

Abstract
Background
The SMXA-5 mouse is an animal model of high-fat diet-induced fatty liver. The major QTL for fatty liver, Fl1sa on chromosome 12, was identified in a SM/J × SMXA-5 intercross. The SMXA-5 genome consists of the SM/J and A/J genomes, and the A/J allele of Fl1sa is a fatty liver-susceptibility allele. The existence of the responsible genes for fatty liver within Fl1sa was confirmed in A/J-12SM consomic mice. The aim of this study was to identify candidate genes for Fl1sa, and to investigate whether the identified genes affect the lipid metabolism.
Results
A/J-12SM mice showed a significantly lower liver triglyceride content compared to A/J mice when fed the high-fat diet for 7 weeks. We detected differences in the accumulation of liver lipids in response to the high-fat diet between A/J and A/J-12SM consomic mice. To identify candidate genes for Fl1sa, we performed DNA microarray analysis using the livers of A/J-12SM and A/J mice fed the high-fat diet. The mRNA levels of three genes (Iah1, Rrm2, Prkd1) in the chromosomal region of Fl1sa were significantly different between the strains. Iah1 mRNA levels in the liver, kidney, and lung were significantly higher in A/J-12SM mice than in A/J mice. The hepatic Iah1 mRNA level in A/J-12SM mice was 3.2-fold higher than that in A/J mice. To examine the effect of Iah1 on hepatic lipid metabolism, we constructed a stable cell line expressing the mouse Iah1 protein in mouse hepatoma Hepa1-6 cells. Overexpression of Iah1 in Hepa1-6 cells suppressed the mRNA levels of Cd36 and Dgat2, which play important roles in triglyceride synthesis and lipid metabolism.
Conclusions
These results demonstrated that Fl1sa on the proximal region of chromosome 12 affected fatty liver in mice on a high-fat diet. Iah1 (isoamyl acetate-hydrolyzing esterase 1 homolog) was identified as one of the candidate genes for Fl1sa. This study revealed that the mouse Iah1 gene regulated the expression of genes related to lipid metabolism in the liver.
Electronic supplementary material
The online version of this article (doi:10.1186/s12863-016-0385-2) contains supplementary material, which is available to authorized users.
Keywords: Genetics, Nutrition, Liver, Lipids, Fatty acids, CD36, Consomic, High-fat diet
Background
Fatty liver is associated with dyslipidemia, type 2 diabetes mellitus, and obesity. The SMXA-5 mouse is an animal model of high fat diet (HFD)-induced type 2 diabetes and fatty liver [1]. The SMXA-5 strain is an SMXA-recombinant inbred (RI) strain established from breeding between SM/J mice and A/J mice [2], and thus the SMXA-5 genome is a mosaic genome derived from SM/J mice and A/J mice. On a commercial chow diet, SM/J mice and A/J mice did not show hyperglycemia and impaired glucose tolerance (IGT), but SMXA-5 mice showed IGT [3]. On a high-carbohydrate diet, SM/J mice and A/J mice were resistant to IGT and fatty liver compared with SMXA-5 mice [1]. On the HFD, although the traits were deteriorated in all strains, both parental strains were without either diabetes or fatty liver. But SMXA-5 mice developed type 2 diabetes and fatty liver on the HFD. It was speculated that the parental strains possess the latent susceptibility loci for type 2 diabetes and fatty liver [1]. We previously performed a search of genetic factors for fatty liver in SMXA-5 mice by quantitative trait locus (QTL) analysis in (SM/J × SMXA-5)F2 intercross mice [4]. We detected several QTLs for liver weight, liver total lipids, liver total cholesterol (TC), and liver triglycerides (TG) on mouse chromosomes 2, 6, 10, 11, 12, and 17. On chromosome 12, a significant QTL for liver TG and highly significant QTLs for relative liver weight/liver lipid content were detected near D12Mit58 (17.1 Mb) and D12Mit270 (32.3 Mb), respectively. Chromosome 12 of SMXA-5 had the A/J-derived genome from the centromere to 54.1 Mb, and the SM/J-derived genome from 54.1 Mb to the telomere. The major QTL for fatty liver on chromosome 12 was designated Fl1sa (fattyliver1in theSMXARI strains). The A/J allele of Fl1sa (on centromere-54.1 Mb) contributed to an increase in lipid accumulation in the liver. The effect of Fl1sa was confirmed in A/J-12SM chromosomal substitution (consomic) mice [4]. On the HFD, A/J-12SM consomic mice that possessed the SM/J allele of Fl1sa showed lower lipid accumulation in the liver than A/J mice.
In this study, we first attempted to determine the time period when the differences in liver TG accumulation between A/J mice and A/J-12SM mice appeared. The effect of the responsible gene(s) for Fl1sa that existed on the A/J-derived region of chromosome 12 (centromere-54.1 Mb) in SMXA-5 emerged after 7 weeks of feeding with the HFD. Secondly, we tried to identify candidate genes involved in the Fl1sa-regulated control of lipid accumulation in the liver by DNA microarray analysis. We identified only three genes that had significantly different expression levels in the liver between A/J-12SM and A/J mice. The function of one of these candidate genes, mammalian Iah1, had not been reported, although the yeast IAH1 protein is known to show esterase activity [5, 6]. Thirdly, to clarify the function of mouse Iah1, we investigated lipid metabolism in the cells stably overexpressing the mouse Iah1 gene.
Results
Phenotypic analyses of A/J-12SM consomic mice at 3, 7, or 11 weeks of feeding with the HFD (Fig. 1 and Table 1)
Fig. 1.
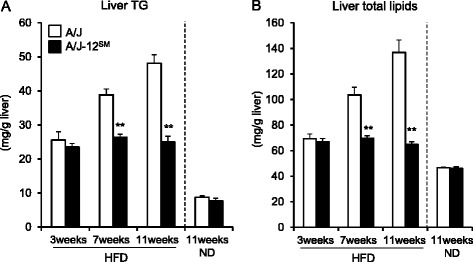
Liver TG and total lipids of A/J mice and A/J-12SM consomic mice at 3, 7, or 11 weeks of feeding with the high-fat diet. a Liver TG and (b) total lipids of A/J mice and A/J-12SM mice at 3, 7, or 11 weeks of feeding with the HFD. ND, 11 weeks of feeding with the ND (n = 8–10; **P < 0.01 versus A/J mice under the same experimental conditions)
Table 1.
Body weight, body mass index, food intake, and body composition of A/J and A/J-12SM strains fed the high-fat diet for 7 weeks
| A/J (n = 9) | A/J-12SM (n = 10) | P-value | |
|---|---|---|---|
| Initial body weight (g)a | 21.6 ± 0.4 | 21.3 ± 0.4 | NS |
| Final body weight (g)b | 36.2 ± 1.0 | 34.4 ± 0.7 | NS |
| Body mass index (g/cm2) | 0.311 ± 0.006 | 0.301 ± 0.006 | NS |
| Food intake (g/week) | 20.3 ± 0.3 | 20.0 ± 0.3 | NS |
| Weights of tissue (g/100 g bw) | |||
| Liver | 4.00 ± 0.06 | 3.55 ± 0.08** | 0.0003 |
| Subcutaneous fatc | 3.30 ± 0.19 | 3.21 ± 0.15 | NS |
| Epididymal fat | 5.21 ± 0.14 | 5.56 ± 0.24 | NS |
| Retroperitoneal fat | 1.45 ± 0.09 | 1.39 ± 0.09 | NS |
| Mesenteric fat | 2.75 ± 0.14 | 2.47 ± 0.12 | NS |
| Blood glucose (mg/dl) | 224.5 ± 5.5 | 192.7 ± 7.8** | 0.0047 |
| Serum insulin (ng/ml) | 1.81 ± 0.23 | 1.08 ± 0.12* | 0.0109 |
Each value is expressed as the mean ± SEM
*P < 0.05, **P < 0.01, significant difference from the value of A/J by Student’s t-test
aInitial body weight was measured at 6 weeks of age (at 0 weeks of feeding with the HFD)
bFinal body weight was measured at 13 weeks of age (at 7 weeks of feeding with the HFD)
cSubcutaneous fat was defined as fat pads below the root of the forefoot on one side of the body
NS not significant
At 11 weeks of feeding with the normal diet (ND), the body weights (A/J, 29.3 ± 0.6 g; A/J-12SM, 27.4 ± 0.9 g), serum TG levels (A/J, 101 ± 8 mg/dl; A/J-12SM, 119 ± 7 mg/dl) and serum TC levels (A/J, 100 ± 3 mg/dl; A/J-12SM, 106 ± 7 mg/dl) were similar between A/J mice and A/J-12SM mice. Liver TG and liver total lipids in A/J-12SM mice were significantly lower than those in A/J mice at 11 weeks of feeding with the HFD (Fig. 1a and b). These results were consistent with our previous study [4]. However, there were no significant differences in liver TG and liver total lipids between A/J mice and A/J-12SM mice at 11 weeks of feeding with the ND (Fig. 1a and b). The levels of liver total lipids in mice fed the ND were markedly lower than those in mice fed the HFD. These results indicate that Fl1sa, the locus for liver lipids accumulation on chromosome 12, affected HFD-induced fatty liver. Therefore, we attempted to determine the time period when the differences of liver TG accumulation between A/J mice and A/J-12SM mice appeared. Fl1sa was detected in (SM/J × SMXA-5)F2 intercross mice at 7 weeks of feeding with the HFD [4]. In this study, we analyzed the traits in A/J-12SM mice and A/J mice at 3 or 7 weeks of feeding with the HFD. Feeding with the HFD increased the content of liver TG and liver total lipids compared to feeding with the ND (Fig. 1a and b), but there were no differences between A/J mice and A/J-12SM mice at 3 weeks of feeding with the HFD. The levels of liver TG and liver total lipids in A/J mice increased from 3 to 7 weeks of feeding, but not in A/J-12SM mice. At 7 weeks of feeding, A/J-12SM mice had significantly lower contents of liver TG and of liver total lipids than A/J mice. At 7 weeks of feeding with the HFD, the body weight, body mass index, total food intake, and the weights of each type of adipose tissues (subcutaneous fat, epididymal fat, retroperitoneal fat, and mesenteric fat) did not differ between A/J mice and A/J-12SM mice (Table 1). The liver weight in A/J-12SM mice was significantly lower than that in A/J mice. In addition, the blood glucose and serum insulin concentrations in A/J-12SM mice were lower than those in A/J mice (Table 1). Serum TG, serum HDL-C, and serum free fatty acids (FFA) concentrations in A/J-12SM mice were similar to those in A/J mice (Fig. 2a and b). However, the serum TC level in A/J-12SM mice was lower than that in A/J mice (Fig. 2a).
Fig. 2.
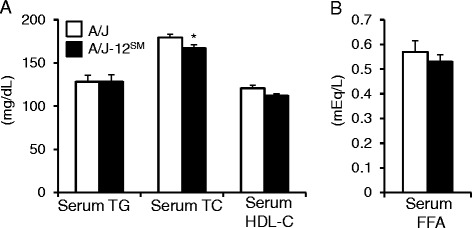
Serum lipids of A/J mice and A/J-12SM consomic mice at 7 weeks of feeding with the high-fat diet. a Serum TG, TC, HDL-C, and (b) serum FFA levels of A/J and A/J-12SM mice at 7 weeks of feeding with the HFD (n = 8–10; *P < 0.05 versus A/J mice)
Hepatic gene expression analyses between A/J and A/J-12SM consomic mice (Table 2 and Fig. 3a)
Table 2.
Genes differentially expressed between the livers of A/J mice and A/J-12SM mice on chromosome 12
| Probe ID | Gene symbol | Description | Position (bp) | GenBank | RefSeq | P-value | Fold change |
|---|---|---|---|---|---|---|---|
| 1454898_s_at | Iah1 | isoamyl acetate-hydrolyzing esterase 1 homolog (S. cerevisiae) | 21316392–21323605 | AU016407 | NM_026347 | 0.00020 | 4.803 |
| 1459274_at | Gpr135 | G protein-coupled receptor 135 | 72069618–72070991 | AV221890 | NM_181752 | 0.00029 | 2.016 |
| 1429119_at | Iah1 | isoamyl acetate-hydrolyzing esterase 1 homolog (S. cerevisiae) | 21316392–21323605 | AK005287 | NM_026347 | 0.00059 | 2.899 |
| 1416951_a_at | Atp6v1d | ATPase, H+ transporting, V1 subunit D | 78842989–78861638 | NM_023721 | NM_023721 | 0.00157 | 0.492 |
| 1443933_at | Tc2n | tandem C2 domains, nuclear | 101645443–101718523 | BB548141 | NM_028924 | 0.00163 | 0.371 |
| 1437608_x_at | Ywhaq | tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, theta polypeptide | 21390071–21417637 | BB414446 | NM_011739 | 0.00183 | 3.096 |
| 1430536_a_at | Erh | enhancer of rudimentary homolog (Drosophila) | 80634022–80644341 | BB071632 | NM_007951 | 0.00218 | 0.827 |
| 1439045_x_at | Tc2n | tandem C2 domains, nuclear | 101645443–101718523 | AV376747 | NM_028924 | 0.00245 | 0.038 |
| 1421139_a_at | Zfp386 | zinc finger protein 386 (Kruppel-like) | 116047724–116063360 | NM_019565 | NM_001004066 ; NM_019565 | 0.00267 | 0.292 |
| 1434437_x_at | Rrm2 | ribonucleotide reductase M2 | 24708241–24714146 | AV301324 | NM_009104 | 0.00340 | 0.546 |
| 1446643_at | 5330409N07Rik | RIKEN cDNA 5330409 N07 gene | 98444816–98448333 | BB022219 | AK030416 | 0.00422 | 1.936 |
| 1418587_at | Traf3 | Tnf receptor-associated factor 3 | 111166370–111267153 | U21050 | NM_011632 | 0.00423 | 0.861 |
| 1420491_at | Eif2s1 | eukaryotic translation initiation factor 2, subunit 1 alpha | 78861819–78887010 | BC016497 | NM_026114 | 0.00473 | 4.125 |
| 1428810_at | 2700097O09Rik | RIKEN cDNA 2700097O09 gene | 55045661–55080110 | AK012621 | NM_028314 | 0.00573 | 1.337 |
| 1447623_s_at | Prkd1 | protein kinase D1 | 50341231–50649223 | AV297026 | NM_008858 | 0.00660 | 0.413 |
| 1449110_at | Rhob | ras homolog gene family, member B | 8497763–8499985 | BC018275 | NM_007483 | 0.00676 | 1.746 |
| 1451146_at | Zfp386 | zinc finger protein 386 (Kruppel-like) | 116047724–116063360 | BC004747 | NM_001004066 ; NM_019565 | 0.00692 | 0.266 |
| 1434700_at | G2e3 | G2/M-phase specific E3 ubiquitin ligase | 51348061–51376986 | BM123748 | NM_001015099 | 0.00809 | 0.636 |
| 1444164_at | Prpf39 | PRP39 pre-mRNA processing factor 39 homolog (yeast) | 65036333–65063386 | BG068268 | NM_177806 | 0.00821 | 0.378 |
| 1447341_at | Esyt2 | extended synaptotagmin-like protein 2 | 116281222–116373096 | BE456208 | NM_028731 | 0.00859 | 1.141 |
| 1454609_x_at | Irf2bpl | interferon regulatory factor 2 binding protein-like | 86880703–86884814 | BB770958 | NM_145836 | 0.00928 | 1.464 |
| 1420830_x_at | Ywhaq | tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, theta polypeptide | 21390071–21417637 | NM_011739 | NM_011739 | 0.00944 | 1.573 |
| 1421430_at | Rad51b | RAD51 homolog B | 79297351–79508656 | NM_009014 | NM_009014 | 0.00982 | 2.596 |
| 1436438_s_at | Dcaf5 | DDB1 and CUL4 associated factor 5 | 80335847–80436601 | BM234499 | NM_177267 | 0.00984 | 1.410 |
Bold genes exist in the chromosomal region of Fl1sa. The fold change was calculated by the gene expression level in A/J-12SM relative to that in A/J mice
Fig. 3.
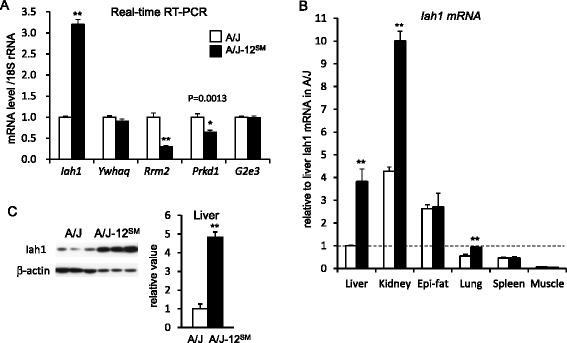
Gene expression levels and protein levels of candidate genes for Fl1sa. a The hepatic mRNA levels of genes identified from a DNA microarray by real-time RT-PCR analyses (n = 9–10; *P < 0.05, **P < 0.01 versus A/J mice). b The tissue distribution of Iah1 mRNA levels in A/J and A/J-12SM mice at 7 weeks of feeding with the HFD (n = 4–5; *P < 0.05 versus A/J mice). c The hepatic levels of Iah1 protein in A/J and A/J-12SM mice at 7 weeks of feeding with the HFD by Western blotting (n = 4; **P < 0.01 versus A/J mice)
Total RNA was extracted from the livers of A/J mice or A/J-12SM consomic mice fed the HFD for 7 weeks. We performed DNA microarray analysis using total RNA from the liver and compared the levels of gene expression between A/J and A/J-12SM. On chromosome 12, we detected 20 genes differentially expressed between A/J mice and A/J-12SM mice (P < 0.01) (Table 2). Five genes (Iah1, Ywhaq, Rrm2, Prkd1, and G2e3) exist in the chromosomal region of Fl1sa (centromere-54.1 Mb). Using a real-time RT-PCR (qPCR) method, we confirmed that the gene expression levels of Iah1 (isoamyl acetate-hydrolyzing esterase 1 homolog (S. cerevisiae), 21.3 Mb), Rrm2 (ribonucleotide reductase M2 subunit, 24.7 Mb), and Prkd1 (protein kinase D1, 50.3 Mb) were significantly different between A/J mice and A/J-12SM mice (Fig. 3a). However, the Ywhaq and G2e3 mRNA levels in A/J-12SM mice did not differ from those in A/J mice. The hepatic Iah1 mRNA level in A/J-12SM mice was 3.2-fold higher than that in A/J mice. In contrast, the hepatic Rrm2 mRNA level in A/J-12SM mice (0.30 ± 0.03) was significantly lower than that in A/J mice (1.00 ± 0.10). The Prkd1 mRNA level in A/J-12SM mice (0.64 ± 0.05) was also significantly lower than that in A/J mice (1.00 ± 0.08).
Tissue distributions of mouse Iah1 expression (Fig. 3b, c)
Among the three genes that were confirmed to show a change of hepatic mRNA levels, we focused on the Iah1 gene as a candidate gene for Fl1sa. We chose this gene because the Iah1 mRNA level in the DNA microarray showed the lowest P-value and the Iah1 protein was expected to have the esterase activity (Table 2).
We compared the Iah1 mRNA level among tissues (liver, kidney, epididymal fat, lung, spleen, and muscle) by using the qPCR method (Fig. 3b). The Iah1 mRNA levels in the liver, kidney, and lung were significantly higher in A/J-12SM mice than in A/J mice. In contrast, the Iah1 mRNA levels in epididymal fat, spleen, and muscle were similar between A/J mice and A/J-12SM mice. In both A/J mice and A/J-12SM mice, the Iah1 mRNA level in the kidney was the highest among the tissues. The hepatic Iah1 protein level in A/J-12SM mice was about 4.8-fold higher than that in A/J mice (Fig. 3c).
Lipid metabolism in Hepa1-6 cells stably overexpressing mouse Iah1 (Fig. 4)
Fig. 4.
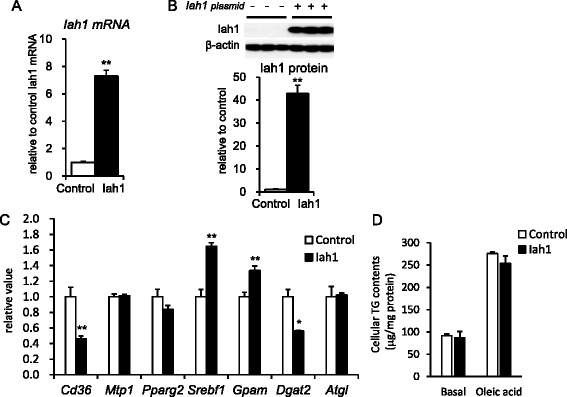
Overexpression of mouse Iah1 in Hepa1-6 cells. a The Iah1 mRNA level in non-transfected (control) and mouse Iah1 cDNA-transfected (Iah1) cells. Iah1 cells were shown to stably overexpress mouse Iah1 protein. b The Iah1 protein level in control cells and mouse Iah1 cells. c The mRNA levels of genes related to lipid metabolism (n = 4–5; *P < 0.05, **P < 0.01 versus A/J mice). d The cellular TG contents in non-transfected (control) and mouse Iah1 cDNA-transfected (Iah1) cells. The treatment medium consisted of serum-free medium plus 0.333 mM oleic acid and was administered to the treatment group for 48 h. The basal group was cultured in serum-free medium without oleic acid. The analyses were performed in duplicate, and at least two independent experiments were performed. The data were analyzed by two-way ANOVA. Oleic acid effect, P < 0.05; Iah1 effect, not significant; Oleic acid × Iah1 (interaction) effect, not significant
We transfected mouse Iah1 cDNA derived from SM/J into Hepa1-6 cells and obtained Hepa1-6 cells stably overexpressing the mouse Iah1 protein (Iah1-cells). The mRNA level and protein level of Iah1 in the non-transfected cells (control cells) could be detected by the qPCR method and western blotting (Fig. 4a and b). The protein level of Iah1 in the Iah1-overexpressed cells was about 42-fold higher than that in the control cells (Fig. 4b). To investigate the effect of stably overexpressing mouse Iah1 protein on lipid metabolism in hepatocytes, we performed an analysis of the mRNA levels of lipid metabolism-related genes (Fig. 4c). The mRNA levels of Cd36 (fatty acid transporter) in the Iah1-cells were significantly lower than those in the control cells. The mRNA levels of Mtp1 (microsomal transfer protein 1) did not differ between the control and the Iah1-cells. The genes Pparg2 (peroxisome proliferator-activated receptor γ), Srebf1 (sterol regulatory element binding protein 1), Gpam (glycerol-3-phosphate acyltransferase, mitochondrial; Gpat, synonymous gene symbol), and Dgat2 (acyl-CoA:diacylglycerol acyltransferase 2) regulate fatty acid and TG synthesis. The mRNA levels of Srebf1 and Gpam were higher in the Iah1-overexpressed cells compared to the control cells. In contrast, the mRNA level of Dgat2 was significantly lower in the Iah1-overexpressed cells than in the control cells. The mRNA level of Atgl (adipose triglyceride lipase), which codes the protein of hepatic major TG lipase, was not changed. In the absence of oleic acid treatment (basal condition), the cellular TG contents did not differ between the control cells and Iah1-cells (Fig. 4d). After treatment with oleic acid, the cellular TG contents in both cell lines were increased. However, overexpression of Iah1did not change the cellular TG contents.
Discussion
We previously mapped a highly significant QTL for fatty liver (Fl1sa) to mouse chromosome 12 in the SMXA-5 mouse, a mouse model for HFD-induced fatty liver and type 2 diabetes. To confirm the effect of this QTL, we constructed A/J-12SM consomic mice. Analysis of the A/J-12SM consomic mice revealed that the SM/J allele on chromosome 12 was resistant to fatty liver [4]. In this study, to elucidate the genetic basis of Fl1sa, we first investigated fatty liver-related traits in A/J-12SM consomic mice (Table 1, Figs. 1 and 2). The phenotypic analysis of A/J-12SM mice showed that the SM/J segment of chromosome 12 contributed to the lower values in liver weight, liver lipids content, blood glucose concentration, and serum insulin concentration. We previously mapped the QTLs for glucose tolerance, non-fasting blood glucose level, and serum insulin levels to the proximal region of chromosome 12 [7]. On these QTLs of chromosome 12, the A/J allele increased the values of each phenotype. Therefore, the results for A/J-12SM consomic mice confirmed that chromosome 12 contained not only genes involved in the regulation of liver lipid metabolism, but also several genes regulating glucose metabolism and insulin sensitivity. Significant differences in the liver lipid contents between A/J mice and A/J-12SM consomic mice appeared in the animals fed an HFD for 11 weeks, but not in those on the ND (Fig. 1). These results showed that the A/J-12SM consomic mice were resistant to HFD-induced fatty liver, and demonstrated that mouse chromosome 12 contained some of the genes responsible for HFD-induced fatty liver. Next, in order to identify these genes, we compared the hepatic gene expression profile between A/J mice and A/J-12SM consomic mice fed the HFD, by using a DNA microarray analysis. We identified three candidate genes (Iah1, Rrm2, Prkd1) which had significantly different levels of expression between A/J mice and A/J-12SM consomic mice within the chromosomal region of Fl1sa (centromere-54.1 Mb). RRM2 is one of the subunits of the ribonucleotide reductase holoenzyme. The ribonucleotide reductase, which is a rate-limiting enzyme in the production of 2’-deoxyribonucleoside 5’-triophosphates (dNTPs), is required for DNA synthesis. In a human study, expression of the Rrm2 gene was shown to be increased in various types of cancer [8]. Protein kinase D1 (PKD1), which is coded by the Prkd1 gene, belongs to the family of stress-activated serine/threonine kinases. PKD1 is activated by the signal via Gq-coupled receptors and protein kinase C, and then phosphorylates various proteins involved in cell growth, apoptosis, adhesion, and angiogenesis [9]. Dysregulation of PKD1 leads to the development of cancer and cardiac hypertrophy [10–12]. The mRNA levels of both Rrm2 and Prkd1 were higher in A/J than in A/J-12SM mice (Fig. 3a), but occurrence of liver cancer in A/J mice has not been reported. At present, the relationship between these genes and lipid metabolism remains uncertain.
Based on the results of the DNA microarray analysis, we focused on the Iah1 gene (Table 2). A previous study reported that mouse Iah1 mRNA was detected in the rostral striatum, kidney, liver, and lung [13]. Fukuda et al. reported that the yeast IAH1 protein has esterase activity for acetate esters such as isoamyl acetate and isobutyl acetate [6]. We found that the Iah1 gene was widely expressed in mouse tissues (liver, kidney, epididymal fat, lung, spleen, muscle) (Fig. 3b), and the mRNA levels in the liver and kidney in A/J mice were especially low compared to those in A/J-12SM mice. However, there has been no report about the function of the mouse Iah1 protein. We focused on the Iah1 mRNA level, because the hepatic Iah1 mRNA level was markedly low in A/J mice (Fig. 3). In this study, we constructed Hepa1-6 cells stably overexpressing the mouse Iah1 protein and found that overexpression of the Iah1 protein suppressed the expression levels of lipid metabolism-related genes such as Cd36 and Dgat2 (Fig. 4). Hepatic Cd36 mRNA expression in C57BL/6 N mice was up-regulated by feeding with the HFD (82 % of calories as fat) for 2 weeks [14]. Diet-induced obesity caused an elevation of hepatic Cd36 expression, and this elevation was correlated with the increase in liver TG storage. In vivo, adenoviral gene delivery of Cd36 to the mouse liver has been shown to lead to an increase in hepatic fatty acid uptake and TG storage in the liver [15]. From our DNA microarray data, the hepatic Cd36 mRNA level in A/J mice was about 2.6-fold higher than that in A/J-12SM mice. In contrast, the mRNA levels of the other genes (Srebf1, Gpam, and Dgat2) regulated by Iah1 overexpression (Fig. 4c) were not different between these two strains (<0.667-fold or >1.5-fold). Our data implied that the elevation of the Cd36 mRNA level in liver was correlated with the increase of hepatic TG accumulation. TG is synthesized via the acylation of diacylglycerol by DGAT enzymes (Dgat1 and Dgat2). Overexpression of the Dgat2 protein in mice has been reported to increase hepatic TG synthesis [16]. Dgat2-knockout mice die soon after birth because of the significant reduction in basal TG synthesis [17]. Dgat2 antisense oligo (ASO) treatment of HFD-induced obese C57BL/6 J mice and ob/ob mice suppresses Dgat2 mRNAs in the liver, resulting in a reduction in hepatic TG [18]. In addition, Dgat2 ASO treatment in rats with diet-induced hepatic steatosis was shown to reduce hepatic diacylglycerol and TG contents, but Dgat1 ASO treatment did not reduce either [19]. These reports indicate that the reduction of the mRNA levels of Cd36 and Dgat2 in liver contributes to the suppression of lipid accumulation in the liver.
We speculate that the elevation of Iah1 gene expression suppresses the TG synthesis and the liver lipid accumulation via the reduction of Cd36 and Dgat2 expression. However, the regulatory mechanisms of Cd36 and Ggat2 gene expressions by Iah1 are unknown. In S. cerevisiae, the IAH1 protein exhibits esterase activity against the ester bonds between alcohol and fatty acid [5, 6]. Therefore, we also speculate that the substrates and/or the metabolites that were catalyzed by the mouse Iah1 protein might regulate the expressions of genes related to lipid metabolism. Although the esterase properties of the mouse Iah1 protein are not yet known, we are currently trying to clarify the physiological substrates of this protein.
Conclusions
This study demonstrated that Fl1sa, a QTL for fatty liver on the proximal region of chromosome 12, was the locus that was affected by feeding with the HFD, but not affected by the ND. We also showed that mouse Iah1 is a candidate gene for Fl1sa. Finally, we showed for the first time that Iah1 regulated the expressions of the Cd36 and Dgat2 genes, which play important roles in TG synthesis and lipid metabolism. In future experiments, to identify the gene responsible for Fl1sa, we will construct several congenic strains. By using these congenic mice, we can finely map the chromosomal region containing the responsible gene. Simultaneously, to determine whether the Iah1 gene is responsible for Fl1sa, we will construct Iah1-knockout mice, which will contribute to the functional analysis of mouse Iah1. Based on our present findings, Iah1 might prove to be a novel mediator of lipid metabolism.
Methods
Animals
The A/J-Chr12SM consomic strain was produced at the Institute for Laboratory Animal Research, Nagoya University School of Medicine. The A/J-12SM consomic mouse strain was established by introduction of the donor SM/J chromosome 12 into the recipient A/J background as previously described [20]. A/J male mice were purchased from Japan SLC (Hamamatsu, Japan). In this study, only male mice were used for the phenotypic analyses. Mice were maintained in a temperature-controlled room (23 ± 2 °C) and 55 ± 5 % humidity with a 12-h light/dark cycle and ad libitum access to food and water under conventional conditions. Mice were weaned at 3 weeks of age in a cage containing five animals or fewer. Until 6 weeks of age, all mice were fed a rodent standard laboratory chow (CE2; Nihon CLEA, Japan). Animal care and all experimental procedures were approved by the Animal Experiment Committee, Graduate School of Bioagricultural Sciences, Nagoya University (approval No. 2008031801, 2011030404, 2012022805), and were conducted according to the Regulations on Animal Experiments of Nagoya University.
Experimental schedule and diet composition
From 6 weeks of age, mice were switched from their standard laboratory diets to the HFD (HFD group), or continued to be fed their standard laboratory diets (ND group). In the ND group, A/J mice and A/J-12SM mice were kept at one animal per cage and fed the standard laboratory chow for 11 weeks. After 11 weeks of feeding with the ND, the mice were killed by decapitation, and the liver was collected at 13:00–14:00 h after 4-h diet deprivation. In the HFD group, A/J mice and A/J-12SM consomic mice were kept at one animal per cage and fed the powdered HFD for 3, 7, or 11 weeks. The composition of the powdered HFD (weight %) was as follows: casein, 20.9; carbohydrate (corn starch: sucrose, 1:1), 36.9; AIN93MX mineral mixture, 3.5; AIN93VX vitamin mixture, 1.0; choline chloride, 0.2; corn oil, 3.5; lard, 30.0; and cellulose (AVICEL type FD-101; Asahi Chemical Industry, Osaka, Japan), 4.0. The content of fat in this HFD was 33.5 % (weight %). After 3, 7, or 11 weeks on the HFD, mice were killed by decapitation, and the serum, liver, and fat pads were collected at 13:00–14:00 h after 4-h diet deprivation. After 7 weeks on the HFD, the measurement of BMI was performed as in our previous report [21]. BMI was calculated as body weight (g) divided by the square of the anal-nasal length (cm).
Hepatic lipid analysis
Frozen livers were homogenized with chloroform:methanol (2:1), and the liver lipids were extracted into organic solvents. A portion of this extract was dried, and the hepatic contents of TG and TC were measured by the triglyceride E-test (Wako, Tokyo, Japan) and the cholesterol E-test (Wako), respectively. This extract was also used to measure total liver lipids according to the method of Folch et al. [22].
Serum glucose, insulin, and lipids
The serum glucose concentration was measured by a glucose oxidase method (Glucose-B test Kit; WAKO, Tokyo, Japan). Serum insulin, TG, TC, HDL-C, and non-esterified free fatty acid concentrations were measured at the end of the experiment (after 7 weeks of feeding with the HFD) by using a Mouse Insulin ELISA kit (Morinaga Institute of Biological Sciences Inc., Japan), Triglyceride-E kit (WAKO Pure Chemical Industries, Japan), Cholesterol-E kit (WAKO Pure Chemical Industries), HDL-Cholesterol-E kit (WAKO Pure Chemical Industries), and NEFA C-kit (WAKO Pure Chemical Industries), respectively.
Microarray experiments
Liver samples were collected from A/J male mice and A/J-12SM male mice fed the HFD for 7 weeks. Total RNA was isolated from the liver using TRI reagent (Molecular Research Center Inc.) and cleaned using an RNeasy Mini kit (Qiagen). We extracted the total RNA from nine mice per strain. Extracted total RNA quality was assessed with an Agilent Bioanalyzer (Agilent Technologies). cDNA synthesis and cRNA labeling reactions were performed with a One-Cycle cDNA Synthesis Kit and IVT Labeling Kit (Affymetrix). In order to minimize the experimental variation, we used three arrays for each strain and obtained data from three replicates. Each biotinylated cRNA was prepared from the pooled RNA samples of three mice. The biotinylated cRNAs were hybridized with a probe array (Mouse Genome 430 2.0 Array) by using a Hybridization, Wash and Stain kit (Affymetrix). Scanning was performed using a Scanner 3000 with GeneChip Operating Software (GCOS) (Affymetrix). Raw data were normalized with the MAS5.0 algorithm by using Expression Console Software (Affymetrix). The microarray data have been deposited in the NCBI Gene Expression Omnibus (GEO) (GSE67340). Data were analyzed by using GeneSpring GX7.3 software (Agilent Technologies).
Real-time RT-PCR
Total RNA was extracted from liver and Hepa1-6 cells using TRI reagent. To eliminate DNA contamination, the RNA was treated with DNase (TURBO DNA-free; Life Technologies). Subsequently, the cDNA was synthesized by using a High Capacity Reverse Transcription kit (Life Technologies). Gene expression was quantified by real-time PCR using an ABI 7300 real-time PCR System with Thunderbird qPCR Mix or Thunderbird SYBR qPCR Mix (Toyobo, Tokyo, Japan). TaqMan primers and probes were used to determine the mRNA levels of mouse Iah1 (TaqMan Gene Expression Assays, Mm00509467_m1; Applied Biosystems) and 18S rRNA (Pre-developed TaqMan Assay Reagents, Eukaryotic 18S rRNA 4319413E; Applied Biosystems). The primers used for the SYBR Green assay are shown in Additional file 1. The level of each mRNA was normalized to that of the corresponding 18S rRNA.
Western blotting
The tissues and cells were homogenized in lysis buffer (10 mM Tris (pH 7.4), 150 mM NaCl, 1 % Nonidet P-40, 0.5 % sodium deoxycholate, 0.1 % SDS) containing protease inhibitor cocktail (Complete Mini, Roche Applied Science) with a homogenizer (HG-30; Hitachi). The homogenates were centrifuged at 10,000 × g for 5 min and the supernatant was obtained as protein extracts. An aliquot of protein (4 μg) was subjected to SDS-PAGE on 10 % acrylamide gel, and the proteins in the gel were transferred onto PVDF membranes (Hybond P; GE Healthcare). The membranes were incubated for 30 min at room temperature with Blocking One (Nacalai Tesque) and incubated overnight at 4 °C with the first antibody, rabbit polyclonal anti-mouse Iah1 (1:20,000) and anti-β-actin (1:10,000; #4967; Cell Signaling Technology Inc.), then washed with TBS buffer containing 0.1 % Tween 20. The membranes were incubated with the horseradish peroxidase-conjugated goat anti-rabbit IgG antibody (1:20,000 or 1:10,000; #7074; Santa Cruz Biotechnology, USA) for 1 h at room temperature and washed. Each antibody was diluted with Can Get Signal (Toyobo). The membranes were autographed with a West Dura Western Blot Detection kit using the ECL method (Thermo Fisher Scientific). Each protein on the band was quantified with Image J software.
Hepa1-6 cell culture and overexpression of mouse Iah1
Hepa1-6 cells, a mouse hepatocyte cell line (RBRC-RCB1638 RIKEN BRC Cell Bank), were grown in high-glucose Dulbecco’s modified Eagle’s medium (DMEM; Wako Pure Chemical) supplemented with 10 % fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin at 37 °C in a 5 % CO2-humidified incubator.
Iah1 cDNA of SM/J mice was amplified with the KOD-plus (TOYOBO) using the following primers: an upper primer containing the start codon, CTTTCTACCATGTCGCTGTGC; a lower primer containing the stop codon, CTAATAGTCTCCATCTCCCAGCAG. The cDNA fragment was subcloned into pcDNA3.3 TOPO plasmid (Thermo Fisher Scientific). The Iah1-expressing plasmid vector with CMV promoter contained Iah1 cDNA (759 bp) constructed from SM/J mice. To construct the mouse Iah1-expressing stable cell line, Hepa1-6 cells were seeded on 12-well plates (3 × 105 cells per well) on day 0. After 24 h, the Iah1-expressing plasmid vector (1.4 μg) was transfected using Lipofectamine 2000 reagent (Invitrogen). After 28 h of transfection, the transfected cells were subcultured on a 60 mm dish (6000 cells per dish) and incubated for 1 week. For the selection of neomycin-resistant cells, cells were maintained in medium containing 500 μg/ml G418 disulfate salt (Sigma Aldrich). Three weeks after colony selection with G418, we obtained the Iah1 cDNA-expressing stable cell line.
To assay the effect of overexpression of Iah1 on various mRNA levels, cells were plated on 12-well plates (2 × 105 cells per well) on day 0. After 72 h, the cells were harvested and used for the assay of protein or the extraction of total RNA.
To examine the effect of overexpression of Iah1 on the intracellular accumulation of triglycerides, cells were seeded on 12-well plates (2.5 × 105 cells per well) at day 0. At day 1, the culture medium was replaced with serum-free medium containing 0.333 mM oleic acid (oleic acid-BSA conjugated solution, finally containing 1 % BSA, (Sigma)) or serum-free medium alone. The treatment medium without oleic acid-BSA was supplemented with 1 % BSA. After 2 days, the cells were homogenized in lysis buffer (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 2 mM EDTA, 1 % Triton X-100, and 0.5 % cholate). The supernatants were obtained by centrifugation at 9730 × g for 10 min at 4 °C. Their triglycerides and protein concentration were measured by a Triglyceride-E test and a DC protein assay (Bio-Rad Laboratories, Japan), respectively. The triglycerides content in cells was normalized to that of protein content.
Statistical analysis
The results are expressed as the means with their standard errors. Mean values, except for the data in Fig. 4d, were compared using Student’s t-test when the variances of each group were equal. When the variances of each group were unequal, the significance of differences was determined using Welch’s test. The data from Fig. 4d were analyzed by two-way ANOVA. Differences with P < 0.05 were regarded as significant. General statistical analyses were also performed using StatView version 5.0 software (SAS Institute).
Abbreviations
Atgl, Adipose triglyceride lipase; AUC, Area under the curve; BMI, Body mass index; Cd36, CD36 antigen; Dgat2, Acyl-CoA:diacylglycerol acyltransferase 2; FFA, Free fatty acid; Gpam, Glycerol-3-phosphate acyltransferase, mitochondrial; HFD, High-fat diet; Iah1, isoamyl acetate-hydrolyzing esterase 1 homolog (S. cerevisiae); Mtp1, Microsomal transfer protein 1; ND, Normal diet; Pparg2, Peroxisome proliferator-activated receptor γ; Srebf1, Sterol regulatory element binding protein 1
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (C) (No. 25450166) from the Japan Society for the Promotion of Sciences, and a grant from the Uehara Memorial Foundation and Kao Research Council for the Study of Healthcare Sciences.
Availability of data and materials
The dataset supporting the conclusions of this article is available in the GEO repository [http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE67340].
Authors’ contributions
MK performed the experiments and wrote the manuscript. MS and KT participated in acquisition of data using Hepa1 cells. CT and ST participated in acquisition of data using DNA microarray and quantitative real-time PCR. TK and YK participated in the design of the experiment using Hepa1 cells. TO and AM contributed to the interpretation of data. FH contributed to the design of the experiments, interpreted the data and edited the manuscript. All authors have read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent to publish
Not applicable.
Ethics approval and consent to participate
Animal care and all experimental procedures were approved by the Animal Experiment Committee, Graduate School of Bioagricultural Sciences, Nagoya University (approval No. 2008031801, 2011030404, 2012022805), and were conducted according to the Regulations on Animal Experiments of Nagoya University.
Additional file
The primers for SYBR Green assay in real-time PCR. (DOCX 14 kb)
References
- 1.Kobayashi M, Io F, Kawai T, Nishimura M, Ohno T, Horio F. SMXA-5 mouse as a diabetic model susceptible to feeding a high-fat diet. Biosci Biotechnol Biochem. 2004;68(1):226–30. doi: 10.1271/bbb.68.226. [DOI] [PubMed] [Google Scholar]
- 2.Nishimura M, Hirayama N, Serikawa T, Kanehira K, Matsushima Y, Katoh H, Wakana S, Kojima A, Hiai H. The SMXA: a new set of recombinant inbred strain of mice consisting of 26 substrains and their genetic profile. Mamm Genome. 1995;6(12):850–7. doi: 10.1007/BF00292434. [DOI] [PubMed] [Google Scholar]
- 3.Anunciado RV, Horio F, Ohno T, Tanaka S, Nishimura M, Namikawa T. Characterization of hyperinsulinemic recombinant inbred (RI) strains (SMXA-5 and SMXA-9) derived from normoinsulinemic SM/J and A/J mice. Exp Anim. 2000;49(2):83–90. doi: 10.1538/expanim.49.83. [DOI] [PubMed] [Google Scholar]
- 4.Kumazawa M, Kobayashi M, Io F, Kawai T, Nishimura M, Ohno T, Horio F. Searching for genetic factors of fatty liver in SMXA-5 mice by quantitative trait loci analysis under a high-fat diet. J Lipid Res. 2007;48(9):2039–46. doi: 10.1194/jlr.M700222-JLR200. [DOI] [PubMed] [Google Scholar]
- 5.Fukuda K, Yamamoto N, Kiyokawa Y, Yanagiuchi T, Wakai Y, Kitamoto K, Inoue Y, Kimura A. Balance of activities of alcohol acetyltransferase and esterase in Saccharomyces cerevisiae is important for production of isoamyl acetate. Appl Environ Microbiol. 1998;64(10):4076–8. doi: 10.1128/aem.64.10.4076-4078.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fukuda K, Kiyokawa Y, Yanagiuchi T, Wakai Y, Kitamoto K, Inoue Y, Kimura A. Purification and characterization of isoamyl acetate-hydrolyzing esterase encoded by the IAH1 gene of Saccharomyces cerevisiae from a recombinant Escherichia coli. Appl Microbiol Biotechnol. 2000;53(5):596–600. doi: 10.1007/s002530051662. [DOI] [PubMed] [Google Scholar]
- 7.Kobayashi M, Io F, Kawai T, Kumazawa M, Ikegami H, Nishimura M, Ohno T, Horio F. Major quantitative trait locus on chromosome 2 for glucose tolerance in diabetic SMXA-5 mouse established from non-diabetic SM/J and A/J strains. Diabetologia. 2006;49(3):486–95. doi: 10.1007/s00125-005-0121-3. [DOI] [PubMed] [Google Scholar]
- 8.Dawany NB, Dampier WN, Tozeren A. Large-scale integration of microarray data reveals genes and pathways common to multiple cancer types. Int J Cancer. 2011;128(12):2881–91. doi: 10.1002/ijc.25854. [DOI] [PubMed] [Google Scholar]
- 9.Steinberg SF. Regulation of protein kinase D1 activity. Mol Pharmacol. 2012;81(3):284–91. doi: 10.1124/mol.111.075986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guha S, Tanasanvimon S, Sinnett-Smith J, Rozengurt E. Role of protein kinase D signaling in pancreatic cancer. Biochem Pharmacol. 2010;80(12):1946–54. doi: 10.1016/j.bcp.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.LaValle CR, George KM, Sharlow ER, Lazo JS, Wipf P, Wang QJ. Protein kinase D as a potential new target for cancer therapy. Biochim Biophys Acta. 2010;1806(2):183–92. doi: 10.1016/j.bbcan.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Avkiran M, Rowland AJ, Cuello F, Haworth RS. Protein kinase d in the cardiovascular system: emerging roles in health and disease. Circ Res. 2008;102(2):157–63. doi: 10.1161/CIRCRESAHA.107.168211. [DOI] [PubMed] [Google Scholar]
- 13.Ghate A, Befort K, Becker JA, Filliol D, Bole-Feysot C, Demebele D, Jost B, Koch M, Kieffer BL. Identification of novel striatal genes by expression profiling in adult mouse brain. Neuroscience. 2007;146(3):1182–92. doi: 10.1016/j.neuroscience.2007.02.040. [DOI] [PubMed] [Google Scholar]
- 14.Inoue M, Ohtake T, Motomura W, Takahashi N, Hosoki Y, Miyoshi S, Suzuki Y, Saito H, Kohgo Y, Okumura T. Increased expression of PPARgamma in high fat diet-induced liver steatosis in mice. Biochem Biophys Res Commun. 2005;336(1):215–22. doi: 10.1016/j.bbrc.2005.08.070. [DOI] [PubMed] [Google Scholar]
- 15.Koonen DP, Jacobs RL, Febbraio M, Young ME, Soltys CL, Ong H, Vance DE, Dyck JR. Increased hepatic CD36 expression contributes to dyslipidemia associated with diet-induced obesity. Diabetes. 2007;56(12):2863–71. doi: 10.2337/db07-0907. [DOI] [PubMed] [Google Scholar]
- 16.Millar JS, Stone SJ, Tietge UJ, Tow B, Billheimer JT, Wong JS, Hamilton RL, Farese RV, Rader DJ. Short-term overexpression of DGAT1 or DGAT2 increases hepatic triglyceride but not VLDL triglyceride or apoB production. J Lipid Res. 2006;47(10):2297–305. doi: 10.1194/jlr.M600213-JLR200. [DOI] [PubMed] [Google Scholar]
- 17.Stone SJ, Myers HM, Watkins SM, Brown BE, Feingold KR, Elias PM, Farese RV. Lipopenia and skin barrier abnormalities in DGAT2-deficient mice. J Biol Chem. 2004;279(12):11767–76. doi: 10.1074/jbc.M311000200. [DOI] [PubMed] [Google Scholar]
- 18.Yu XX, Murray SF, Pandey SK, Booten SL, Bao D, Song XZ, Kelly S, Chen S, McKay R, Monia BP, et al. Antisense oligonucleotide reduction of DGAT2 expression improves hepatic steatosis and hyperlipidemia in obese mice. Hepatology. 2005;42(2):362–71. doi: 10.1002/hep.20783. [DOI] [PubMed] [Google Scholar]
- 19.Choi CS, Savage DB, Kulkarni A, Yu XX, Liu ZX, Morino K, Kim S, Distefano A, Samuel VT, Neschen S, et al. Suppression of diacylglycerol acyltransferase-2 (DGAT2), but not DGAT1, with antisense oligonucleotides reverses diet-induced hepatic steatosis and insulin resistance. J Biol Chem. 2007;282(31):22678–88. doi: 10.1074/jbc.M704213200. [DOI] [PubMed] [Google Scholar]
- 20.Ohno T, Hata K, Baba T, Io F, Kobayashi M, Horio F, Nishimura M. Establishment of consomic strains derived from A/J and SM/J mice for genetic analysis of complex traits. Mamm Genome. 2012;23(11–12):764–9. doi: 10.1007/s00335-012-9435-x. [DOI] [PubMed] [Google Scholar]
- 21.Kobayashi M, Ohno T, Tsuji A, Nishimura M, Horio F. Combinations of nondiabetic parental genomes elicit impaired glucose tolerance in mouse SMXA recombinant inbred strains. Diabetes. 2003;52(1):180–6. doi: 10.2337/diabetes.52.1.180. [DOI] [PubMed] [Google Scholar]
- 22.Folch J, Lees M, Sloane Stanlley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226(1):497–509. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The dataset supporting the conclusions of this article is available in the GEO repository [http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE67340].