

Abstract
Context
Under normal physiological conditions, leptin regulates body weight by creating a balance between food intake and energy expenditure. However, in obesity, serum leptin levels increase and become defective to retain energy balance.
Evidence Acquisition
Elevated serum leptin levels are regarded as an established marker of obesity. It is also reported that obese asthmatic patients have maximum serum leptin levels compared to other groups such as non-obese asthmatics, and normal obese and non obese subjects without asthma. In addition to having an appetite suppressing effect, leptin also regulates certain acute-phase protein expressions including α-1 antitrypsin (A1AT) in the liver.
Results
A1AT is a protease inhibitor that counterbalances the activity of the neutrophil elastase (NE) enzyme. A1AT reductions in obese-leptin resistant subjects lead to increased NE activity. The overactivity of NE degrades lung tissue proteins, which may lead to pulmonary disorders including asthma.
Conclusions
On the basis of prior studies, it could be hypothesized that, in obese asthmatic patients, the highest degree of leptin failure/resistance might lead to the creation of an imbalance between NE and its inhibitor A1AT. To ascertain this, large scale prospective studies are warranted to assess the comparative serum leptin and A1AT levels and NE activity in asthmatic non-obese and obese patients, simultaneously. Such studies might help to devise novel interventional therapies for the treatment of pulmonary-related problems including asthma, chronic obstructive pulmonary disorder (COPD), and other lung defects in susceptible obese subjects in the future.
Keywords: Leptin Resistance, Obesity, Asthma, Chronic Obstructive Pulmonary Disorder (COPD), α-1 Antitrypsin (A1AT), Neutrophil Elastase (NE)
1. Context
Obesity is characterized by an excessive accumulation of fat in the adipose tissue (1, 2). A product of the obese (Ob) gene, called leptin, is released primarily from adipocytes and plays a key role in regulating body weight (3). In most obese subjects, leptin fails to perform its physiological functions in spite of its high serum levels (4, 5).
Obesity is linked with several disorders including cardiovascular diseases (CVDs), certain types of cancer (6), type 2 diabetes, and it raises the risk of pulmonary defects (7).
Recently, Arteaga-Solis et al. demonstrated that leptin resistance leads to increased parasympathetic tone, which in turn causes bronchoconstriction and obesity-associated asthma (8). Another previous study described a link between hypoventilation and adiposity (9). Phipps et al. proposed that hyperleptinemia is a cause of respiratory failure in obese leptin-resistant subjects (10). It has been suggested that a derangement of leptin expression in adipocytes may affect the lungs and promote asthma (11). Interestingly, a previous study reported the highest levels of serum leptin occur in asthmatic obese patients compared to the other human groups including non-obese controls, obese subjects, and asthmatic non obese patients (12). Serum leptin elevation has already been shown to be an established marker of leptin resistance in severe obesity (4). This might lead to the creation of an imbalance between α-1 antitrypsin (A1AT) and NE activity (13, 14). A severe deficiency of serum A1AT is important in the development of chronic obstructive pulmonary disorder (COPD) and asthma, which might be due to the degradation of lung tissue elastin by NE-enhanced activity (15, 16). Briefly, on the basis of the facts described above, it could be postulated that leptin resistance with a protease-antiprotease imbalance may be vital for the obesity predisposition to pulmonary disorders including, among others, asthma and COPD.
This review is divided into two parts. The first part highlights body weight regulation and the disruption of leptin’s physiological function in obesity. The second part summarizes a possible link between obesity and pulmonary disorders.
2. Evidence Acquisition
2.1. Body Weight Regulation by the Leptin Hormone
Leptin was first identified in 1994 in the obese (ob/ob) mouse model. It is a 16 kDa non glycated protein consisting of 167 amino acids and is primarily expressed in adipose tissues (17). It is encoded by the obese gene (Ob gene), located on chromosome number 7 in humans, and is responsible for regulating the balance between food intake and energy expenditure (18). During starvation, leptin levels go down, which increases appetite and decreases energy consumption. On the other hand, with sufficient energy stores, leptin inhibits appetite and permits the utilization of energy stores (19). Leptin regulates energy expenditure and food intake by communicating with the central nervous system (CNS) via its receptor (Ob-Rb) located in the hypothalamus (20). The hypothalamus is the key site for leptin detection as it contains two types of neurons: type 1 expresses appetite suppressing peptides derived from pro-opiomelanocortin (POMC) precursors, whereas type 2 produces appetite stimulating peptides such as neuropeptide Y (NPY) and agouti-related peptide (AgRP). Leptin suppresses appetite by counteracting NPY and AgRP, whereas leptin activates POMC mRNA expression, which enhances the release of a potent appetite-suppressing peptide, alpha-melanocyte stimulating hormone (α-MSH) (21, 22). Mechanisms of leptin action and dysfunction are shown in Figures 1 and 2, respectively.
Figure 1. An Overview of the Regulation of Food Intake and Energy Expenditure by Leptin’s Actions in the Brain.
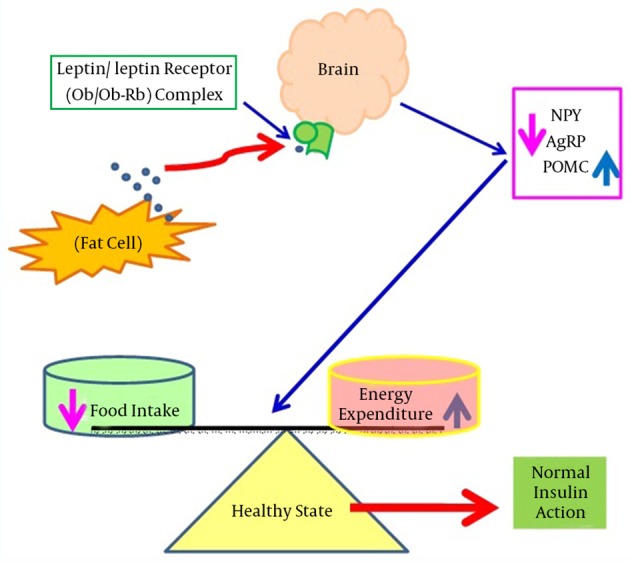
This shows shows that during normal physiological states, leptin binds with its receptors in the brain and suppresses appetite by counteracting NPY and AgRP, however, leptin also induces POMC mRNA expression. Abbreviations: AgRP, agouti-related peptide; mRNA, messenger ribonucleic acid; NPY, neuropeptide Y; Ob, leptin; Ob-Rb, leptin receptor; POMC, pro-opiomelanocortin.
Figure 2. Consequences of the Impairment of Leptin Signaling.

This figure shows that in obesity, leptin cannot bind with its receptors situated in the brain (hypothalamus), resulting in adiposity signals arrived due to the stimulation of NPY and AgRP expression with a concomitant decrease of POMC mRNA expression. The leptin failure leads to severe obesity that is associated with various disorders including insulin resistance, T2D, CVD, hypertension, and asthma. Abbreviations: AgRP, agouti-related peptide; CVD, cardiovascular disease; NPY, neuropeptide Y; Ob, leptin; Ob-Rb, leptin receptor; POMC, pro-opiomelanocortin; T2D, type 2 diabetes.
2.2. Obesity and Leptin Dysfunction
Different causes underlie the disruption of leptin’s physiological functions. Some studies have shown that obese (Ob/Ob) mice are leptin deficient, and diabetic (db/db) mice have mutated leptin receptors contributing to leptin dysfunctions (23, 24). A rare genetic disorder has been reported in obese humans, which might be due to a mutation in the leptin gene and can be treated with exogenous leptin administration. It has been identified that 12 Pakistani, 5 Turkish, 1 Austrian, and 2 Egyptian obese subjects are leptin deficient because of leptin gene mutations (25). On the other hand, several previous studies have reported that most obese individuals have elevated serum leptin levels, which are positively correlated with body mass index (BMI) (26, 27). Despite having increased serum leptin levels, leptin fails to retain its appetite-suppressing effect of reducing weight in obese subjects. Approximately 30 fold higher leptin concentrations were required for weight reduction in obesity (28). The failure of leptin to function in severely obese subjects may be due to extracellular circulating factors. An interaction between circulating leptin and serum leptin interacting proteins (SLIPs) contributes to leptin failures (4, 29). This leptin appetite suppressing effect, which is markedly impaired in obese subjects, is shown in Figure 2.
2.3. Induction of A1AT Expression by Leptin in Hepatocytes
Leptin is structurally identical to the granulocyte colony-stimulating factor (GCSF). GCSF is a member of the interleukin-6 (IL-6) family that includes IL-6 and oncostatin-M (OSM) cytokines (30, 31). In addition to leptin’s control of energy homeostasis by stimulating its hypothalamic receptor, the Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling is essential in peripheral tissues, especially in the liver. This is vital because it affects differential expression of the target genes of acute phase proteins (APPs), including A1AT (13). Using a wide range of techniques, it has been shown that OSM induces a functional response after 24 hours via STAT3 binding to the STAT sequence (32). Moreover, different reports using gel shift and luciferase assays have identified that a perfect consensus for STAT present in the 3'-A1AT enhancer region was capable of binding the transcription factor STAT3 (33). For other cell types such as monocytes and macrophages, lipopolysaccharides (LPS) up-regulate the A1AT gene, which is also stimulated in the lung epithelial tissues in response to OSM (34, 35). In HepG2 cells, the A1AT gene regulation occurs at the transcriptional stage, which is mediated primarily via hepatocyte promoters containing the STAT3 sequence (32). The A1AT levels were stimulated up to three folds by two IL-6 and OSM in HepG2 cell lines (35). It was suggested that the short and long leptin receptor isoforms were expressed in HepG2 hepatic cells, which supports the hypothesis that, like OSM cytokines, leptin might act to regulate few gene targets in hepatocytes (36). Recently, Jiang’s laboratory demonstrated that the expression of the A1AT gene was leptin-dependent in mouse models and leptin stimulation increases the A1AT levels both at the mRNA and protein levels via the JAK/STAT3 pathway in cultured hepatic Hep1-6 cell lines (13). A specific tyrosine residue, Tyr 1138, in the intracellular domain of the leptin receptor (Ob-Rb) mediates the activation of STAT3 (37). The binding of the leptin-ligand causes the Ob-Rb to undergo homo-oligomerization and subsequently binds to JAK2. Only Ob-Rb possesses the STAT-binding site. In vivo studies have demonstrated that STAT3 is the major transcriptional factor in signaling. Binding of Ob-Rb with JAK2 leads to the JAK2 autophosphorylation and the phosphorylation of Tyr985, tyr1077, and Tyr 1138 on the Ob-Rb receptor. The phosphorylation of Tyr1138 recruits STAT3 proteins to the Ob-Rb-JAK2 complex. Tyrosine-phosphorylated STAT3 molecules can dimerize and translocate into the nucleus to activate the transcription of target genes in peripheral tissues such as vascular endothelial cells and HepG2 liver cells (35, 38). An induction of A1AT expression by leptin is illustrated in Figure 3.
Figure 3. Stimulation of A1AT Expression by the Leptin-JAK/STAT3 Pathway in Peripheral Tissues Including Hepatocytes.
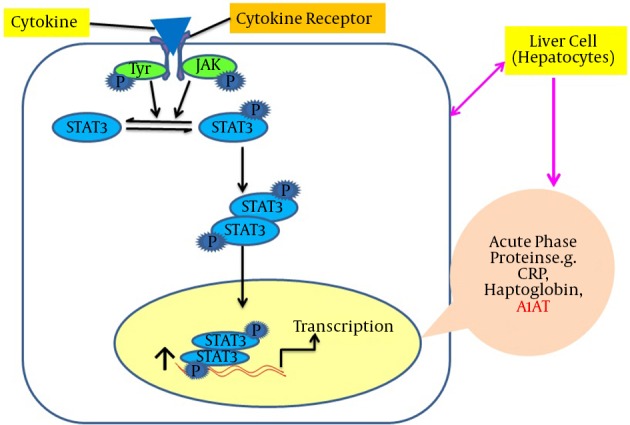
This shows shows that leptin binds with its receptors on hepatocytes, successively STAT3 molecules dimerize and translocate into the nucleus and bind with the promoter region of the serpin gene in order to up-regulate the acute phase protein expression, including A1AT. Abbreviations: A1AT, alpha-1-antitrypsin; Jak-STAT3, Janus kinase–signal transducer and activator of transcription.
3. Results
In obesity, the disruption of leptin-mediated signaling occurs that may lead to lung function failure by concomitant increases in body weight. Olson et al. suggested that leptin stimulates lung ventilation, whereas leptin deficiencies lead to hypoventilation in obese subjects (11). The relationship between obesity and asthma is complex and involves several mechanisms (9). A few recent studies have indicated the highest levels of leptin occur in the sera of asthmatic obese patients relative to the other groups such as asthmatic non obese patients, normal obese subjects, and non-obese subjects without asthma or other pulmonary complications. Canoz et al. report 14.5, 40, and 76% increases in serum leptin in asthmatic obese patients compared to obese subjects without asthma and non-obese subjects, with and without asthma, respectively (12). Wahab and colleagues have indicated significant increases in serum leptin levels in obese asthmatic patients (25.8 ± 11.1 ng/mL) compared with non-obese asthmatic patients (8.8 ± 11.1 ng/mL) (39). Another study has shown the maximum serum leptin levels in obese asthmatic patients (19.37 ± 14.04 ng/mL) is elevated compared with non-obese asthmatic patients (6.37 ± 2.46 ng/mL) and healthy controls (6.50 ± 3.51 ng/mL) (40). Mahmoud et al. demonstrated maximum serum leptin levels in COPD cases, both in incitement and static conditions, relative to the other groups. Their calculations indicated that, compared to the non obese control subjects, there were 90.34, 68.75, and 64% increases in serum leptin levels in obese with COPD, obese without COPD, and non-obese with COPD cases, respectively (41). Therefore, these previous investigations clearly indicate that leptin resistance is linked to respiratory/pulmonary-related complications and may contribute to the development of a unique asthma phenotype in obese patients.
An elegant study reported that leptin stimulates A1AT expression at both the mRNA and protein levels via the JAK2-STAT3 pathway in liver HepG 2 and Hep 1-6 cell lines. Alternatively, serum levels of A1AT are reduced in obese leptin-resistant subjects with parallel increases in neutrophil elastase (NE) activity (13). Elevated serum NE levels are also related to airway constriction in obese subjects (42). In the same obese subjects, increases in C-reactive protein (CRP) have also been reported (42), which induce leptin resistance via CRP-leptin adduct formation (29). An imbalance of A1AT and NE leads to lung tissue impairments (14, 43). Leptin’s actions on the brain and hepatocytes are summarized in Figure 4.
Figure 4. Summary of Leptin’s Actions in the Brain and Liver.

4. Conclusions
4.1. Hypothesis and Outlook for Further Research
In short, previously published data raise the possibility that the protease-antiprotease balance is leptin dependent, and due to the leptin resistance protective capacity of A1AT, it could be arrested to counteract NE activity. Consequently, the degradation of lung tissues, especially elastin, occurs by NE overactivity, which could lead to pulmonary related problems in the susceptible obese population. This hypothesis is summarized as: leptin resistance in asthmatic obese patients may reduce serum alpha 1 antitrypsin levels. In turn, NE enhances, which can lead to the development of pulmonary-related complications including asthma and COPD due to the degradation of proteins in lung tissues.
Exploring mechanisms underlying the derangement of protease antiprotease counterparts will aid in devising personalized interventional therapy for particular obese patients prone to lung complications. This option is preferred over generalizing treatment for all patients suffering from respiratory complications, including non-obese subjects.
Footnotes
Authors Contributions:Abdul Rehman Khan conceived the idea and wrote the initial drafts; Fazli Rabbi Awan helped in writing and revision of the manuscript.
References
- 1.Farooqi S, O'Rahilly S. Genetics of obesity in humans. Endocr Rev. 2006;27(7):710–8. doi: 10.1210/er.2006-0040. [DOI] [PubMed] [Google Scholar]
- 2.Yang W, Kelly T, He J. Genetic epidemiology of obesity. Epidemiol Rev. 2007;29:49–61. doi: 10.1093/epirev/mxm004. [DOI] [PubMed] [Google Scholar]
- 3.Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395(6704):763–70. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- 4.Martin SS, Qasim A, Reilly MP. Leptin resistance: a possible interface of inflammation and metabolism in obesity-related cardiovascular disease. J Am Coll Cardiol. 2008;52(15):1201–10. doi: 10.1016/j.jacc.2008.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Myers MJ, Leibel RL, Seeley RJ, Schwartz MW. Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metab. 2010;21(11):643–51. doi: 10.1016/j.tem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goodpaster BH, Krishnaswami S, Harris TB, Katsiaras A, Kritchevsky SB, Simonsick EM, et al. Obesity, regional body fat distribution, and the metabolic syndrome in older men and women. Arch Intern Med. 2005;165(7):777–83. doi: 10.1001/archinte.165.7.777. [DOI] [PubMed] [Google Scholar]
- 7.Boulet LP. Asthma and obesity. Clin Exp Allergy. 2013;43(1):8–21. doi: 10.1111/j.1365-2222.2012.04040.x. [DOI] [PubMed] [Google Scholar]
- 8.Arteaga-Solis E, Zee T, Emala CW, Vinson C, Wess J, Karsenty G. Inhibition of leptin regulation of parasympathetic signaling as a cause of extreme body weight-associated asthma. Cell Metab. 2013;17(1):35–48. doi: 10.1016/j.cmet.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Castro-Rodriguez JA. [Relationship between obesity and asthma]. Arch Bronconeumol. 2007;43(3):171–5. doi: 10.1016/s1579-2129(07)60043-3. [DOI] [PubMed] [Google Scholar]
- 10.Phipps PR, Starritt E, Caterson I, Grunstein RR. Association of serum leptin with hypoventilation in human obesity. Thorax. 2002;57(1):75–6. doi: 10.1136/thorax.57.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olson AL, Zwillich C. The obesity hypoventilation syndrome. Am J Med. 2005;118(9):948–56. doi: 10.1016/j.amjmed.2005.03.042. [DOI] [PubMed] [Google Scholar]
- 12.Canoz M, Erdenen F, Uzun H, Muderrisoglu C, Aydin S. The relationship of inflammatory cytokines with asthma and obesity. Clin Invest Med. 2008;31(6):E373–9. doi: 10.25011/cim.v31i6.4924. [DOI] [PubMed] [Google Scholar]
- 13.Mansuy-Aubert V, Zhou QL, Xie X, Gong Z, Huang JY, Khan AR, et al. Imbalance between neutrophil elastase and its inhibitor alpha1-antitrypsin in obesity alters insulin sensitivity, inflammation, and energy expenditure. Cell Metab. 2013;17(4):534–48. doi: 10.1016/j.cmet.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stockley RA. Neutrophils and protease/antiprotease imbalance. Am J Respir Crit Care Med. 1999;160(5 Pt 2):S49–52. doi: 10.1164/ajrccm.160.supplement_1.13. [DOI] [PubMed] [Google Scholar]
- 15.Ranes J, Stoller JK. A review of alpha-1 antitrypsin deficiency. Semin Respir Crit Care Med. 2005;26(2):154–66. doi: 10.1055/s-2005-869536. [DOI] [PubMed] [Google Scholar]
- 16.Gadek JE, Pacht ER. The protease-antiprotease balance within the human lung: implications for the pathogenesis of emphysema. Lung. 1990;168 Suppl:552–64. doi: 10.1007/BF02718178. [DOI] [PubMed] [Google Scholar]
- 17.Huang L, Li C. Leptin: a multifunctional hormone. Cell Res. 2000;10(2):81–92. doi: 10.1038/sj.cr.7290038. [DOI] [PubMed] [Google Scholar]
- 18.Friedman JM. A tale of two hormones. Nat Med. 2010;16(10):1100–6. doi: 10.1038/nm1010-1100. [DOI] [PubMed] [Google Scholar]
- 19.Bates SH, Myers MJ. The role of leptin receptor signaling in feeding and neuroendocrine function. Trends Endocrinol Metab. 2003;14(10):447–52. doi: 10.1016/j.tem.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 20.Harvey J, Ashford ML. Leptin in the CNS: much more than a satiety signal. Neuropharmacology. 2003;44(7):845–54. doi: 10.1016/s0028-3908(03)00076-5. [DOI] [PubMed] [Google Scholar]
- 21.Enriori PJ, Evans AE, Sinnayah P, Cowley MA. Leptin resistance and obesity. Obesity (Silver Spring). 2006;14 Suppl 5:254S–8S. doi: 10.1038/oby.2006.319. [DOI] [PubMed] [Google Scholar]
- 22.Patel HR, Qi Y, Hawkins EJ, Hileman SM, Elmquist JK, Imai Y, et al. Neuropeptide Y deficiency attenuates responses to fasting and high-fat diet in obesity-prone mice. Diabetes. 2006;55(11):3091–8. doi: 10.2337/db05-0624. [DOI] [PubMed] [Google Scholar]
- 23.Mark AL, Correia ML, Rahmouni K, Haynes WG. Loss of leptin actions in obesity: two concepts with cardiovascular implications. Clin Exp Hypertens. 2004;26(7-8):629–36. doi: 10.1081/ceh-200031948. [DOI] [PubMed] [Google Scholar]
- 24.Palmer G, Aurrand-Lions M, Contassot E, Talabot-Ayer D, Ducrest-Gay D, Vesin C, et al. Indirect effects of leptin receptor deficiency on lymphocyte populations and immune response in db/db mice. J Immunol. 2006;177(5):2899–907. doi: 10.4049/jimmunol.177.5.2899. [DOI] [PubMed] [Google Scholar]
- 25.Paz-Filho G, Mastronardi C, Delibasi T, Wong ML, Licinio J. Congenital leptin deficiency: diagnosis and effects of leptin replacement therapy. Arq Bras Endocrinol Metabol. 2010;54(8):690–7. doi: 10.1590/s0004-27302010000800005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu FB, Chen C, Wang B, Stampfer MJ, Xu X. Leptin concentrations in relation to overall adiposity, fat distribution, and blood pressure in a rural Chinese population. Int J Obes Relat Metab Disord. 2001;25(1):121–5. doi: 10.1038/sj.ijo.0801480. [DOI] [PubMed] [Google Scholar]
- 27.Maachi M, Pieroni L, Bruckert E, Jardel C, Fellahi S, Hainque B, et al. Systemic low-grade inflammation is related to both circulating and adipose tissue TNFalpha, leptin and IL-6 levels in obese women. Int J Obes Relat Metab Disord. 2004;28(8):993–7. doi: 10.1038/sj.ijo.0802718. [DOI] [PubMed] [Google Scholar]
- 28.Stanley S, Wynne K, McGowan B, Bloom S. Hormonal regulation of food intake. Physiol Rev. 2005;85(4):1131–58. doi: 10.1152/physrev.00015.2004. [DOI] [PubMed] [Google Scholar]
- 29.Chen K, Li F, Li J, Cai H, Strom S, Bisello A, et al. Induction of leptin resistance through direct interaction of C-reactive protein with leptin. Nat Med. 2006;12(4):425–32. doi: 10.1038/nm1372. [DOI] [PubMed] [Google Scholar]
- 30.Baumann H, Gauldie J. The acute phase response. Immunol Today. 1994;15(2):74–80. doi: 10.1016/0167-5699(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 31.Moshage H. Cytokines and the hepatic acute phase response. J Pathol. 1997;181(3):257–66. doi: 10.1002/(sici)1096-9896(199703)181:3<257::aid-path756>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 32.Morgan K, Marsters P, Morley S, van Gent D, Hejazi A, Backx M, et al. Oncostatin M induced alpha1-antitrypsin (AAT) gene expression in Hep G2 cells is mediated by a 3' enhancer. Biochem J. 2002;365(Pt 2):555–60. doi: 10.1042/BJ20011312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morgan K, Scobie G, Marsters P, Kalsheker NA. Mutation in an alpha1-antitrypsin enhancer results in an interleukin-6 deficient acute-phase response due to loss of cooperativity between transcription factors. Biochim Biophys Acta. 1997;1362(1):67–76. doi: 10.1016/s0925-4439(97)00064-1. [DOI] [PubMed] [Google Scholar]
- 34.Barbey-Morel C, Pierce JA, Campbell EJ, Perlmutter DH. Lipopolysaccharide modulates the expression of alpha 1 proteinase inhibitor and other serine proteinase inhibitors in human monocytes and macrophages. J Exp Med. 1987;166(4):1041–54. doi: 10.1084/jem.166.4.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sallenave JM, Tremblay GM, Gauldie J, Richards CD. Oncostatin M, but not interleukin-6 or leukemia inhibitory factor, stimulates expression of alpha1-proteinase inhibitor in A549 human alveolar epithelial cells. J Interferon Cytokine Res. 1997;17(6):337–46. doi: 10.1089/jir.1997.17.337. [DOI] [PubMed] [Google Scholar]
- 36.Zhou J, Lei W, Shen L, Luo HS, Shen ZX. Primary study of leptin and human hepatocellular carcinoma in vitro. World J Gastroenterol. 2008;14(18):2900–4. doi: 10.3748/wjg.14.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang R, Barouch LA. Leptin signaling and obesity: cardiovascular consequences. Circ Res. 2007;101(6):545–59. doi: 10.1161/CIRCRESAHA.107.156596. [DOI] [PubMed] [Google Scholar]
- 38.Singh P, Hoffmann M, Wolk R, Shamsuzzaman AS, Somers VK. Leptin induces C-reactive protein expression in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2007;27(9):e302–7. doi: 10.1161/ATVBAHA.107.148353. [DOI] [PubMed] [Google Scholar]
- 39.Abdul Wahab A, Maarafiya MM, Soliman A, Younes NB, Chandra P. Serum Leptin and Adiponectin Levels in Obese and Nonobese Asthmatic School Children in relation to Asthma Control. J Allergy (Cairo). 2013;2013:654104. doi: 10.1155/2013/654104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zivanovic SS, Saranac L, Kamenov B, Bjelakovic B, Petrovic S. Leptin levels in obese and non-obese children with asthma. Eur Respir J. 2011;38(Suppl 55):p4267. [Google Scholar]
- 41.Mahmoud AE, Omar MM, Abdelghaffar Hibah NA, Issa HA. Leptin hormone in obese and non-obese stable and exacerbated cases of chronic obstructive pulmonary disease. Egypt J Chest Dis Tuberc. 2015;64(3):557–65. doi: 10.1016/j.ejcdt.2015.03.011. [DOI] [Google Scholar]
- 42.El-Eshmawy MM, El-Adawy EH, Mousa AA, Zeidan AE, El-Baiomy AA, Abdel-Samie ER, et al. Elevated serum neutrophil elastase is related to prehypertension and airflow limitation in obese women. BMC Womens Health. 2011;11:1. doi: 10.1186/1472-6874-11-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang P, Bamlet WR, Sun Z, Ebbert JO, Aubry MC, Krowka MJ, et al. Alpha1-antitrypsin and neutrophil elastase imbalance and lung cancer risk. Chest. 2005;128(1):445–52. doi: 10.1378/chest.128.1.445. [DOI] [PubMed] [Google Scholar]