

Abstract
The ability of anti-heat shock protein 90 (Hsp90) drugs to attenuate NF-κB-mediated transcription is the major basis for their anti-inflammatory properties. While the molecular mechanisms underlying this effect are not clear, they appear to be distinct in human endothelial cells. We now show for the first time that type 2 sirtuin (Sirt-2) histone deacetylase binds human NF-κB target gene promoter and prevents the recruitment of NF-κB proteins and subsequent assembly of RNA polymerase II complex in human lung microvascular endothelial cells. Hsp90 inhibitors stabilize the Sirt-2/promoter interaction and impose a “transcriptional block,” which is reversed by either inhibition or downregulation of Sirt-2 protein expression. Furthermore, this process is independent of NF-κB (p65) Lysine 310 deacetylation, suggesting that it is distinct from known Sirt-2-dependent mechanisms. We demonstrate that Sirt-2 is recruited to NF-κB target gene promoter via interaction with core histones. Upon inflammatory challenge, chromatin remodeling and core histone H3 displacement from the promoter region removes Sirt-2 and allows NF-κB/coactivator recruitment essential for RNA Pol II-dependent mRNA induction. This novel mechanism may have important implications in pulmonary inflammation.
Keywords: human lung endothelial cells, NF-κB, Sirt-2, histone h3, Hsp90
exposure to bacteria through either inhalation or systemically causes acute lung injury (ALI) in animals and humans (15, 22). Lipopolysaccharide (LPS) binds to endothelial Toll-like receptor 4 (TLR-4) and induces IKBα phosphorylation leading to ubiquitination and proteasomal degradation. NF-κB exists in a complex with the inhibitory IKBα in the cytoplasm. Following IKBα degradation, NF-κB is released and translocates into the nucleus, where it binds its cognate promoters (10, 13). NF-κB regulates the expression of proinflammatory cytokines and cell adhesion molecules on the surface of endothelial cells (6).
NF-κB binding to the cognate promoter and transcriptional activation of target genes are controlled by a complex set of transcriptional co-regulators. NF-κB coactivators serve as bridges between the NF-κB inducer and the RNA polymerase II transcription initiation complexes (14, 28, 30), whereas NF-κB co-repressors destabilize the NF-κB activator complex at the target promoter (8). NF-κB responsive promoters exhibit a unique histone modification pattern of hyperacetylated core histone H3 at K9. This allows rapid chromatin remodeling and eviction of nucleosomes from the enhancer region that unmasks obscured NF-κB enhancer elements (19). We have demonstrated that inhibition of this process by hsp90 inhibitor suppresses gene expression (31).
The family of Sirtuins negatively impact NF-κB signaling (24, 33). Sirtuins deacetylate histones and nonhistone proteins with equal efficiency (14). Seven types of sirtuins serve as epigenetic transcriptional silencers (7, 18). Sirtuins attenuate NF-κB signaling via deacetylation of either NF-κB subunits or histone proteins at target promoter region (2, 17). Type 2 sirtuin (Sirt-2) is a tubulin deacetylase and predominantly present in the cytoplasm (21). It interacts with p65 (NF-κB subunit) and deacetylates it at K310. This attenuates agonist-mediated NF-κB target gene expression (26).
The most common mechanism of NF-κB regulation is by posttranslational modifications (10). The phosphorylation of p65 by MSK-1 or PKA kinase at Ser276 is essential for its interaction with the p300/CBP coactivator (9, 25, 36). This promotes the acetylation of NF-κB by the histone acetylases associated with p300/CBP. p65 acetylation at Ser276 and Ser536 enhances its DNA binding efficiency and inhibits its interaction with IKBα (32). Previously, we reported that, in human lung endothelial cells, the inhibition of NF-κB by hsp90 inhibitors is independent of p65 Ser276 phosphorylation (31); a more direct mechanism appears to be involved. For example, the physical exclusion of NF-κB from the promoter by glucocorticoid receptors effectively suppresses NF-κB activation in dexamethasone-treated pancreatic cancer cells (20). However, this mechanism seems rather unlikely, since sirtuins lack functional DNA binding domains (27, 34), although sirt-2 binds the NEDD4 promoter at the transcription start site and thus blocks retinoic acid receptor-mediated gene induction (16).
An important regulator of immune function and NF-κB signaling pathway is the 90 kDa heat shock protein (Hsp90). We have earlier reported that the prototype hsp90 inhibitor, 17-AAG (17-N-allylamino-17-demethoxygeldanamycin), attenuates NF-κB transcription in human endothelial cells (31). We now provides evidence of the involvement of Sirt-2 in this process.
MATERIALS AND METHODS
Reagents and antibodies.
Escherichia coli endotoxin (LPS) L-3137 was purchased from Sigma-Aldrich (St. Louis, MO). Hsp90 inhibitor 17-AAG was from Selleck Chemicals (Houston, TX). Sirtuin inhibitors Sirtinol, AGK2, and EX527 were purchased from ENZO Life Sciences (Farmingdale, NY). Anti-histone H3 mouse mAB (14269), acetyl-histone H3 (Lys9) rabbit pAB (9649), and anti-PARP rabbit pAB (9542) antibodies were purchased from Cell Signaling Technology (Danvers, MA). ChIP grade anti-p65 rabbit pAB (ab7970), anti-p65 (acetyl K310) rabbit pAB (52175), anti-Sirt-2 rabbit pAB (ab67299), anti-Sirt-1 rabbit pAB (ab32441), anti-T7 goat pAB (ab9138), and anti-Flag goat pAB (ab1257), antibodies were purchased from Abcam (Cambridge, MA). Chromatin immunoprecipitation (ChIP)-grade anti-RNA polymerase II mouse mAB (39097) antibody was purchased from Active Motif (Carslbad, CA). Anti-beta-actin mouse mAB (A2228) was purchased from Sigma Aldrich. Anti-Hsp90 mouse mAB (6140419) was purchased from BD Biosciences (San Jose, CA). Anti-alpha-tubulin mouse mAB (909603) was from CRP (Covance Research Products, Denver, PA). Secondary antibodies IRDye 800CW and IRDye 680RD were purchased from Li-Cor (Lincoln, NE).
Cell culture.
Human lung microvascular endothelial cells (HLMVEC) were isolated and cultured as described in Ref. 3. HeLa human cervical carcinoma cells were a kind gift from Dr. Andrei Pakhomov (Frank Reidy Research Center for Bioelectrics, Old Dominion University) and were cultured in high glucose Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 IU penicillin, and 100 μg/ml streptomycin (CellGro Mediatech, Manassas, VA). B16F10 mouse melanoma cells were a kind gift from Dr. Loree Heller (Frank Reidy Research Center for Bioelectrics, Old Dominion University) and were cultured in McCoy's 5A (Iwakata & Grace Modification) supplemented with 25 μg/ml gentamicin (Cellgro, Mediatech) and 10% FBS.
Western blotting and coimmunoprecipitation.
Western blotting and coimmunoprecipitation were performed as described (31). Briefly, treated cells were lysed in RIPA lysis buffer supplemented with protease inhibitor cocktail V and phosphatase inhibitors (Sigma Aldrich). Either 3–5 μg normal IgG or antibody of interest was used for immunoprecipitation for 1,000 μg lysate. The immune complex was collected with Protein A/G plus agarose beads (Santa Cruz Biotechnologies, Dallas, TX) and washed with lysis buffer. The immune complex was then resolved on SDS-PAGE and transferred to the nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA). Protein interaction was detected by immunoblotting with either normal IgG or antibodies of interest. The signal was developed by using Quick Western Kit IRDye 680RD (LI-Cor BioSciences, Lincoln, NE) and developed using Li-Cor Odyssy CLx.
Immunofluorescence.
We seeded 50 × 103 HLMVEC/well of eight-well MiniCell cell culture plates (Millipore, Billerica, MA) and grew them overnight. The cells were treated with 1 EU/ml LPS for 1 h with and without 5 μg/ml 17-AAG (16 h pretreatment). The cells were then fixed with 4% paraformaldehyde (Sigma Aldrich) for 10 min, permeabilized for 15 min in 0.5% Triton X-100 at room temperature, and blocked with 3% BSA containing phosphate-buffered saline for 1 h at room temperature, followed by 5 μg/ml anti-rabbit Sirt-2 antibody (ab67299) from Abcam. After being washed, the cells were probed with 1:500 Alexa Fluor 488 anti-Rabbit IgG (H + L) from Molecular Probes (Invitrogen, Eugene, OR) for 1 h at room temperature, washed and mounted with a drop of Prolong Gold Antifade Reagent with DAPI from Molecular Probes, Invitrogen. Cells were observed using a FLUOVIEW FV1Oi confocal microscope (Olympus American, Melville, NY).
NF-κB luciferase reporter assay.
NF-κB firefly luciferase reporter adenovirus was purchased from Vector Biolabs (Philadelphia, PA). Green fluorescent protein (GFP)-expressing adenovirus was generated and characterized as in (35). HLMVEC were cotransduced with NF-κB-Luc adenovirus [10 multiplicities of infection (MOI)] and GFP adenovirus (100 MOI) in 96-well plates for 3 days and then treated with 10 EU/ml LPS for 4 h in the presence and absence of 17-AAG (5 μg/ml, 16 h pretreatment). Equal amounts of the lysate were used in triplicate for determining GFP fluorescence (485/528 nm). Luminescence was measured using the Bright Glo Luciferase reagent (Promega, Madison, WI) in a FluoStar Omega plate reader (LabTech, Cary, NC) and normalized to GFP fluorescence.
Flag-Sirt-2 and T7-p65 overexpression in B16F10 mouse melanoma cells.
B16F10 were transfected with pcDNA3.1-Flag Sirt-2 or empty pcDNA3.1-Flag/HA vector purchased from Addgene (Cambridge, MA). Cells were grown in 100 mm dishes and transfected with 10 μg plasmid with 60 μl Effectene transfection reagent (QIAGEN, Valencia, CA). Next day, the cells were transduced with T7-p65 adenoviral particles as described in Ref. 13. After 3 days, the cells were treated with LPS (1 EU/ml) for 1 h in the presence or absence of 17-AAG (16 h). Flag/Sirt-2 and T7-p65 expression levels were assessed by Western blotting.
RNAi-mediated Sirt-2 downregulation in human lung endothelial cells.
HLMVEC were transduced with either control GFP or Sirt-2 shRNA lentiviral particles (10 MOI) from Santa Cruz Biotechnology in 24-well tissue culture plates with 4 μg/ml polybrene from Santa Cruz Biotechnology in culture medium. On the 4th day, the cells were treated with LPS (1 EU/ml) for 4 h in the presence and absence of 17-AAG (5 μg/ml, 16 h). The downregulation of Sirt-2 mRNA expression was determined by TaqMan real-time quantitative (q)PCR (Applied Biosystems) and Sirt-2 protein expression by Western blot analysis.
Sirt-2 activity assay.
Sirt-2 activity assay was performed with immunoprecipitated Sirt-2 using SIRT-Glo Assay kit (Promega). Briefly, HLMVEC were grown in 150 mm dishes to 90% confluence and treated with drugs for 16 h followed by LPS (1 EU/ml) for 30 min. Cells were lysed in chilled Sirt assay buffer containing protease inhibitors and centrifuged to discard the pellet. We immunoprecipitated 1,000 μg of cell lysate with 5 μg anti Sirt-2 antibody (ab67299) (Abcam) and 25 μl of magnetic protein-G beads (Active Motif) overnight at 4°C. The immune complex was washed three times with 1 ml of chilled NP-40 buffer (10 mM Tris·HCl pH 7.6, 150 mM NaCl, and 0.1% NP-40) and finally washed with 200 μl of Sirt assay buffer. Sirt activity assay was performed with the immunoprecipitated Sirt-2 according to the supplier's instruction. Reactions were performed in triplicate, and the deacetylase activity that was inhibited by 1 mM AGK2 (Sirt-2-specific inhibitor) was considered as Sirt-2 activity.
Real-time qPCR.
Total cellular RNA was extracted with using RNeasy Plus Micro Kit from Qiagen (Valencia, CA) and reverse transcribed using random hexamer primers (High-Capacity cDNA Reverse Transcription Kit from Applied Biosystems, Carlsbad, CA). Real-time qPCR was performed using human IKBα and GAPDH TaqMan probes (Applied Biosystems) using StepOnePlus Real-Time PCR System (Applied Biosystems). All reactions were performed in triplicate; actin or GAPDH were used as internal standards.
ChIP assay.
ChIP assays were performed using IT-Express ChIP Assay Kit from Active Motif according to the supplier's instructions. Briefly, confluent HLMVEC were fixed in 1% formaldehyde and sonicated in 350 μl of shearing buffer. Approximately 14–21 μg sonicated chromatin was immunoprecipitated with 2–3 μg ChIP-grade antibody overnight at 4°C. The immune complex was captured with magnetic protein-G beads. The eluted DNA was used for amplification of human IKBα promoter region using forward: 5′-GGAATTTCCAAGCCAGTCAG-3′ and reverse: 5′-GAAGGACTTTCCAGCCACTC-3′ primers. Human VEGF promoter primers forward: 5′-CCTCAGTTCCCTGGCAACATCTG-3′ and reverse: 5′-GAAGAATTTGGCACCAAGTTTGT-3′ were used as negative control. PCR steps of 30–35 cycles were performed at 50°C depending on the ChIP antibody using HotStart IT Taq 2X PCR master mix from Affymetrix (Santa Clara, CA). The amplified PCR products were resolved on ethidium bromide agarose gel. The images were analyzed using ChemiDoc XRS+ System from (Bio-Rad). We also used ImageJ (National Institute of Health, Bethesda, MD) to determine DNA band intensity. The normalized DNA band intensity was represented as fold change in the form of a bar graph generated by GraphPad Prism 5 (La Jolla, CA).
In vivo experiments.
We purchased 6 to 8 wk old male C57BL/6 mice from Harlan (Indianapolis, IN). Mice were maintained under pathogen-free conditions with a 12:12 h light-dark cycle. All animal care and experimental procedures were approved by the Animal Care Committee of Old Dominion University. Three to six animals per group received either vehicle (saline) or 1.6 mg/kg body wt LPS intratracheally; 24 h later, mice were injected with 17-AAG (10 mg/kg body wt) alone or in combination with Sirtinol (10 mg/kg body wt) dissolved in 10% DMSO, intraperitoneally. Mice were killed 72 h following LPS treatment, and bronchoalveolar lavage fluid was collected to determine cellularity, as we have previously described (4).
Statistics.
Data represent three to six independent experiments and are reported as means ± SE. Differences among groups were analyzed by one-way ANOVA and post hoc tested with the Tukey-Kramer multiple-comparison test. The t-test for independent samples was used for comparisons between two groups. Significance was accepted at P < 0.05. Statistical calculations were performed using GraphPad Prism 5.
RESULTS
Sirt-2-specific inhibitors selectively potentiate LPS-mediated NF-κB activation and target gene expression in HLMVEC.
HLMVEC were cotransduced with adeno-NF-κB luciferase reporter (10 MOI) and adeno-GFP viruses (100 MOI) and then treated with LPS (10 EU/ml) for 4 h in the presence or absence of either a Sirt-1 inhibitor (10 μM EX527) or one of two Sirt-2 inhibitors (50 μM sirtinol or 10 μM AGK2) (pretreated for 16 h). Sirt-2, but not Sirt-1, inhibitors potentiated the NF-κB promoter activation by LPS (Fig. 1A). Additionally, HLMVEC were treated with 1 EU/ml LPS for 4 h in the presence and absence of the inhibitors. Real-time qRT-PCR analysis revealed that only Sirt-2 inhibitors potentiated the LPS-induced IKBα mRNA induction (Fig. 1B). These data suggest that Sirt-2 is the major isoform that acts as suppressor of LPS-mediated NF-κB transcriptional activation in HLMVEC. To confirm its functional significance, Sirt-2 expression was downregulated in HLMVEC using shRNA lentiviral particles. Sirt-2 expression in GFP- and Sirt-2 shRNA-transduced HLMVEC. There was significant reduction in Sirt-2 expression and potentiation of LPS-induced IKBα mRNA expression in Sirt-2 knock-down cells (Fig. 1, C–E). These data confirm that Sirt-2 predominantly acts as the negative regulator of LPS-induced NF-κB transcriptional activation HLMVEC.
Fig. 1.

Type 2 sirtuin (Sirt-2) suppresses NF-κB transcriptional activation in human lung microvascular endothelial cells (HLMVEC). A: HLMVEC were cotransduced with adeno-green fluorescent protein (GFP) and adeno-NF-κB reporter luciferase viral particles. The cells were then treated with 10 EU/ml LPS for 4 h in the absence or presence of the Sirt-1-specific inhibitor (10 μM EX527) or of either one of the Sirt-2 selective inhibitors (50 μM Sirtinol or 10 μM AGK2) (16 h pretreatment). Luciferase activity was determined in triplicate in equal volumes of cell lysates using a luminometer, normalized to the respective GFP fluorescence, and plotted as fold change from untreated controls (± SE, n = 3). B: HLMVEC were treated with 1 EU/ml LPS for 4 h in the absence or presence of sirtuin inhibitors as in A. Total RNA was extracted and reverse transcribed as discussed in materials and methods. The bar graph (± SE, n = 3) represents fold change in IKBα mRNA expression from control. HLMVEC were transduced with either GFP control or Sirt-2 shRNA lentiviral particles, as discussed in materials and methods. C and D: the shRNA lentivirus-mediated downregulation of Sirt-2 protein and Sirt-2 mRNA expression by Western blotting and TaqMan quantitative (q)RT-PCR, respectively. In E, 1 EU/ml LPS significantly induced more IKBα mRNA expression compared with control in wild-type cells and the downregulation of Sirt-2 significantly potentiated IKBα mRNA induction by LPS compared with LPS-treated wild-type cells (± SE, n = 3).
Hsp90 inhibitor suppresses NF-κB activation via Sirt-2 in human lung endothelial cells.
The effect of hsp90 inhibition in human lung endothelial cells is unique (31). The hsp90 inhibitor 17-AAG does not prevent the LPS-mediated NF-κB nuclear translocation but attenuates NF-κB activation and target gene expression. To study whether Sirt-2 is involved in this process, we treated HLMVEC with LPS along with 5 μg/ml 17-AAG and sirtuin inhibitors for 16 h. Sirtinol potentiated the LPS-induced NF-κB activation and reversed the suppression of LPS-induced NF-κB reporter activation by 17-AAG (Fig. 2A). Furthermore, qRT-PCR analysis showed that only Sirt-2 inhibitors (Sirtinol and AGK2) reversed the inhibitory effect of 17-AAG on NF-κB target gene (IKBα) mRNA induction by LPS (1 EU/ml, 4 h), whereas the Sirt-1 inhibitor EX527 did not (Fig. 2B). Moreover, Sirt-2 knock-down partially but significantly recovered IKBα mRNA expression in the presence of 17-AAG (Fig. 2C). The Sirt-2-dependent effect is also observed in the induction of IL-8 mRNA (another well-known NF-κB target gene) by LPS and 17-AAG in HLMVEC (data not shown).
Fig. 2.

Sirt-2 inhibition reverses the repression of LPS-mediated NF-κB activation and target gene expression by 17-AAG. A: adeno-GFP and adeno-NF-κB reporter luciferase viral particles were cotransduced as in Fig. 1. Cells were treated with LPS for 4 h in the absence and presence of 5 μg/ml 17-AAG and 50 μM Sirtinol (Sirt-2 selective inhibitor, together for 16 h, pretreatment). Luciferase activity (± SE, n = 3) was significantly increased by LPS; 17-AAG suppressed the LPS effect, whereas Sirtinol potentiated the LPS effect and partially recovered the 17-AAG effect on LPS treatment in HLMVEC. B: TaqMan qRT-PCR analysis (± SE, n = 3) showed that LPS induced IKBα mRNA expression; 17-AAG suppressed the LPS effect, the sirtuin inhibitor potentiated the LPS and reversed the 17-AAG effect in HLMVEC. B (i), (ii), and (iii): 50 μM Sirtinol-, 10 μM AGK2-, and 10 μM EX527-treated cells, respectively. C: Sirt-2 expression was downregulated by Sirt-2 shRNA lentiviral particles in HLMVEC; cells were then treated with LPS in the presence and absence of 17-AAG. Bar graph (± SE) represents fold change in IKBα mRNA expression in 1 of 3 independent experiments. LPS induced IKBα mRNA expression in wild-type and potentiated IKBα mRNA expression in Sirt-2 knock-down HLMVEC; 17-AAG suppressed and partially but significantly reversed the effect of 17-AAG in Sirt-2 knock-down HLMVEC.
Hsp90 inhibitor suppresses LPS-induced inflammation via Sirt-2, in vivo.
To confirm the above observations in vivo, we employed the intratracheal LPS mouse model of acute lung injury (ALI) that is characterized by severe alveolo-capillary inflammation. Figure 3 shows that LPS (1.6 mg/kg body wt intratracheally) induced profound infiltration of immune cells into C57BL/6 mouse lungs, as reflected in increased cellularity of the bronchoalveolar lavage fluid (BALF). 17-AAG (10 mg/kg body wt intraperitoneal, 24 h posttreatment) significantly reduced this hypercellularity. However, concurrent posttreatment with Sirt-2 inhibitor (Sirtinol, 10 mg/kg body wt intraperitoneal) potentiated BALF hypercellularity and significantly reversed the effect of 17-AAG. These data confirm that Sirt-2 plays a critical role in the regulation of LPS-induced lung inflammation in vivo.
Fig. 3.
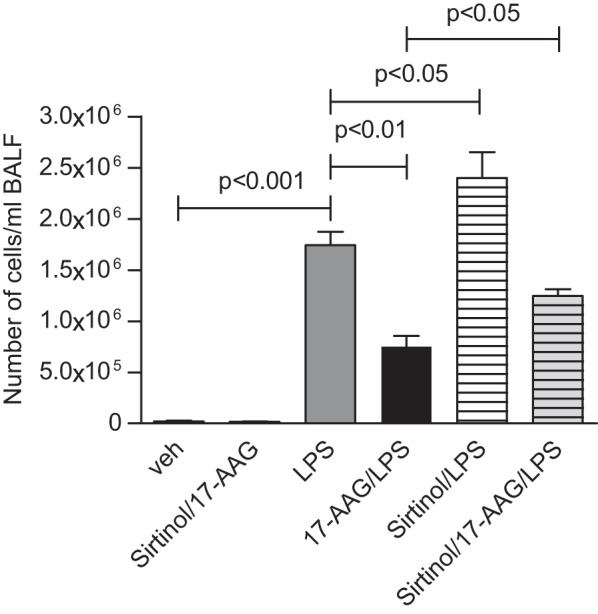
In vivo potentiation of LPS-induced lung inflammation by the Sirt-2-specific inhibitor, Sirtinol, and reversal of the 17-AAG effect. C57BL/6 mice (3–8 animals/group) received 1.6 mg LPS/kg body wt intratracheally. We treated the mice, 24 h later, with 17-AAG (10 mg/kg body wt ip) either alone or in combination with Sirtinol (10 mg/kg body wt). We determined lung inflammation 72 h after LPS treatment from the degree of immune cell infiltration into the lung by bronchoalveolar lavage (BAL). Means ± SE of BAL fluid (BALF) cellularity/ml.
Hsp90 inhibition induces Sirt-2 nuclear accumulation without affecting Sirt-2 activity.
A leucine-rich nuclear export signal of Sirt-2 causes the localization of Sirt-2 both in the cytoplasm and in the nucleus (17), suggesting that a nucleo-cytoplasmic shuttling mechanism exists for Sirt-2. Hsp90 regulates the function of several nuclear import-export proteins (23), so we investigated whether 17-AAG may perturb Sirt-2 cellular distribution. While a major portion of Sirt-2 was expressed in the cytosolic fraction, a small but significant amount was present in the nuclear fraction of HLMVEC. LPS or 17-AAG alone did not change Sirt-2 distribution (Fig. 4A), but there was a small but significant induction of nuclear Sirt-2 by LPS and 17-AAG together (Fig. 4, A and B). This observation was confirmed in immunocytochemistry/immunofluorescence studies (Fig. 4C). It remains unclear how 17-AAG and LPS affected this process. However, a more abundant nuclear Sirt-2 should promote a robust transcriptional suppression. Next, to determine whether LPS and 17-AAG influence Sirt-2 activity, HLMVEC were treated with 1 EU/ml LPS for 30 min in the presence and absence of 5 μg/ml 17-AAG (16 h, pretreatment). LPS induced total Sirt-2 activity (Fig. 4D), and that was unaffected by 17-AAG. We also overexpressed Flag-tagged human Sirt-2 in HeLa cells and performed coimmunoprecipitation-immunoblotting (co-IP/IB) analysis. No direct interaction between Sirt-2 and hsp90 was observed (Fig. 4E), indicating that Sirt-2 is not a client of hsp90 and that it is thus unlikely that hsp90 inhibition by 17-AAG would directly influence Sirt-2 activity. In a parallel study, we overexpressed Sirt-2 and PCAF (a bonafied Sirt-2 interacting protein) in HeLa cells and observed their interaction, confirming that the absence of hsp90 in Sirt-2 immune complex was not due to inappropriate techniques (data not shown).
Fig. 4.

Heat shock protein (Hsp)90 inhibition does not alter LPS-induced total Sirt-2 activity but promotes nuclear accumulation of Sirt-2 in the presence of LPS. HLMVEC were treated with LPS for 1 h in the presence and absence of 17-AAG (16 h, pretreatment). A: cytosolic and nuclear fractions were prepared and cellular distribution of Sirt-2 was determined by Western blot analysis. Top: PARP nuclear expression served as nuclear loading control; bottom: α-Tubulin served as loading control. B: fold changes in nuclear and cytosolic Sirt-2 expression (± SE, n = 3). The nuclear accumulation of Sirt-2 was significant only in the presence of both 17-AAG and LPS. C: immunofluorescence staining of Sirt-2; representative ICC/IF image of Alexa 488-stained (green) Sirt-2 (top), counterstained with DAPI (blue, middle), and a merged image (bottom). D: HLMVEC were treated with 1 EU/ml LPS for 30 min in the presence and absence of 5 μg/ml 17-AAG (16 h pretreatment). Sirt-2 was immunoprecipitated from the whole cell lysate, and Sirt-2 activation was determined, as discussed in materials and methods. Bar graph (± SE, n = 3) represents fold Sirt-2 activity changes from untreated controls. Total Sirt-2 activity was induced by LPS and 17-AAG/LPS. 17-AAG did not affect LPS-induced total Sirt-2 activity. D: i: B16F10 mouse melanoma cells were cooverexpressed with T7-tagged p65 adenovirus and Flag-tagged Sirt-2 and treated as indicated. Cell lyaste was immunoprecipitated with anti-Flag antibody and immunoblotted with anti-T7 antibody. ii: HLMVEC were infected with T7-tagged p65 adenovirus and treated as indicated. Cell lysate was immunoprecipitated with anti-T7 antibody and immunoblotted with anti-Sirt-2 antibody. Sirt-2 and p65 did not coimmunoprecipitate. E: to investigate whether Sirt-2 is a client protein of hsp90, we performed coimmunoprecipitation (co-IP) analysis using Flag-Sirt-2 overexpressing HeLa cells, as described in materials and methods. Representative co-IP/immunoblots (IB) indicate that there is no direct association between Sirt-2 and hsp90.
Sirt-2 is not involved in the deacetylation of p65 in HLMVEC.
Sirt-2 has been reported to influence NF-κB activation via deacetylation of K310 of p65 (21). To investigate whether this also occurs in HLMVEC, we overexpressed T7-tagged p65 adenovirus particles (31) in HLMVEC and then treated them with 1 EU/ml LPS for 1 h in the presence or absence of 5 μg/ml 17-AAG (16 h pretreatment). LPS induced p65 acetylation that was attenuated by 17-AAG (Fig. 5A). Additionally, similarly treated HLMVEC were fixed in 1% formaldehyde and sonicated to obtain 200 bp - 1.5 Kb chromatin fragments. We immunoprecipitated 14–21 μg chromatin with anti-p65 and acetyl-p65 K310 antibodies. The eluted chromatin fragment was PCR-amplified with the HIF-1 binding element of VEGF promoter serving as negative control. Acetylated p65K310 bound to the IKBα promoter; LPS induced p65 promoter binding but did not further increase acetylated p65K310 binding; 17-AAG reduced both the basal as well as the LPS-induced binding of both p65 and acetylated p65 (Fig. 5, B and C). Next, we tested whether Sirt-2 interacted with p65. T7-/p65 adenovirus and Flag-tagged Sirt-2 were co-overexpressed in B16F10 mouse melanoma cells and HLMVEC. Cells were then treated as before; T7-p65 was immunoprecipitated and immunoblotted with anti-Flag antibody. No interaction was observed between Sirt-2 and p65 (Fig. 5D). Thus, it appears unlikely that Sirt-2 influences NF-κB activation through p65 K310 deacetylation. We have previously demonstrated that 17-AAG suppresses LPS-induced CREB-binding protein (CBP) histone acetylase (HAT) activity, without affecting LPS-induced histone deacetylase (HDAC) activity. 17-AAG also disrupted the LPS-induced interaction between p65 and CBP coactivator in HLMVEC. These observations suggest that p65-interacting HAT coactivators, rather than Sirt-2, were the major contributors to LPS-induced p65 acetylation/deacetylation.
Fig. 5.

p65 K310 acetylation is not involved in Sirt-2-dependent NF-κB regulation. A: T7-tagged p65 was overexpressed in HLMVEC using adenoviral particles. T7-p65 was immunoprecipitated (IP) with anti-T7 antibody and immunoblotted (IB) with anti-Acetyl lysine antibody. The immunocomplex was washed and resolved on SDS-PAGE. There was induction of lysine acetylation of p65 by LPS that was attenuated by 17-AAG. B: to investigate whether LPS and 17-AAG affect Acetyl-lysine310-p65 recruitment to the IKBα promoter, chromatin Immunoprecipitation (ChIP) assays were performed, as described in materials and methods. B: a representative image of PCR products resolved on ethidium bromide agarose gel indicating that LPS does not induced Acetyl-lysine310-p65 binding. C: quantification of data presented in B (± SE, n = 3) expressed as fold changes from vehicle. LPS did not enhance Acetyl-lysine310-p65 binding to the IKBα promoter.
Sirt-2 is recruited to NF-κB target gene (IKBα) promoter and prevents transcription in HLMVEC.
The Sirt-2 structure does not contain a DNA-binding domain (27, 34), yet Sirt-2 binds near the transcription start site of the NEDD4 promoter (16). Thus, Sirt-2 could influence IKBα promoter activation either directly or indirectly (via an epigenetic mechanism). Using ChIP analysis, under basal conditions, we found that Sirt-2 is recruited to the IKBα promoter region (−178 to +65 bp). As expected, Sirt-1 did not bind to the promoter, and its effect on IKBα induction was negligible (Fig. 6A). LPS caused core histone H3 displacement and Sirt-2 dissociation from the promoter (Fig. 6, A, C, and D). This was followed by p65 recruitment at the same promoter region (Fig. 6A), which contains three critical NF-κB binding sites (12). Finally, there was an enhanced binding of RNA Polymerase II (Fig. 6, A and E), indicating activation of IKBα gene transcription. 17-AAG prevented the LPS-induced dissociation of core histone H3 and Sirt-2 from the IKBα promoter (Fig. 6, A, C, and D). We therefore propose that the stability of Sirt-2/promoter interaction could be the major factor determining the recruitment of transcription factors and formation of RNA polymerase II complex at the IKBα promoter. The mutual exclusion of Sirt-2 and p65 from the IKBα promoter could be due to the lack of physical interaction between these two proteins. We also speculated that Sirt-2 and p65 are parts of two different complexes with opposing influences on IKBα promoter activation. Previously we have shown that, upon LPS treatment, p65 forms a complex with CBP coactivator at the IKBα promoter and that the activator complex is destabilized and removed by 17-AAG (31). Thus, we propose that an as yet unidentified factor forms a repressor complex with Sirt-2.
Fig. 6.

Direct association of Sirt-2 with the IKBα promoter is responsible for the activation of NF-κB-dependent transcription in HLMVEC. To investigate how Sirt-2 affects IKBα promoter activity and the recruitment of other factors, we performed ChIP assays with treated HLMVEC, as in Fig. 4. A: a representative image of PCR products resolved on ethidium bromide agarose gel. B–E: the quantification of data shown in A (± SE, n = 3) expressed as fold change in promoter binding of p65, Sirt-2, histone H3, and RNA Pol II, respectively. LPS induced p65 and RNA Pol II promoter binding but reduced Sirt-2 and histone H3 binding to the IKBα promoter. 17-AAG blocked the LPS induced p65 binding, restored Sirt-2 and histone H3 binding and induced RNA Pol II binding to the IKBα promoter.
Sirt-2 inhibitor reverses the 17-AAG effect and restores p65 and RNA pol II binding to the NF-κB target gene (IKBα) promoter.
To understand how Sirt-2 inhibitors affect Sirt-2 promoter recruitment we performed ChIP assays. Sirtinol destabilized Sirt-2 promoter binding and reversed the 17-AAG-induced binding stabilization (Fig. 7, A and C). Sirtinol also reversed the 17-AAG effect and induced the displacement of core histone H3 from the IKBα promoter (Fig. 7, A and D). Furthermore, Sirtinol promoted p65 (Fig. 7, A and B) and RNA polymerase II (Fig. 7, A and E) recruitment to the promoter and reversed the suppressive effect of 17-AAG. Thus, the cosuppressors recruited by 17-AAG at the NF-κB target gene (IKBα) promoter appears to require Sirt-2 activity. We have shown that LPS removes Sirt-2 from the IKBα promoter and, at the same time, induces Sirt-2 activation in HLMVEC. While these observations may seem paradoxical, we believe that Sirt-2 activation could be part of a negative feedback mechanism and that a prolific cosuppressor complex is assembled only in the presence of 17-AAG; this complex requires Sirt-2 activity to assemble on the IKBα promoter and prevent the recruitment and assembly of the NF-κB coactivator complex. The destabilization of p65-coactivators complex by 17-AAG further enhances cosuppressor complex recruitment. In the absence of 17-AAG and of cosuppressors, the NF-κB coactivator complex is assembled unhindered at the promoter, in response to LPS.
Fig. 7.

The Sirt-2 selective inhibitor, sirtinol, counters the 17-AAG effect and stabilizes Sirt-2 promoter binding in HLMVEC. We performed ChIP assays with HLMVEC treated with 17-AAG and LPS and increasing concentrations of sirtinol. A: a representative image of PCR products resolved on ethidium bromide agarose gel. B–E: the quantification of data from A (± SE, n = 3), expressed as fold changes in promoter binding of p65, Sirt-2, histone H3, and RNA Pol II, respectively. LPS induced p65 and RNA Pol II promoter binding but reduced Sirt-2 and histone H3 binding to the IKBα promoter. 17-AAG blocked the LPS effects and this was restored by sirtinol, in a concentration-dependent manner. F: total HLMVEC lysate was immunoprecipitated with anti-Sirt-2 antibody and immunoblotted with anti-histone H3 antibody. We observed coimmunoprecipitation of Sirt-2 and histone H3.
It is not clear how Sirt-2 is recruited at the NF-κB target gene promoters under basal conditions. To investigate a possible interaction between histone H3 and Sirt-2, co-IP/IB analysis was performed. Sirt-2 physically interacts with histone H3 (Fig. 7F). This suggests that Sirt-2 may be recruited to the IKBα promoter via its interaction with core histone H3.
Results from these studies are summarized in Fig. 7: Sirt-2 is involved in the suppression of NF-κB activation and NF-κB target gene (IKBα) induction by inflammatory stimuli (e.g., LPS) in HLMVEC. Under basal, unstimulated conditions, Sirt-2 is recruited to the promoter via interaction with core histone H3 and occupies the enhancer region of the promoter. Inflammatory stimuli paradoxically enhance Sirt-2 activity, but this is counteracted the rapid induction in NF-κB activity. Thus, Sirt-2 appears to be a novel NF-κB negative feedback regulator in HLMVEC. LPS induces p65 nuclear transport and interaction with p300/CBP coactivators. This interaction also enables p65 lysine acetylation by p300/CBP HAT. Together, these events promote the displacement of core histone H3 and Sirt-2 from the promoter region. The p65 coactivator complex in turn recruits RNA Pol II to initiate gene transcription. In the presence of hsp90 inhibitor, there is no p65 coactivator complex formation and the hypoacetylated p65 alone is not sufficient to dissociate Sirt-2 from the promoter. Additionally, the enhanced nuclear accumulation and the elevated Sirt-2 activity in the presence of both LPS and 17-AAG further stabilize Sirt-2/promoter interaction.
DISCUSSION
Hsp90 inhibitors are promising therapeutic agents because of their ability to target multiple signaling pathways simultaneously. These targets are mostly, but not exclusively, hsp90 client proteins, which number nearly 300 (23). The expression and activity of these proteins are under the control of hsp90, which, in turn, is intimately coupled to the 26S proteasome (11). The inhibition of hsp90/client interaction by anti-hsp90 drugs leads to the destabilization, polyubiquitination, and degradation of the client protein. Several key kinases and important intermediates of NF-κB signaling pathway, such as RIP kinase, TAK-1 kinase, and IκB kinases are hsp90 clients (23). Consequently, the NF-κB signaling pathway is highly sensitive to hsp90 inhibitors. Hsp90 inhibition in most cell types leads to disruption in NF-κB signaling and prevents the nuclear translocation of NF-κB proteins. Thus, anti hsp90 drugs have potential as anti-inflammatory drugs.
Severe bacterial infection causes hyperactivation of NF-κB in the pulmonary endothelium. Hsp90 inhibition by 17-AAG prevents LPS induced hyperpermeability and other pulmonary complications in mice (5). We have previously shown that, unlike in other cell types, in human pulmonary vascular endothelium 17-AAG prevents NF-κB transcriptional activation without blocking NF-κB nuclear translocation (31). Until now, the mechanisms behind this observation remained unclear.
Of the seven sirtuin isoforms (Sirt-1 to -7), types 1, 2, and 6 have been implicated in the regulation of NF-κB via direct deacetylation of either p65 or acetyl histone H3 K9 (2, 17). Our data confirm that Sirt-2 is the key regulator of NF-κB activation in human lung endothelial cells. Sirt-2 did not affect p65 lysine acetylation and did not perturb NF-κB signaling. However, it regulated the recruitment and binding of essential factors at the target promoter. We discovered that Sirt-2 binds to NF-κB target gene (IKBα) promoter under basal conditions. The binding of Sirt-2 was associated with the suppression of IKBα promoter activity either basally or in the presence of 17-AAG. Other sirtuin isoforms, Sirt-1 and Sirt-6, did not bind to IKBα promoter (data not shown), and thus, their influence was minimal. Either pharmacologic inhibition or shRNA-mediated Sirt-2 downregulation potentiated the effect of inflammatory challenge and reversed the suppression of IKBα expression by 17-AAG.
We also observed that Sirt-2 is not an hsp90 client protein and did not physically interact with hsp90. Thus, we did not observe drastic changes in Sirt-2 activity by hsp90 inhibition. However, hsp90 inhibitors induced accumulation of Sirt-2 in the nucleus in stimulated (inflamed) cells. This suggested that perhaps Sirt-2 nuclear import is facilitated by the combined action of inflammation and hsp90 inhibition. The enhancement of nuclear Sirt-2 accumulation in the presence of both LPS and 17-AAG likely helped sustain its inhibitory function at NF-κB target promoters.
The major NF-κB negative feedback regulatory system is via IKBα induction (29). The instant induction of IKBα by NF-κB activation causes IKBα accumulation in the nucleus. IKBα-NF-κB interaction is much more stringent than NF-κB-DNA interaction and effectively removes NF-κB from the promoter. This provides an efficient mechanism to terminate NF-κB signaling. Thus, the induction of Sirt-2 activity by LPS adds another dimension to endothelial NF-κB signaling. LPS paradoxically induces both NF-κB signaling and Sirt-2 activity. As Sirt-2 inhibition or knock-down potentiated NF-κB activation, we propose that Sirt-2 activation is a component of a negative feedback regulatory mechanism of NF-κB signaling. Our study also suggested that Sirt-2 activity is essential for keeping the NF-κB target promoter in the closed conformation.
Sirtuins do not possess a DNA binding motif, and thus it is not clearly understood how Sirt-2 is recruited to the IKBα promoter in the basal state. Perhaps chromatin-associated proteins may be responsible for Sirt-2 binding. Although NF-κB-responsive promoters have hyperacetylated core histone H3 K9, the position of nucleosome at the enhancer region largely determines the recruitment of NF-κB subunits and subsequent assembly of RNA Pol II initiation complex at the IKBα promoter (31). We have previously demonstrated that, under basal conditions, core histone H3 masks the NF-κB enhancer elements of the IKBα promoter. Upon inflammatory stimulation, core histone H3 is displaced from the promoter to unmask the enhancer region and permit NF-κB binding. This is similar to the pattern of Sirt-2 binding to the IKBα promoter. Sirt-2 has been shown to interact with hyperacetylated histones H3 and H4 (21). Thus, we speculate that Sirt-2 binds to the IKBα promoter via its interaction with core histone H3. Whether Sirt-2 requires other factors in addition to its interaction to occupy the IKBα enhancer region is not clear.
Alternatively, Sirt-2 is recruited to the promoter via a basal co-repressor complex. The human IKBα promoter exists in a closed conformation in the unstimulated state. We have demonstrated (MNase assay) that the IKBα promoter region becomes accessible to MNase only in the presence of LPS (31). This suggests that a basal suppressor complex is initially assembled at the promoter possibly to prevent nonspecific gene activation. Additionally, we have observed the existence of an SWI/SNF suppressor complex at the IKBα promoter, under basal conditions (unpublished data). Hsp90 inhibitors revert the open promoter conformation to the closed one, which no longer is accessible to MNase, even in the presence of LPS.
The inability to displace core histone H3 from the promoter in the presence of hsp90 inhibitor reduces IKBα mRNA expression. Thus, hsp90 inhibition perhaps favors the reassembly of the Sirt-2/repressor complex at the promoter, which cannot be displaced by LPS. One reason for that could be the destabilization of NF-κB/p300/CBP coactivator complex by 17-AAG (31). This destabilization affects the acetylation-dependent NF-κB/DNA interaction, as well as the recruitment of other transcription factors to sustain RNA Pol II-dependent transcription. The main difference between the basal and stimulated states is the Sirt-2 activity status: total Sirt-2 activity is lowered in the basal state compared with LPS stimulation. Inhibition of Sirt-2 activity displaces the repressor complex and promotes NF-κB binding. However, this mechanism only partly recovers NF-κB activation and IKBα mRNA expression. It is still not clear whether Sirt-2 inhibition restores the disruption of NF-κB/p300/CBP interaction by hsp90 inhibitors. The proposed mechanism of hsp90-dependent NF-κB transcriptional activation and the role of Sirt-2 is summarized in Fig. 8.
Fig. 8.

Schematic representation of suppression of inflammation-induced NF-κB target gene promoter activation by hsp90 inhibitors via Sirt-2.
Pathological NF-κB activation in the pulmonary microvascular endothelium is under the control of hsp90. We now demonstrate a novel mechanism, one that is unique to human pulmonary endothelium, namely the involvement of Sirtuins and especially Sirt-2. As discussed elsewhere, hsp90 inhibition blocks IKB kinase activation and restores inhibitory IKBα protein in several other cell types (31). This is considered the universal negative regulatory mechanism that blocks NF-κB target gene expression (10, 29). However, in HLMVEC, hsp90 inhibition does not prevent NF-κB nuclear translocation in response to LPS. As the availability of active NF-κB proteins in the nucleus becomes sufficient to induce target promoters, human lung microvascular endothelia appear to have evolve a secondary regulatory mechanism, via Sirt-2. As we demonstrate here, Sirt-2 plays the crucial role of the NF-κB transcriptional inhibitor in the lung microvasculature. Our in vivo data also confirm the physiological role of Sirt-2 in LPS-induced lung inflammation in mice, which has been directly correlated to abnormal NF-κB induction (1, 15, 22). It is intriguing to consider that drugs that modulate Sirt-2 function could have therapeutic potential for sepsis and acute lung inflammatory diseases.
GRANTS
This work was funded by National Heart, Lung, and Blood Institute Grant HL-093460.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
G.S.T., C.B., N.B., B.W.G., M.A.C., J.R.N., D.J.F., and J.D.C. conception and design of research; G.S.T., C.B., N.B., B.W.G., and M.A.C. performed experiments; G.S.T., C.B., N.B., B.W.G., M.A.C., J.R.N., D.J.F., and J.D.C. analyzed data; G.S.T., C.B., N.B., B.W.G., M.A.C., J.R.N., D.J.F., and J.D.C. interpreted results of experiments; G.S.T., C.B., and N.B. prepared figures; G.S.T. drafted manuscript; G.S.T. and J.D.C. edited and revised manuscript; G.S.T., C.B., N.B., B.W.G., M.A.C., J.R.N., D.J.F., and J.D.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Drs. Loree Heller and Ardrei Pakhomov of Old Dominion University, Norfolk, VA, for kindly providing B16F10 Mouse melanoma cells and HeLa human cervical cancer cells, respectively.
REFERENCES
- 1.Abraham E. Nuclear factor-kappaB and its role in sepsis-associated organ failure. J Infect Dis 178: S364–S369, 2003. [DOI] [PubMed] [Google Scholar]
- 2.Bagul PK, Deepthi N, Sultana R, Banerjee SK. Resveratrol ameliorates cardiac oxidative stress in diabetes through deacetylation of NFkB-p65 and histone 3. J Nutr Biochem 26: 1298–1307, 2015. [DOI] [PubMed] [Google Scholar]
- 3.Catravas JD, Snead C, Dimitropoulou C, Chang AS, Lucas R, Verin AD, Black SM. Harvesting, identification and barrier function of human lung microvascular endothelial cells. Vascul Pharmacol 52: 175–181, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chatterjee A, Dimitropoulou C, Drakopanayiotakis F, Antonova G, Snead C, Cannon J, Venema RC, Catravas JD. Heat shock protein 90 inhibitors prolong survival, attenuate inflammation, and reduce lung injury in murine sepsis. Am J Respir Crit Care Med 176: 667–675, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chatterjee A, Snead C, Yetik-Anacak G, Antonova G, Zeng J, Catravas JD. Heat shock protein 90 inhibitors attenuate LPS-induced endothelial hyperpermeability. Am J Physiol Lung Cell Mol Physiol 294: L755–L763, 2008. [DOI] [PubMed] [Google Scholar]
- 6.Dauphinee SM, Karsan A. Lipopolysaccharide signaling in endothelial cells. Lab Invest 86: 9–22, 2006. [DOI] [PubMed] [Google Scholar]
- 7.Favero G, Franceschetti L, Rodella LF, Rezzani R. Sirtuins, aging, and cardiovascular risks. Age (Dordr) 37: 9804, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao Z, Chiao P, Zhang X, Zhang X, Lazar MA, Seto E, Young HA, Ye J. Coactivators and corepressors of NF-kappaB in IkappaB alpha gene promoter. J Biol Chem 280: 21091–21098, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sci USA 94: 2927–2932, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev 18: 2195–2224, 2004. [DOI] [PubMed] [Google Scholar]
- 11.Imai J, Maruya M, Yashiroda H, Yahara I, Tanaka K. The molecular chaperone Hsp90 plays a role in the assembly and maintenance of the 26S proteasome. EMBO J 22: 3557–3567, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ito CY, Kazantsev AG, Baldwin AS Jr. Three NF-kappa B sites in the I kappa B-alpha promoter are required for induction of gene expression by TNF alpha. Nucleic Acids Res 22: 3787–3792, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med 13: 460–469, 2007. [DOI] [PubMed] [Google Scholar]
- 14.Kim JW, Jang SM, Kim CH, An JH, Kang EJ, Choi KH. New molecular bridge between RelA/p65 and NF-kappaB target genes via histone acetyltransferase TIP60 cofactor. J Biol Chem 287: 7780–7791, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X, Su J, Cui X, Li Y, Barochia A, Eichaker PQ. Can we predict the effects of NF-kappaB inhibition in sepsis? Studies with parthenolide and ethyl pyruvate. Expert Opin Investig Drugs 18: 1047–1060, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu PY, Xu N, Malyukova A, Scarlett CJ, Sun YT, Zhang XD, Ling D, Su SP, Nelson C, Chang DK, Koach J, Tee AE, Haber M, Norris MD, Toon C, Rooman I, Xue C, Cheung BB, Kumar S, Marshall GM, Biankin AV, Liu T. The histone deacetylase SIRT2 stabilizes Myc oncoproteins. Cell Death Differ 20: 503–514, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu TF, Vachharajani V, Millet P, Bharadwaj MS, Molina AJ, McCall CE. Sequential actions of SIRT1-RELB-SIRT3 coordinate nuclear-mitochondrial communication during immunometabolic adaptation to acute inflammation and sepsis. J Biol Chem 290: 396–408, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinez-Redondo P, Vaquero A. The diversity of histone versus nonhistone sirtuin substrates. Genes Cancer 4: 148–163, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Natoli G. NF-kappaB and chromatin: ten years on the path from basic mechanisms to candidate drugs. Immunol Rev 246: 183–192, 2012. [DOI] [PubMed] [Google Scholar]
- 20.Newton R, Holden NS. Separating transrepression and transactivation: a distressing divorce for the glucocorticoid receptor? Mol Pharmacol 72: 799–809, 2007. [DOI] [PubMed] [Google Scholar]
- 21.North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell 11: 437–444, 2003. [DOI] [PubMed] [Google Scholar]
- 22.Orfanos SE, Mavrommati I, Korovesi I, & Roussos C. Pulmonary endothelium in acute lung injury: from basic science to the critically ill. Intensive Care Med 30: 1702–1714, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Picard D. Hsp90 Interactors. http://www.picard.ch/downloads/Hsp90interactors.pdf (last accessed on Dec. 21, 2015). [Google Scholar]
- 24.Poulose N, Raju R. Sirtuin regulation in aging and injury. Biochim Biophys Acta 1852: 2442–55, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reber L, Vermeulen L, Haegeman G, Frossard N. Ser276 phosphorylation of NF-kB p65 by MSK1 controls SCF expression in inflammation. PLoS One 4: e4393, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rothgiesser KM, Erener S, Waibel S, Luscher B, Hottiger MO. SIRT2 regulates NF-kappaB dependent gene expression through deacetylation of p65 Lys310. J Cell Sci 123: 4251–4258, 2010. [DOI] [PubMed] [Google Scholar]
- 27.Sanders BD, Jackson B, Marmorstein R. Structural basis for sirtuin function: what we know and what we don't. Biochim Biophys Acta 1804: 1604–1616, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sapountzi V, Logan IR, Robson CN. Cellular functions of TIP60. Int J Biochem Cell Biol 38: 1496–1509, 2006. [DOI] [PubMed] [Google Scholar]
- 29.Scott ML, Fujita T, Liou HC, Nolan GP, Baltimore D. The p65 subunit of NF-kappa B regulates I kappa B by two distinct mechanisms. Genes Dev 7: 1266–1276, 1993. [DOI] [PubMed] [Google Scholar]
- 30.Sheppard KA, Rose DW, Haque ZK, Kurokawa R, McInerney E, Westin S, Thanos D, Rosenfeld MG, Glass CK, Collins T. Transcriptional activation by NF-kappaB requires multiple coactivators. Mol Cell Biol 19: 6367–6378, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thangjam GS, Dimitropoulou C, Joshi AD, Barabutis N, Shaw MC, Kovalenkov Y, Wallace CM, Fulton DJ, Patel V, Catravas JD. Novel mechanism of attenuation of LPS-induced NF-kappaB activation by the heat shock protein 90 inhibitor, 17-N-allylamino-17-demethoxygeldanamycin, in human lung microvascular endothelial cells. Am J Respir Cell Mol Biol 50: 942–952, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wan F, Lenardo MJ. Specification of DNA binding activity of NF-kappaB proteins. Cold Spring Harb Perspect Biol 1: a000067, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Winnik S, Auwerx J, Sinclair DA, Matter CM. Protective effects of sirtuins in cardiovascular diseases: from bench to bedside. Eur Heart J 36: 3404–3412, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuan H, Marmorstein R. Structural basis for sirtuin activity and inhibition. J Biol Chem 287: 42428–42435, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Q, Malik P, Pandey D, Gupta S, Jagnandan D, Belin de Chantemele E, Banif B, Marrero MB, Rudic RD, Stepp DW, Fulton DJ. Paradoxical activation of endothelial nitric oxide synthase by NADPH oxidase. Arterioscler Thromb Vasc Biol 28: 1627–1633, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell 1: 661–671, 1998. [DOI] [PubMed] [Google Scholar]