is required for thermotherapy-induced apoptosis in human gastric cancer cells. World J Gastroenterol. 2012;18:7348–56. doi: 10.3748/wjg.v18.i48.7348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan JY, Tian FM, Hu WN, Zhang JH, Cai HF, Li N. Apoptosis of human gastric cancer cells line SGC 7901 induced by garlic-derived compound S-allylmercaptocysteine (SAMC) Eur Rev Med Pharmacol Sci. 2013;17:745–51. [PubMed] [Google Scholar]
- 19.Xia HH, He H, De Wang J, Gu Q, Lin MC, Zou B, et al. Induction of apoptosis and cell cycle arrest by a specific c-Jun NH2-terminal kinase (JNK)inhibitor, SP-600125, in gastrointestinal cancers. Cancer Lett. 2006;241:268–74. doi: 10.1016/j.canlet.2005.10.031. [DOI] [PubMed] [Google Scholar]
- 20.Shibata W, Maeda S, Hikiba Y, Yanai A, Sakamoto K, Nakagawa H, et al. c-Jun NH2-terminal kinase 1 is a critical regulator for the development of gastric cancer in mice. Cancer Res. 2008;68:5031–9. doi: 10.1158/0008-5472.CAN-07-6332. [DOI] [PubMed] [Google Scholar]
- 21.Arden KC. Multiple roles of FOXO transcription factors in mammalian cells point to multiple roles in cancer. Exp Gerontol. 2006;41:709–17. doi: 10.1016/j.exger.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 22.Ju Y, Xu T, Zhang H, Yu A. FOXO1-dependent DNA damage repair is regulated by JNK in lung cancer cells. Int J Ocol. 2014;44:1284–92. doi: 10.3892/ijo.2014.2269. [DOI] [PubMed] [Google Scholar]
- 23.Kim JH, Kim MK, Lee HE, Cho SJ, Cho YJ, Lee BL, et al. Constitutive phosphorylation of the FOXO1A transcription factor as a prognostic variable in gastric cancer. Mod Pathol. 2007;20:835–42. doi: 10.1038/modpathol.3800789. [DOI] [PubMed] [Google Scholar]
- 24.Li F, Liu B, Gao Y, Liu Y, Xu Y, Tong W, et al. Upregulation of microRNA-107 induces proliferation in human gastric cancer cells by targeting the transcription factor FOXO1. FEBS Lett. 2014;588:538–44. doi: 10.1016/j.febslet.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 25.Kim SY, Ko YS, Park J, Choi Y, Park JW, Kim Y, et al. Forkhead transcription factor FOXO1 Inhibits angiogenesis in gastric cancer in relation to SIRT1. Cancer Res Treat. 2016;46:345–54. doi: 10.4143/crt.2014.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maaike CW, van den Berg MC, van Gogh IJ, Smits AM, van Triest M, Dansen TB, et al. The small GTPase RALA controls c-Jun N-terminal kinase-mediated FOXO activation by regulation of a JIP1 scaffold complex. J Biol Chem. 2013;288:21729–41. doi: 10.1074/jbc.M113.463885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Essers MA, Weijzen S, de Vries-Smits AM, Saarloos I, de Ruiter ND, Bos JL, et al. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 2004;23:4802–12. doi: 10.1038/sj.emboj.7600476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kwon IK, Wang R, Thangaraju M, Shuang H, Liu K, Dashwood R, et al. PKG inhibits TCF signaling in colon cancer cells by blocking β-catenin expression and activating FOXO4. Oncogene. 2010;29:3423–34. doi: 10.1038/onc.2010.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karger S, Weidinger C, Krause K, Sheu SY, Aigner T, Gimm O, et al. FOXO3a: a novel player in thyroid carcinogenesis? Endocr Relat Cancer. 2009;16:189–99. doi: 10.1677/ERC-07-0283. [DOI] [PubMed] [Google Scholar]
- 30.de Keizer PL, Packer LM, Szypowska AA, Riedl-Polderman PE, van den Broek NJ, de Bruin A, et al. Activation of forkhead box O transcription factors by oncogenic BRAF promotes p21cip1-dependent senescence. Cancer Res. 2010;70:8526–36. doi: 10.1158/0008-5472.CAN-10-1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Chen WR, Xing D. A pathway from JNK through decreased ERK and Akt activities for FOXO3a nuclear translocation in response to UV irradiation. J Cell Physiol. 2012;227:1168–78. doi: 10.1002/jcp.22839. [DOI] [PubMed] [Google Scholar]
- 32.Lee HS, Lee HK, Kim HS, Yang HK, Kim WH. Tumour suppressor gene expression correlates with gastric cancer prognosis. J Pathol. 2003;200:39–46. doi: 10.1002/path.1288. [DOI] [PubMed] [Google Scholar]
- 33.Lee HS, Lee HK, Kim HS, Yang HK, Kim YI, Kim WH. MUC1, MUC2, MUC5AC, and MUC6 expressions in gastric carcinomas. Cancer. 2001;92:1427–34. doi: 10.1002/1097-0142(20010915)92:6<1427::AID-CNCR1466>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 34.Takamaru H, Yamamoto E, Suzuki H, Nojima M, Maruyama R, Yamano HO, et al. Aberrant methylation of RASGRF1 is associated with an epigenetic field defect and increased risk of gastric cancer. Cancer Prev Res (Phila) 2012;5:1203–12. doi: 10.1158/1940-6207.CAPR-12-0056. [DOI] [PubMed] [Google Scholar]
- 35.Rojo F, Gonzalez-Navarrete I, Bragado R, Dalmases A, Menendez S, Cortes-Sempere M, et al. Mitogen-activated protein kinase phosphatase-1 in human breast cancer independently predicts prognosis and is repressed by doxorubicin. Clin Cancer Res. 2009;15:3530–9. doi: 10.1158/1078-0432.CCR-08-2070. [DOI] [PubMed] [Google Scholar]
- 36.Matsuyoshi S, Shimada K, Nakamura M, Ishida E, Konishi N. Bcl-2 phosphorylation has pathological significance in human breast cancer. Pathobiology. 2006;73:205–12. doi: 10.1159/000096022. [DOI] [PubMed] [Google Scholar]
- 37.Wang Z, Ma B, Li H, Xiao X, Zhou W, Liu F et al. Protein 4.1N acts as a potential tumor suppressor linking PP1 to JNK-c-Jun pathway regulation in NSCLC. Oncotarget. 2015; [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- 38.Wang SY, Gao K, Deng DL, Cai JJ, Xiao ZY, He LQ et al. TLE4 promotes colorectal cancer progression through activation of JNK/c-Jun signaling pathway. Oncotarget. 2015; [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- 39.Tava F, Luinetti O, Ghigna MR, Alvisi C, Perego M, Trespi E, et al. Type or extension of intestinal metaplasia and immature/atypical “indefinite-for-dysplasia” lesions as predictors of gastric neoplasia. Hum Pathol. 2006;37:1489–97. doi: 10.1016/j.humpath.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 40.Lee KM, Lee DS, Yang JM, Ahn BM, Lee EH, Yoo JY, et al. Effect of Helicobacterpylori on gastric epithelial cell kinetics and expression of apoptosis-related proteins in gastric carcinogenesis. Korean J Gastroenterol. 2003;42:12–9. [PubMed] [Google Scholar]
- 41.Lee KH, Lee HE, Cho SJ, Cho YJ, Lee HS, Kim JH, et al. Immunohistochemical analysis of cell cycle-related molecules in gastric carcinoma: prognostic significance, correlation with clinicopathological parameters, proliferation and apoptosis. Pathobiology. 2008;75:364–72. doi: 10.1159/000164221. [DOI] [PubMed] [Google Scholar]
- 42.Altan B, Yokobori T, Ide M, Mochiki E, Toyomasu Y, Kogure N et al. Nuclear PRMT1 expression is associated with poor prognosis and chemosensitivity in gastric cancer patients. Gastric Cancer. 2015; [Epub ahead of print] [DOI] [PubMed]
- 43.Nocito A, Kononen J, Kallioniemi OP, Sauter G. Tissue microarrays (TMAs) for high-throughput molecular pathology research. Int J Cancer. 2001;94:1–5. doi: 10.1002/ijc.1385. [DOI] [PubMed] [Google Scholar]
- 44.Torhorst J, Bucher C, Kononen J, Haas P, Zuber M. Kochli OR et al. Tissue microarrays for rapid linking of molecular changes to clinical endpoints. Am J Pathol. 2001;159:2249–56. doi: 10.1016/S0002-9440(10)63075-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kallioniemi OP, Wagner U, Kononen J, Sauter G. Tissue microarray technology for high-throughput molecular profiling of cancer. Hum Mol Genet. 2001;10:657–62. doi: 10.1093/hmg/10.7.657. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All the data supporting our findings are contained within the present manuscript.