

Abstract
Sepsis induces an acute inflammatory response in the liver, which can lead to organ failure and death. Given the anti-inflammatory effects of exercise, we hypothesized that habitual physical activity could protect against acute sepsis-induced liver inflammation via mechanisms, including heat shock protein (HSP) 70/72. Male C57BL/6J mice (n = 80, ∼8 wk of age) engaged in physical activity via voluntary wheel running (VWR) or cage control (SED) for 10 wk. To induce sepsis, we injected (2 mg/kg ip) LPS or sterile saline (SAL), and liver was harvested 6 or 12 h later. VWR attenuated increases in body and epididymal adipose tissue mass, improved glucose tolerance, and increased liver protein content of PEPCK (P < 0.05). VWR attenuated increases in LPS-induced IL-6 signaling and mRNA expression of other inflammatory markers (TNF-α, chemokine C-C motif ligand 2, inducible nitric oxide synthase, IL-10, IL-1β) in the liver; however, this was not reflected at the whole body level, as systemic markers of inflammation were similar between SED and VWR. Insulin tolerance was greater in VWR compared with SED at 6 but not 12 h after LPS. The protective effect of VWR occurred in parallel with increases in the liver protein content of HSP70/72, a molecular chaperone that can protect against inflammatory challenges. This study provides novel evidence that physical activity protects against the inflammatory cascade induced by LPS in the liver and that these effects may be mediated via HSP70/72.
Keywords: lipopolysaccharide, inflammation, liver, physical activity, exercise
sepsis is a continuum of clinical events defined by the presence of infection and systemic inflammation (18). The worldwide incidence of sepsis was recently estimated at 437 cases per 100,000 person years between 2003 and 2015, with an associated fatality rate of 17% (11). This puts a significant burden on the health care system, and in the United States alone, the direct hospital costs associated with sepsis are over $24 billion (17).
The infiltration of a host with both gram-negative and gram-positive bacterial species is often considered to be the cause of acute sepsis (34). LPS from Escherichia coli, a component of gram-negative bacteria, is often used to produce these effects in rodent models. Upon exposure to LPS, an inflammatory cascade is activated both systemically and at a tissue-specific level (1). Initial signaling involves a Toll-like receptor 4 (TLR4)-myeloid differentiation primary response gene 88 (MyD88) interaction that then propagates the inflammatory response (24). Increases in circulating inflammatory factors, such as TNF-α, IL-6, and IL-1β, occur rapidly and are regulated by the dose and duration of exposure (21, 29, 40). Clearance of inflammatory markers begins as early as 8 h postexposure, and most markers return to baseline levels by 16 h (10). In parallel with the systemic inflammatory response, acute exposure to LPS induces profound inflammation within the liver. LPS activates JAK, a STAT signaling complex, which includes phosphorylation of STAT3, and is a well-known marker of IL-6 signaling (31, 36, 38). Further, SOCS3 is induced by activation of the JAK/STAT signaling cascade and acts as an inhibitor of this pathway (6). This inflammatory cascade leads to increases in circulating markers of liver damage, such as aspartate aminotransferase (AST) and alanine aminotransferase (ALT) (12), and morphological alterations to the liver (39).
A number of different mechanisms have been shown to modulate the inflammatory response to LPS-induced inflammation. Of interest, the transforming growth factor β (TGF-β) superfamily protein follistatin (FST) offers protection via binding to activin A (16, 37), proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibits LPS uptake and clearance (35), and heat shock protein (HSP) 70/72 acts a molecular chaperone (2, 8, 23). Since physical activity, in the form of voluntary wheel running (VWR), leads to increases in the protein content of HSP70/72 in skeletal muscle (14), VWR may be able to protect against LPS-induced inflammation. Somewhat surprisingly, Martin et al. (22) demonstrated that 10 wk of VWR did not protect against increases in the hepatic expression of TNF-α, IL-6, and IL-1β when assessed 24 h after a 0.33 mg/kg injection of LPS. However, at 24 h post-LPS, others have shown circulating inflammatory markers are at near-baseline levels (10), and this dose of LPS leads to less than peak inflammatory responses (21). With these points in mind, a protective effect of physical activity may have been missed.
In this report, we assessed whether VWR could protect against LPS-induced liver inflammation in C57BL/6J mice at 6 and 12 h after a 2 mg/kg injection, which we consider to correspond to late peak and early resolution of the inflammatory response (10, 21). We hypothesized that VWR would protect against the deleterious effects of LPS on the liver and that this would be associated with increases in HSP70/72 and follistatin and reductions in PCSK9. Indeed, here, we show that VWR provides modest protection against the inflammatory cascade induced by LPS within the liver at both 6 and 12 h postexposure and that this occurs, in parallel with increases in HSP70/72.
METHODS
Ethics.
All procedures adhered to the guidelines of the Canadian Council on Animal Care and were approved by the University of Guelph Animal Care Committee.
Animals.
Eight-week-old male C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME). These experiments were completed in two rounds of n = 40 each. In round 1, mice were exposed to saline (SAL) or LPS for 6 h, and in round 2, mice were exposed to SAL or LPS for 12 h. After acclimation, mice were housed in shoebox cages as sedentary controls (SED group; n = 20 per experiment) or in cages outfitted with a 14-cm diameter running wheel (VWR group, n = 20 per experiment) and rotation counter (VDO M3 wired bike computer; Mountain Equipment Co-Op, Vancouver, Canada). All mice were housed individually on a 12:12-h light-dark cycle (∼9 AM to 9 PM), with access to food and water ad libitum. Body mass and food intake (amount of food in hopper of cage) were measured weekly.
Glucose tolerance test.
During the final week of the intervention, and at least 48 h prior to LPS injections, an intraperitoneal glucose tolerance test (GTT) was performed. Following a 6-h fast, and with wheels removed from cages, mice were injected intraperitoneally with a weight-adjusted bolus (2 g/kg) of d-glucose. Tail blood glucose was measured immediately prior to the glucose injection and at 15, 30, 45, 60, 90, and 120 min afterward using a hand-held Freestyle Lite glucometer (Abbott Laboratories; Abbott Park, IL). The glucose area under the curve (AUC) was then calculated.
LPS.
LPS was purchased from Sigma-Aldrich (St. Louis, MO) (0111:B4, L2630). After 10 wk of VWR or SED control, a dose of 2 mg/kg of LPS was injected intraperitoneally into awake, nonanesthetized mice starting at 9 or 6 AM (6- and 12-h experiments, respectively), and control mice were injected with SAL. A 2 mg/kg injection of LPS has been shown to induce peak cytokine concentrations in serum (21) and leads to impaired insulin action (30). Food was provided ad libitum prior to and during LPS exposure, and mice that were in the VWR group had their wheels locked 24 h prior to injection. After the LPS injection, mice were monitored for signs of shock, kept warm using a heating blanket, and, if necessary, supplemented with saline.
Insulin stimulation and tissue collection.
At 6 or 12 h post-LPS injection, we measured tail blood glucose, as LPS is known to reduce blood glucose levels in mice (30). To assess LPS-mediated impairments in insulin action, we conducted an insulin tolerance test (ITT) using a 0.5 U/kg ip insulin injection, as previously described (30), and blood glucose was monitored for 20 min, which is the time in which blood glucose declines in a nearly linear manner (30). Mice were then anesthetized with pentobarbital sodium (∼5 mg/100 g body wt), blood was collected for serum analysis, and the liver was removed and frozen in liquid nitrogen for storage at −80°C or fixed in 10% neutral buffered formalin (for 12-h experiments only).
Western blot analysis.
Whole liver tissue was homogenized in a 30× cocktail of cell lysis buffer, supplemented with phenylmethylsulfonyl fluoride, and protease inhibitor cocktail (Sigma-Aldrich). Western blotting procedures were completed similar to that which we have previously described (3). Nitrocellulose membranes were incubated with indicated primary antibodies overnight and then with corresponding secondary antibody for 1 h. These membranes were then imaged using enhanced chemiluminescence signals on a FluorChem HD imaging system (Alpha Innotect, Santa Clara, CA). Antibodies used were obtained from AbCam (Cambridge, MA; tubulin cat. no. 7291, FST cat. no. 64490, GAPDH cat. no. 8245, PCSK9 cat. no. 31762, and TLR4 cat. no. 83444), Cayman (Ann Arbor, MI; PEPCK cat. no. 10004943), Cell Signaling (Beverly, MA; MyD88 cat. no. 4283; pSTAT3 cat. no. 9138, STAT3 cat. no. 8678, pSTAT1 cat. no. 7649, STAT1 cat. no. 9172, and SOCS3 cat. no. 2932), Enzo Life Sciences (Farmingdale, NY; HSP70/72 cat. no. ADI-SPA-810), and Santa Cruz Biotechnology (Santa Cruz, CA; G6Pase cat. no. 25840).
Real-time quantitative PCR.
Liver RNA extraction was completed using a Qiagen RNeasy mini-kit (Qiagen, Germantown, MD). cDNA was synthesized using Superscript II reverse transcriptase kit (cat. no. 18064-014), purchased from Thermo Fisher Scientific (Waltham, MA) and diluted 1:15 for all genes of interest. PCR plates were prepared using Quanta mix, water, TaqMan gene expression assay for the gene of interest (Table 1), and 5 μl of cDNA. Relative differences were compared using the 2−ΔΔCT method (20). The endogenous control gene, GAPDH, did not change between the experimental groups.
Table 1.
TaqMan PCR primers
| Gene | Assay ID |
|---|---|
| TNF-α | Mm00443258_m1 |
| IL-6 | Mm00446190_m1 |
| IL-1β | Mm00434228_m1 |
| Ccl2 | Mm00441242_m1 |
| Nos2 | Mm00440502_m1 |
| IL-10 | Mm01288386_m1 |
| socs3 | Mm00545913_s1 |
| FST | Mm00514982_m1 |
All gene expression assays were purchased from Thermo Fisher Scientific (cat. no. 4331182).
Histology.
Liver tissue that was fixed in 10% neutral buffered formalin was dehydrated with xylene overnight prior to paraffin wax embedding. Tissue sections were cut at 5 μm, mounted (1.2-mm Superfrost Slides, VWR Mississauga ON, Canada), and then stained with hematoxylin and eosin. Images were obtained using an Olympus FSX 100 light microscope (Olympus, Tokyo Japan) at ×40.
Serum analysis.
Blood was collected from mice after euthanasia and tissue removal, centrifuged at 4,000 g for 15 min at 4°C, and then serum was collected and stored at −80°C. Serum AST and ALT were measured as previously described (19, 28). Measurement of TNF-α, IL-6, IL-1β, Ccl2, Nos2, IL-10, SOCS3 (MCYTOMAG-70K-06), and FST (MAGPMAG-24K-01) were completed using the Milliplex MAP system (Billerica, MA). Samples were diluted twofold, and for IL-6 and Ccl2, data points that fell above the standard curve were extrapolated using the five-parameter logistic curve-fitting method.
Statistical analysis.
Data were analyzed using repeated-measures two-way ANOVA (GTT), three-way ANOVA (ITT), unpaired t-test (body mass, AUC, and fold changes), and two-way ANOVA (all other measures). Data were first assessed for normality and homogeneity of variance, and when not met for two-way ANOVA tests, the data were transformed (log10). For t-tests a Mann-Whitney U-test or Welch's correction was used. The uncorrected Fisher's least significant difference post hoc test was used to compare two-way ANOVA interactions. All data are presented as means ± SE, and a significance level of P < 0.05 was used. Statistical tests were completed using SigmaPlot version 11.0 (San Jose, CA) and Graph Pad version 6.2 (La Jolla, CA).
RESULTS
VWR attenuates body mass and adipose tissue gain, improves glucose tolerance, and increases markers of hepatic glucose production.
We first wanted to ensure the effectiveness of VWR; thus, we assessed differences in body mass, glucose homeostasis, and levels of gluconeogenic enzymes previously shown to be increased with exercise (13). Mice in the VWR group averaged ∼3–5 km/night over the 10-wk period (Fig. 1A), and despite greater food intake than SED mice (Fig. 1B), they had attenuated gains in body mass (Fig. 1C). VWR also improved glucose tolerance (Fig. 1D) and reduced epididymal white adipose tissue mass (Fig. 1E). Further, the protein content of PEPCK was increased ∼50% in VWR compared with SED mice (P < 0.05), whereas there were no differences in hepatic G6Pase protein content (Fig. 1F).
Fig. 1.

Effects of voluntary wheel running on distance run (A), food intake (B), body mass (C), blood glucose during a GTT (D), epididymal adipose tissue mass (E), and protein content of PEPCK and G6Pase (F). Values are pooled from 6- and 12-h experiments, unless indicated. Two-way ANOVA main effect for VWR, §P < 0.05. #P < 0.05, main effect of time. Unpaired t-test between sedentary (SED) and voluntary wheel running (VWR) rats, *P < 0.05.
Voluntary wheel running attenuates LPS-mediated induction of liver inflammatory markers.
To assess the inflammatory cascade induced by LPS within the liver, we measured the mRNA expression of well-known inflammatory markers. At 6 h post-LPS, VWR mice injected with LPS had an attenuated response compared with SED mice for IL-6 (P = 0.001) (Fig. 2B). In addition, VWR tended to lower both TNF-α and Ccl2 (two-way ANOVA main effect of VWR, P = 0.08 and P = 0.09, respectively) (Fig. 2, A and D). We then compared the level of induction above saline (SAL) control within SED and VWR mice injected with LPS and found that VWR mice had attenuated increases in TNF-α, IL-6, Ccl2, Nos2, and IL-10 (P < 0.05) (Fig. 2, A–F, insets). At 12 h post-LPS, VWR mice injected with LPS had an attenuated increase in IL-1β compared with SED mice (P = 0.002) (Fig. 3C). In addition, there was a main effect of VWR for TNF-α, IL-1β, and Ccl2 (Fig. 3, A, C, D). When comparing the level of induction above SAL control within LPS-treated mice, we observed attenuations for IL-1β and Nos2 in VWR mice (Fig. 3, C and E). Together, these data suggest that VWR attenuates inflammatory response to LPS within the liver when assessed at both 6 and 12 h post-LPS.
Fig. 2.
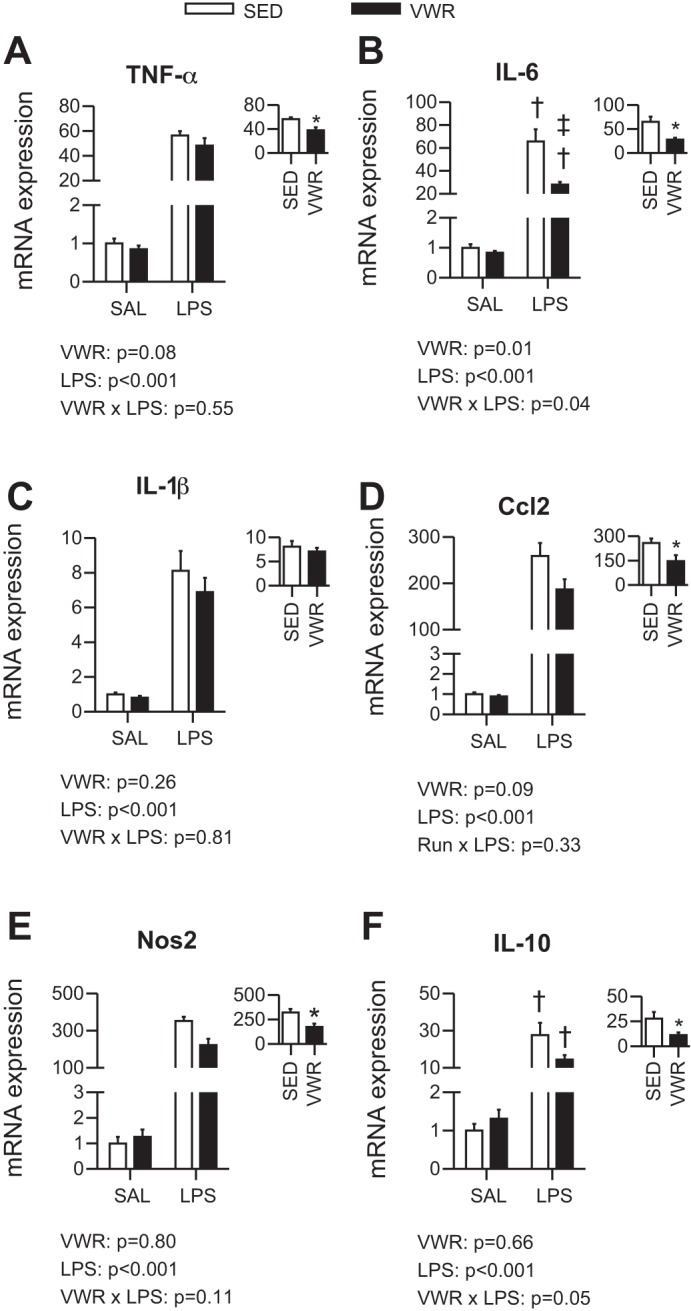
Effect of voluntary wheel running on 6-h exposure to LPS for mRNA expression of TNF-α (A), IL-6 (B), IL-1β (C), Ccl2 (D), Nos2 (E), and IL-10 (F). Text indicates P values for two-way ANOVA, and when an interaction was observed, †P < 0.05 indicates LPS group significantly different within SED or VWR and ‡P < 0.05 indicates VWR group different within Sal or LPS. Inset indicates fold change relative to saline (SAL) control and *P < 0.05 for t-test between SED and VWR groups.
Fig. 3.
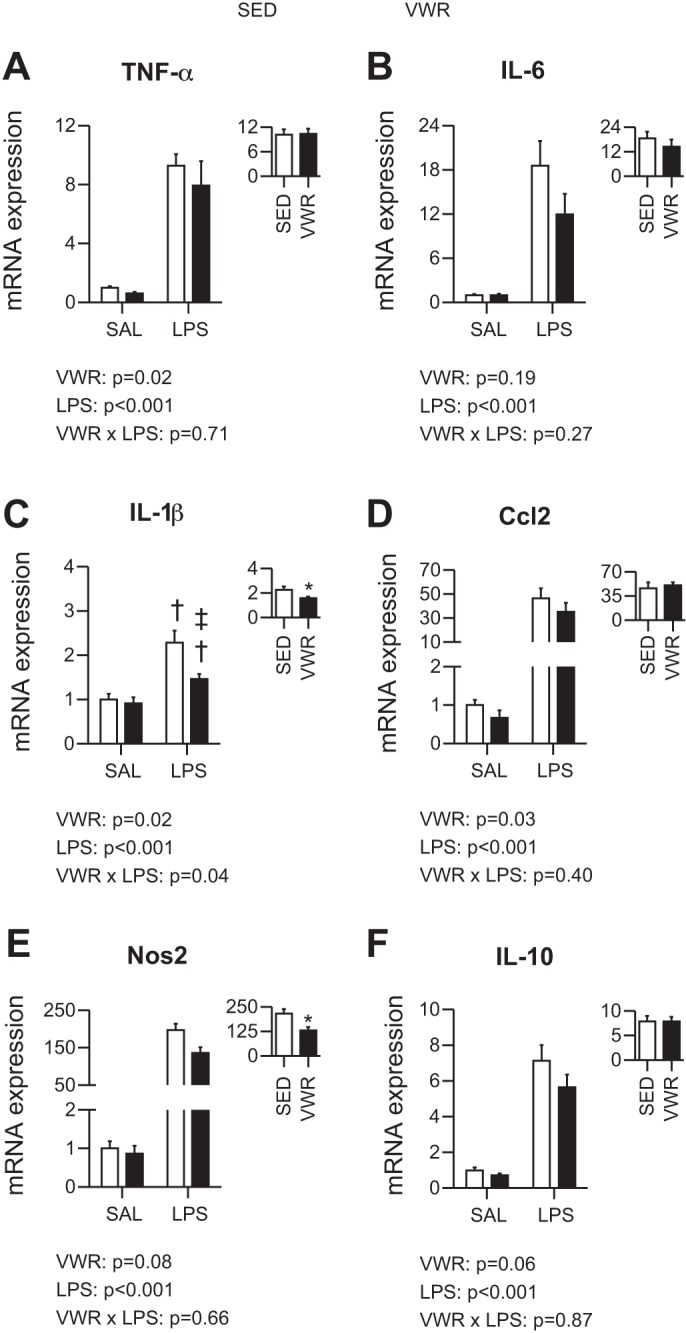
Effect of voluntary wheel running on 12-h exposure to LPS for mRNA expression of TNF-α (A), IL-6 (B), IL-1β (C), Ccl2 (D), Nos2 (E), and IL-10 (F). Text indicates P values for two-way ANOVA, and when an interaction was observed, †P < 0.05 indicates LPS group significantly different within SED or VWR and ‡P < 0.05 indicates VWR group different within Sal or LPS. Inset: fold change relative to SAL control and *P < 0.05 for t-test between SED and VWR groups.
Next, we wanted to determine whether VWR attenuated the induction of IL-6 signaling after LPS exposure. In mice injected with LPS, the phosphorylation of STAT3 was increased (P < 0.001), which was modestly attenuated by VWR at 12 h post-LPS (two-way ANOVA main effect, P < 0.001) (Fig. 4, A and B). The phosphorylation of STAT1 was also increased after LPS; however, as this was undetectable in SAL controls, we compared the level of induction only between LPS-injected mice. At 6 h post-LPS, there was no protective effect of VWR against increases in the phosphorylation of STAT1 (P = 0.95); however, at 12 h post-LPS, VWR offered mild protection (P = 0.10) (Fig. 4, C and D). At 6 h post-LPS, SOCS3 mRNA expression was not different between SED and VWR mice injected with LPS; however, VWR mice injected with SAL had a higher mRNA expression compared with SED mice (P < 0.001) (Fig. 4E). In contrast, the fold change compared with SED control was attenuated at both 6 and 12 h post-LPS by VWR (P < 0.05) (Fig. 4, E and F). These results did not translate to the protein level, as the content of hepatic SOCS3 was not different with VWR or LPS (data not shown).
Fig. 4.
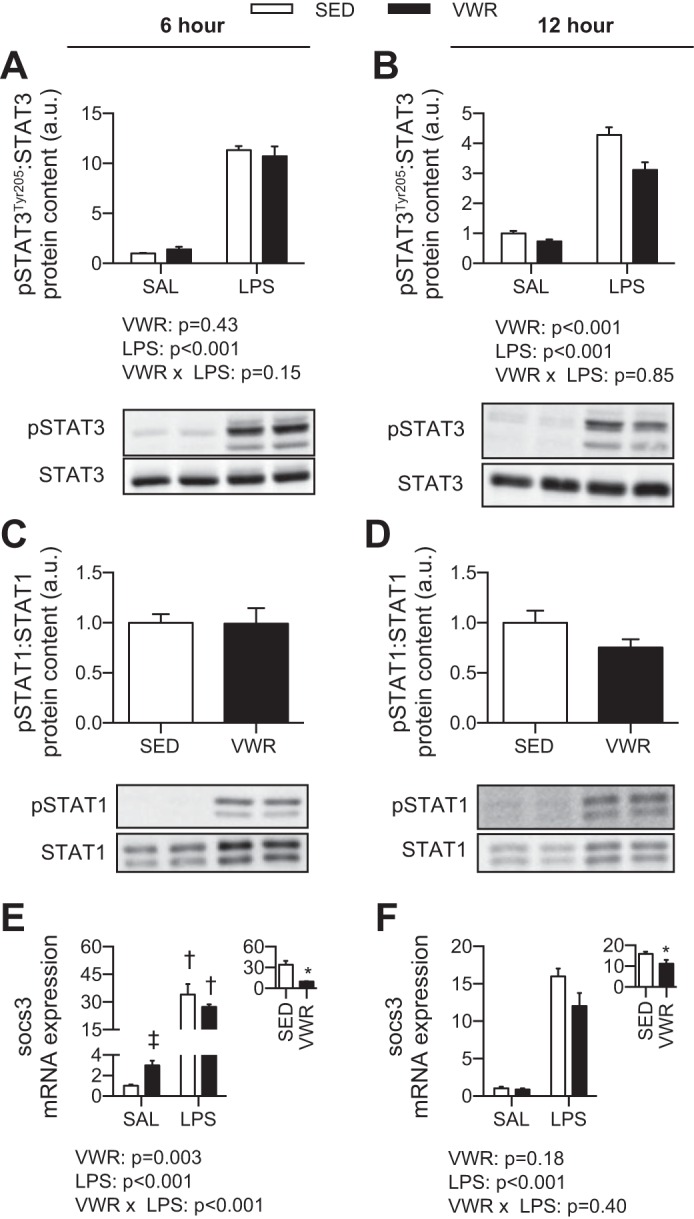
Effect of voluntary wheel running and LPS on the phosphorylation of STAT3 (A) and STAT1 (B) and the mRNA expression of SOCS3 (C). Two-way ANOVA P values are indicated below figures, with post hoc testing of interaction. †P < 0.05 indicates LPS different within SED or VWR. ‡P < 0.05 indicates VWR different within SAL or LPS group. Inset: fold change compared with SAL control. *P < 0.05 for t-test between SED and VWR groups.
To determine whether the beneficial effect at the mRNA and protein level in the liver corresponded to clinically relevant markers of liver damage, we measured serum AST and ALT, and both increased in response to LPS (two-way ANOVA main effect of LPS, P < 0.001). However, this increase was similar in SED and VWR mice for AST (SED SAL: 94.4 ± 11.3 U/l; VWR SAL: 97.7 ± 9.4 U/l; SED LPS: 166.0 ± 27.8 U/l; VWR LPS: 185.8 ± 18.7 U/l) and ALT (SED SAL: 25.9 ± 2.8 U/l; VWR SAL: 21.8 ± 1.2 U/l; SED LPS, 41.8 ± 1.0 U/l; VWR LPS: 50.0 ± 8.7 U/l). As further confirmation of this finding, we wanted to determine whether LPS exposure led to morphological alterations in the liver and whether VWR could protect against this. There were no gross histological alterations observed (data not shown).
We next wanted to determine whether the attenuation of LPS-induced liver inflammation occurred in parallel with attenuated levels of the circulating inflammatory markers IL-6, Ccl2, IFN-γ, TNF-α, IL-1β, and IL-10. There was a significant induction of all inflammatory markers in mice injected with LPS; however, as the levels in mice injected with SAL were below the level of detection, we compared SED and VWR mice injected with LPS independent of SAL controls. Despite an attenuated induction of IL-1β in VWR mice after 6 h of LPS exposure (P = 0.02), there were no other differences in circulating inflammatory markers, suggesting a direct role for VWR in attenuating LPS-induced liver inflammation (Fig. 5, A and B).
Fig. 5.
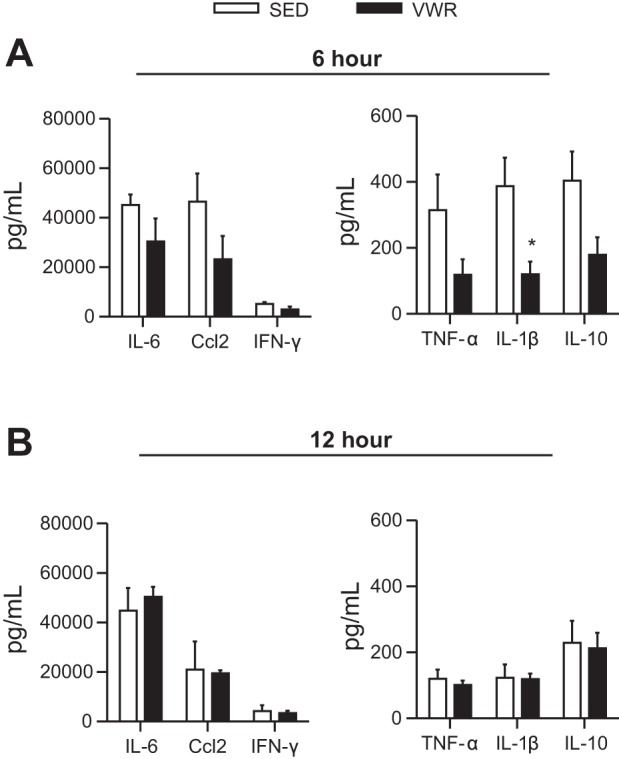
Effect of voluntary wheel running on serum levels of inflammatory markers at 6 h (A) and 12 h (B) after LPS exposure. SAL-injected mice had levels lower than the limit of detection; therefore, we used an unpaired t-test between SED and VWR groups, *P < 0.05.
The protective effects of voluntary wheel running on LPS-induced inflammation are associated with increases in HSP70/72.
As VWR conferred a protective effect against LPS induced inflammation in the liver, we next sought to elucidate potential mechanisms that could be mediating this effect. First, we assessed members of the LPS signaling complex and found no differences in TLR4 protein content with VWR or LPS (Fig. 6A); however, MyD88 protein content was increased at both 6 and 12 h post-LPS (∼1.7 and 1.3 fold, respectively), which was similar between SED and VWR groups (Fig. 6B). Next, we assessed hepatic FST and found that the mRNA expression was robustly elevated in mice injected with LPS. At 6 h post-LPS, VWR attenuated the LPS response compared with SED mice, and at 12 h post-LPS, there was a two-way ANOVA interaction that revealed the induction of FST post-LPS was lower in VWR mice (P = 0.029) (Fig. 6C). Circulating levels of FST, although induced by LPS, did not differ between SED and VWR mice (Fig. 6D). Further, there were no differences in the hepatic protein content of FST with VWR or LPS (data not shown). Third, we measured PCSK9, and neither VWR nor LPS had an effect (Fig. 6E). Lastly, we measured the protein content of HSP70/72 and in both the 6- and 12-h experiments, higher levels of HSP70/72 were observed in VWR mice regardless of SAL or LPS treatment (6-h main effect of VWR, P < 0.01; 12-h main effect of VWR, P < 0.001) (Fig. 6F).
Fig. 6.

Potential mechanisms mediating the beneficial effect of wheel running on LPS-induced inflammation. TLR4 protein content in liver (A), MyD88 protein content in liver (B), FST mRNA expression in liver (C), circulating FST in serum (D), HSP70/72 protein content in liver (E), and PCSK9 protein content in liver (F). Two-way ANOVA P values indicated in text and when a significant interaction occurred, †P < 0.05 indicates LPS different within SED or VWR group and ‡P < 0.05 indicates VWR different within LPS group. Inset: displays fold change compared with SAL control and *P < 0.05 for t-test between groups.
LPS-mediated changes in glucose homeostasis are modestly altered by voluntary wheel running.
As a final outcome measure, we wanted to determine whether VWR could protect against LPS-induced hypoglycemia and insulin resistance. At both 6 and 12 h post-LPS, blood glucose values were reduced to ∼40% of SAL control, with no protective effect of VWR (data not shown). Upon insulin injection, the blood glucose response (i.e., reduction) was impaired in mice injected with LPS, and when comparing the AUC of the blood glucose response VWR moderately attenuated the impairment at 6 but not 12 h post-LPS (main effect of VWR, P = 0.004) (Fig. 7, A and B). Together, this suggests that VWR is not able to offer significant protection against LPS-mediated reductions in blood glucose and insulin resistance, especially with a prolonged exposure to LPS.
Fig. 7.
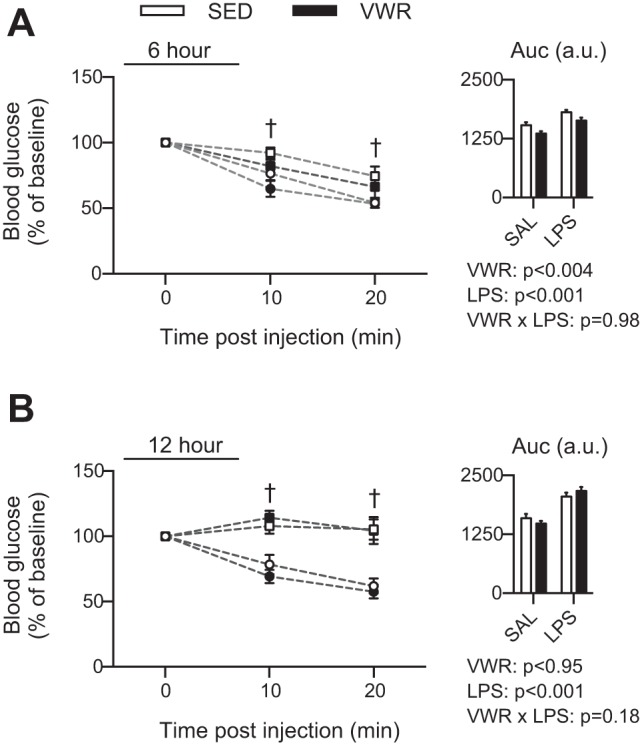
Time course of blood glucose response to an insulin injection at 6 h (A) and 12 h (B) after LPS injections. Insulin tolerance test (ITT) curve shows SAL-injected (circles) and LPS injected (squares), and data were normalized to baseline levels. Inset indicates area under curve (AUC). †A three-way ANOVA interaction post hoc indicates LPS different from SED for indicated time points at P < 0.001.
DISCUSSION
In this study, we demonstrate that 10 wk of VWR attenuates the inflammatory response to LPS within the liver of male C57BL/6J mice. In a time-dependent manner, VWR led to attenuations in the liver mRNA expression of known inflammatory markers (24). At 6 h post-LPS, there were attenuations in IL-6 and FST, while at 12 h post-LPS, IL-1β was attenuated, which together is indicative of a reduced inflammatory load. These effects occurred despite only minor alterations to circulating levels of inflammatory markers, which supports the idea that the protective effect of VWR was directly in the liver. In addition, there was modest prevention of insulin resistance in response to LPS in VWR mice at 6 h post-exposure, but not at 12 h post-exposure. Despite these protective effects, VWR had little influence on AST and ALT, which likely indicates that VWR is not able to overcome the robust effect of a supraphysiological LPS challenge on these measures.
The effects of VWR on modulating LPS-induced liver inflammation have produced mixed results. For example, Martin et al. (22) used a 0.33 mg/kg injection of LPS and did not show a protective effect of VWR on increases in the liver mRNA expression of inflammatory markers TNF-α, IL-6, and IL-1β when assessed at 24 h post-LPS. In contrast, here, we show a protective effect of VWR on the mRNA expression of IL-6 at 6 h post-LPS, and for IL-1β at 12 h post-LPS. In support of our data, others have demonstrated that 4 wk of treadmill exercise attenuates the deleterious effects of a 10 mg/kg injection of LPS on mean arterial pressure, heart rate, and leukocytopenia in nonanesthetized rats over 72 h (4). As we used a 2 mg/kg injection of LPS, which has been shown to induce a peak inflammatory response (21), but does not lead to significant mortality, as would higher doses (32), and measured mRNA expression in the liver at time points prior to significant resolution of the systemic inflammatory response (10), the discrepancies between our study and that by Martin et al. (22) are likely attributed to dose and time. However, the protective effect that we observed for the mRNA expression of inflammatory markers was not found for protein content (e.g., pSTAT3). This may be due to a different time or dose response in the liver at the protein level vs. that systemically (10).
In addition to liver, it is possible, that VWR may also confer a protective effect against LPS-induced markers of inflammation in other tissues. A recent report demonstrated that individuals who were exercise-trained responded differently than untrained subjects to LPS, and this occurred in a tissue-dependent manner (25). Trained subjects had a slightly delayed and attenuated systemic TNF-α and IL-6 response, increased adipose tissue IL-6 mRNA expression, and a heightened skeletal muscle TNF-α and IL-6 mRNA expression to a 2-h LPS challenge (25). As humans are more sensitive to LPS than mice but have a similar cytokine response (5), the findings of previous studies along with ours, suggest that physical activity and exercise training are likely able to attenuate the acute inflammatory response induced by large doses of LPS.
Exposure to LPS leads to alterations in glucose and insulin homeostasis. In this study, mice injected with LPS were hypoglycemic, as evident from a 40% reduction in blood glucose levels at 6 and 12 h postinjection, which has been previously shown by other groups (27, 30). In addition, LPS exposure induced insulin resistance that was attenuated by VWR at 6 but not 12 h, suggesting that VWR is not able to overcome the effect of a more prolonged exposure. Whether this attenuation occurred directly due to VWR or to attenuation in inflammation is not known. Ellingsgaard et al. (9) recently demonstrated that an acute injection of IL-6 induces increases in insulin secretion, and IL-6 is known to have a part in regulation of glucose homeostasis in response to LPS (33). Therefore, it is possible that attenuated insulin resistance at 6 h post-LPS occurs secondary to alterations in inflammatory levels. Our data would suggest that despite attenuated LPS induced-inflammation in the liver of VWR mice, there were minimal differences in insulin and glucose homeostasis.
In this study, we were interested in determining potential mechanisms through which VWR may protect against LPS induced inflammation. In VWR mice, we detected increases in the liver protein content of HSP70/72, a protein considered to have dual roles as a molecular chaperone and in buffering against metabolic stressors (2). Unlike other studies that observed increases in HSP70/72 protein content in the skeletal muscle after LPS exposure (7), we did not observe an effect of LPS in the liver but only of VWR. Increases in the liver protein content of HSP70/72 have recently been shown to protect against LPS induced inflammation. For example, adenoviral overexpression of HSP70/72 in the liver attenuated LPS-induced increases in serum IL-6, serum TNF-α, and hepatic NF-κB p65 expression in the nucleus (8). In addition, others (23) have used heat stress to induce HSP72 prior to an acute injection (10 mg/kg) of LPS in normal or cirrhotic rats, and this protected against increases in AST, ALT, and TNF-α in serum, while also preventing neutrophil infiltration in the liver. Together, our data, and that from previous studies (8, 23), suggest that HSP70/72 likely contributes to the beneficial effect of VWR on LPS-induced inflammation.
In addition to HSP70/72, we were also interested in exploring the potential role of the TLR4-MyD88 signaling complex and FST in mediating the protective effects of VWR. While others have shown that swim training reduces the protein content of TLR4 and its association with MyD88 in livers from rats fed a high-fat diet (26), we did not detect reductions in the content of these proteins in response to LPS injection in mice given access to a running wheel. Next, we wanted to determine the role of FST, a member of the TGFβ superfamily that has been shown to confer protection against LPS-induced inflammation. FST mRNA expression in the liver and circulating levels peak ∼5 h post-exposure to LPS (37), and the direct treatment of mice with FST prior to LPS exposure attenuates the inflammatory response (16). We show that FST mRNA in the liver is increased at 6 and 12 h post-LPS and that this is moderately attenuated with VWR. These findings suggest that despite a response of FST to LPS, it likely does not play a role in the beneficial effects of VWR. As FST would appear to be increased in response to inflammatory stresses, the blunted induction of FST in liver from VWR mice in our study may be the result of the attenuated induction of liver inflammation in these animals.
Perspectives and Significance
In this study, we demonstrate that habitual physical activity, via 10 wk of VWR, is able to protect against the inflammatory cascade induced by LPS within the liver of mice. The protective effect is primarily evident for mRNA expression of inflammatory cytokines and occurred in parallel with increases in the protein content of HSP70/72. Future work should explore whether the protective effect of habitual physical activity occurs across different doses and durations of LPS exposure and if a similar relationship exists with HSP 70/72 in different tissues.
GRANTS
This work was supported by an National Sciences and Engineering Research Council Discovery grant to D. C. Wright and a VHA-CDA2 award (1 IK2 BX001299) to R. S. Rector. D. C. Wright is a Tier II Canada Research Chair in Lipids, Metabolism, and Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: W.T.P., Z.G.A., R.S.R., and D.C.W. conception and design of research; W.T.P., Z.G.A., C.D.S., R.S.R., and D.C.W. performed experiments; W.T.P., C.D.S., and D.C.W. analyzed data; W.T.P., Z.G.A., C.D.S., R.S.R., and D.C.W. interpreted results of experiments; W.T.P. and D.C.W. prepared figures; W.T.P. and D.C.W. drafted manuscript; W.T.P., Z.G.A., C.D.S., R.S.R., and D.C.W. edited and revised manuscript; W.T.P., Z.G.A., C.D.S., R.S.R., and D.C.W. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Wendy Ward and Amanda Longo at Brock University for providing infrastructure and support for serum analyses and Laura MacRae at the University of Guelph for assistance with data collection. This work was supported, in part, with resources and the use of facilities at the Harry S. Truman Memorial Veterans Hospital in Columbia, MO.
REFERENCES
- 1.Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med 369: 840–851, 2013. [DOI] [PubMed] [Google Scholar]
- 2.Asea A, Kraeft SK, Kurt-Jones EA, Stevenson MA, Chen LB, Finberg RW, Koo GC, Calderwood SK. HSP70 stimulates cytokine production through a CD 14-dependent pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med 6: 435–442, 2000. [DOI] [PubMed] [Google Scholar]
- 3.Buzelle SL, MacPherson RE, Peppler WT, Castellani L, Wright DC. The contribution of IL-6 to β3 adrenergic receptor-mediated adipose tissue remodeling. Physiol Rep 3: 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen HI, Hsieh SY, Yang FL, Hsu YH, Lin CC. Exercise training attenuates septic responses in conscious rats. Med Sci Sports Exerc 39: 435–442, 2007. [DOI] [PubMed] [Google Scholar]
- 5.Copeland S, Warren HS, Lowry SF, Calvano SE, Remick D. Acute inflammatory response to endotoxin in mice and humans. Clin Diagn Lab Immunol 12: 60–67, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Forster I, Clausen BE, Nicola NA, Metcalf D, Hilton DJ, Roberts AW, Alexander WS. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol 4: 540–545, 2003. [DOI] [PubMed] [Google Scholar]
- 7.Cruzat VF, Pantaleão LC, Donato J Jr, de Bittencourt PIH Jr, Tirapegui J. Oral supplementations with free and dipeptide forms of l-glutamine in endotoxemic mice: effects on muscle glutamine-glutathione axis and heat shock proteins. J Nutr Biochem 25: 345–352. [DOI] [PubMed] [Google Scholar]
- 8.Dokladny K, Lobb R, Wharton W, Ma T, Moseley P. LPS-induced cytokine levels are repressed by elevated expression of HSP70 in rats: possible role of NF-κB. Cell Stress Chaperones 15: 153–163, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT, Eppler E, Bouzakri K, Wueest S, Muller YD, Hansen AM, Reinecke M, Konrad D, Gassmann M, Reimann F, Halban PA, Gromada J, Drucker DJ, Gribble FM, Ehses JA, Donath MY. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat Med 17: 1481–1489, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erickson MA, Banks WA. Cytokine and chemokine responses in serum and brain after single and repeated injections of lipopolysaccharide: multiplex quantification with path analysis. Brain Behav Immun 25: 1637–1648, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fleischmann C, Scherag A, Adhikari NK, Hartog CS, Tsaganos T, Schlattmann P, Angus DC, Reinhart K. Assessment of global incidence and mortality of hospital-treated sepsis—current estimates and limitations. Am J Respir Crit Care Med 193: 259–272, 2016. [DOI] [PubMed] [Google Scholar]
- 12.Guo Y, Zhang Y, Hong K, Luo F, Gu Q, Lu N, Bai A. AMPK inhibition blocks ROS-NF-κB signaling and attenuates endotoxemia-induced liver injury. PLos One 9: e86881, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haase TN, Ringholm S, Leick L, Bienso RS, Kiilerich K, Johansen S, Nielsen MM, Wojtaszewski JF, Hidalgo J, Pedersen PA, Pilegaard H. Role of PGC-1α in exercise and fasting-induced adaptations in mouse liver. Am J Physiol Regul Integr Comp Physiol 301: R1501–R1509, 2011. [DOI] [PubMed] [Google Scholar]
- 14.Huey KA, Meador BM. Contribution of IL-6 to the Hsp72, Hsp25, and αβ-crystallin responses to inflammation and exercise training in mouse skeletal and cardiac muscle. J Appl Physiol 105: 1830–1836, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA 304: 1787–1794, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones KL, Mansell A, Patella S, Scott BJ, Hedger MP, de Kretser DM, Phillips DJ. Activin A is a critical component of the inflammatory response, and its binding protein, follistatin, reduces mortality in endotoxemia. Proc Natl Acad Sci USA 104: 16,239–16,244, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lagu T, Rothberg MB, Shieh MS, Pekow PS, Steingrub JS, Lindenauer PK. Hospitalizations, costs, and outcomes of severe sepsis in the United States 2003 to 2007. Crit Care Med 40: 754–761, 2012. [DOI] [PubMed] [Google Scholar]
- 18.Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, Cohen J, Opal SM, Vincent JL, Ramsay G. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med 31: 1250–1256, 2003. [DOI] [PubMed] [Google Scholar]
- 19.Linden MA, Fletcher JA, Morris EM, Meers GM, Laughlin MH, Booth FW, Sowers JR, Ibdah JA, Thyfault JP, Rector RS. Treating NAFLD in OLETF rats with vigorous-intensity interval exercise training. Med Sci Sports Exerc 47: 556–567, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2[-Delta Delta C(T)] method. Methods 25: 402–408, 2001. [DOI] [PubMed] [Google Scholar]
- 21.Luster MI, Germolec DR, Yoshida T, Kayama F, Thompson M. Endotoxin-induced cytokine gene expression and excretion in the liver. Hepatology 19: 480–488, 1994. [PubMed] [Google Scholar]
- 22.Martin SA, Pence BD, Greene RM, Johnson SJ, Dantzer R, Kelley KW, Woods JA. Effects of voluntary wheel running on LPS-induced sickness behavior in aged mice. Brain Behav Immun 29: 113–123, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mikami K, Otaka M, Goto T, Miura K, Ohshima S, Yoneyama K, Lin JG, Watanabe D, Segawa D, Kataoka E, Odashima M, Watanabe S. Induction of a 72-kDa heat shock protein and protection against lipopolysaccharide-induced liver injury in cirrhotic rats. J Gastroenterol Hepatol 19: 884–890, 2004. [DOI] [PubMed] [Google Scholar]
- 24.Morris MC, Gilliam EA, Li L. Innate immune programing by endotoxin and its pathological consequences. Front Immunol 5: 680, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olesen J, Bienso RS, Meinertz S, van Hauen L, Rasmussen SM, Gliemann L, Plomgaard P, Pilegaard H. Impact of training status on LPS-induced acute inflammation in humans. J Appl Physiol 118: 818–829, 2015. [DOI] [PubMed] [Google Scholar]
- 26.Oliveira AG, Carvalho BM, Tobar N, Ropelle ER, Pauli JR, Bagarolli RA, Guadagnini D, Carvalheira JB, Saad MJ. Physical exercise reduces circulating lipopolysaccharide and TLR4 activation and improves insulin signaling in tissues of DIO rats. Diabetes 60: 784–796, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Raetzsch CF, Brooks NL, Alderman JM, Moore KS, Hosick PA, Klebanov S, Akira S, Bear JE, Baldwin AS, Mackman N, Combs TP. Lipopolysaccharide inhibition of glucose production through the Toll-like receptor-4, myeloid differentiation factor 88, and nuclear factor-κB pathway. Hepatology 50: 592–600, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rector RS, Thyfault JP, Uptergrove GM, Morris EM, Naples SP, Borengasser SJ, Mikus CR, Laye MJ, Laughlin MH, Booth FW, Ibdah JA. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J Hepatol 52: 727–736, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rojas M, Woods CR, Mora AL, Xu J, Brigham KL. Endotoxin-induced lung injury in mice: structural, functional, and biochemical responses. Am J Physiol Lung Cell Mol Physiol 288: L333–L341, 2005. [DOI] [PubMed] [Google Scholar]
- 30.Schertzer JD, Tamrakar AK, Magalhaes JG, Pereira S, Bilan PJ, Fullerton MD, Liu Z, Steinberg GR, Giacca A, Philpott DJ, Klip A. NOD1 activators link innate immunity to insulin resistance. Diabetes 60: 2206–2215, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Severgnini M, Takahashi S, Rozo LM, Homer RJ, Kuhn C, Jhung JW, Perides G, Steer M, Hassoun PM, Fanburg BL, Cochran BH, Simon AR. Activation of the STAT pathway in acute lung injury. Am J Physiol Lung Cell Mol Physiol 286: L1282–L1292, 2004. [DOI] [PubMed] [Google Scholar]
- 32.Starr ME, Ueda J, Takahashi H, Weiler H, Esmon CT, Evers BM, Saito H. Age-dependent vulnerability to endotoxemia is associated with reduction of anticoagulant factors activated protein C and thrombomodulin. Blood 115: 4886–4893, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strassmann G, Fong M, Windsor S, Neta R. The role of interleukin-6 in lipopolysaccharide-induced weight loss, hypoglycemia and fibrinogen production, in vivo. Cytokine 5: 285–290, 1993. [DOI] [PubMed] [Google Scholar]
- 34.Vincent JL, Rello J, Marshall J, Silva E, Anzueto A, Martin CD, Moreno R, Lipman J, Gomersall C, Sakr Y, Reinhart K. International study of the prevalence and outcomes of infection in intensive care units. JAMA 302: 2323–2329, 2009. [DOI] [PubMed] [Google Scholar]
- 35.Walley KR, Thain KR, Russell JA, Reilly MP, Meyer NJ, Ferguson JF, Christie JD, Nakada Ta Fjell CD, Thair SA, Cirstea MS, Boyd JH. PCSK9 is a critical regulator of the innate immune response and septic shock outcome. Sci Transl Med 6: 258ra143–258ra143, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang H, Lafdil F, Kong X, Gao B. Signal transducer and activator of transcription 3 in liver diseases: a novel therapeutic target. Int J Biol Sci 7: 536–550, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu H, Chen Y, Winnall WR, Phillips DJ, Hedger MP. Acute regulation of activin A and its binding protein, follistatin, in serum and tissues following lipopolysaccharide treatment of adult male mice. Am J Physiol Regul Integr Comp Physiol 303: R665–R675, 2012. [DOI] [PubMed] [Google Scholar]
- 38.Yamawaki Y, Kimura H, Hosoi T, Ozawa K. MyD88 plays a key role in LPS-induced Stat3 activation in the hypothalamus. Am J Physiol Regul Integr Comp Physiol 298: R403–R410, 2010. [DOI] [PubMed] [Google Scholar]
- 39.Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci USA 94: 2557–2562, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhong J, Deaciuc IV, Burikhanov R, de Villiers WJ. Lipopolysaccharide-induced liver apoptosis is increased in interleukin-10 knockout mice. Biochim Biophys Acta 1762: 468–477, 2006. [DOI] [PubMed] [Google Scholar]