

Abstract
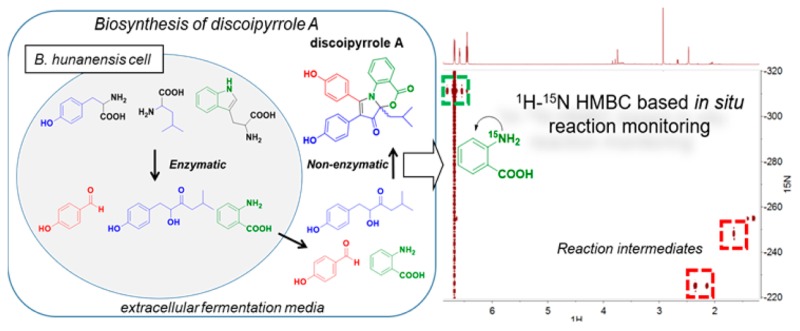
Discoipyrroles A–D (DPA–DPD) are recently discovered natural products produced by the marine bacterium Bacillus hunanensis that exhibit anticancer properties in vitro. Initial biosynthetic studies demonstrated that DPA is formed in the liquid fermentation medium of B. hunanensis from three secreted metabolites through an unknown but protein-independent mechanism. The increased identification of natural products that depend on non-enzymatic steps creates a significant need to understand how these different reactions can occur. In this work, we utilized 15N-labeled starting materials and continuous high-sensitivity 1H–15N HMBC NMR spectroscopy to resolve scarce reaction intermediates of the non-enzymatic discoipyrrole reaction as they formed in real time. This information guided supplemental experiments using 13C- and 18O-labeled materials to elucidate the details of DPA’s non-enzymatic biosynthesis, which features a highly concerted pyrrole formation and necessary O2-mediated oxidation. We have illustrated a novel way of using isotopically enhanced two-dimensional NMR spectroscopy to interrogate reaction mechanisms as they occur. In addition, these findings add to our growing knowledge of how multicomponent non-enzymatic reactions can occur through inherently reactive bacterial metabolites.
Introduction
The discoipyrroles (DPs) are a family of natural products that were isolated from bacterial fermentation of Bacillus hunanensis strain SNA-048. The DPs were shown to be inhibitors of discoidin domain receptor-2 (DDR2)-dependent cell migration of BR5 human foreskin fibroblasts and potent cytotoxins to DDR2 mutant non-small-cell lung cancer cell lines.1
In the initial report of discoipyrrole A (DPA, 1), we demonstrated its production in the aqueous fermentation medium of SNA-048 via a protein-independent multicomponent reaction from three starting materials: 4-hydroxysattabacin (4), anthranilic acid (5), and 4-hydroxybenzaldehyde (6) (Figure 1). The non-enzymatic nature of this reaction was demonstrated through a series of feeding studies in fermentation media depleted of bacteria and proteins via filtration and heating, respectively. We were further able to recapitulate the chemistry in organic solvent via a three-step procedure that was later utilized toward the total synthesis of discoipyrrole D (DPD, 3) reported by the May laboratory.2
Figure 1.

Metabolites found in the fermentation medium of B. hunanensis strain SNA-048.
It is well-established that bacteria most often use complementary enzymes to sequentially catalyze bond formation in natural product biosynthesis and that these proteins are expressed from highly conserved and organized clusters of genes.3 However, there have been an increasing number of cases in which isolated secondary metabolites differ from their predicted structures as a result of non-enzymatic reactions stemming from the serendipitous proximity of complementary reactive metabolites.4 The knowledge gained from interrogating these mechanisms has fueled inventive and effective techniques to generate analogues with improved biological activities.5
Previously described microbial natural products that utilize a non-enzymatic step feature attack at an electrophilic sp2 carbon of a late-stage enzymatic intermediate by a nucleophile.5,6 In the case of 1, however, the self-assembly of 4, 5, and 6 requires the formation of four covalent bonds as well as two oxidations. Deciphering the mechanism by which the discoipyrroles are formed will expand our general knowledge of the types of non-enzymatic reactions to be expected in the milieu of microbial fermentation. This understanding will improve our ability to predict natural products that may undergo these reactions. Specific to the discoipyrroles, further understanding of the mechanism of formation can aid in our medicinal chemistry efforts.
Results and Discussion
Herein we report the details of non-enzymatic discoipyrrole formation obtained by a combination of NMR and mass spectrometric approaches utilizing isotopically labeled substrates (13C, 15N, and 18O). In particular, 15N-labeled substrates were used for highly sensitive NMR experiments to identify 1H–15N heteronuclear correlations of key intermediates. The increased sensitivity of the isotope label allowed for short (∼30 min) experiments capable of detecting low-abundance intermediates in complicated mixtures. This approach could be applied to the study of mechanisms of heteronuclear bond-forming reactions and most notably applied to multicomponent reactions.
We began our studies of discoipyrrole formation by examining the origins of the precursor molecules 4, 5, and 6. 4 resembles members of a growing family of acyloin-containing molecules that are products of condensation between two amino acids. 5 and 6 are known degradation products of tryptophan and tyrosine, respectively (KEGG compounds C00108 and C00633).7
To confirm the amino acid origins of 4, we utilized sequences of known thiamine pyrophosphate (TPP)-dependent acyloin synthases to discover homologous candidate genes in the annotated genome of B. hunanensis strain SNA-048 (Figure S3).8 Particularly of use were the recent findings by Park et al.8a describing the sattabacin-producing acyloin synthase known as Thzk1050 from Thermosporothrix hazakensis. On the basis of the presence of these putative biosynthetic genes in SNA-048, we performed feeding studies with 13C-labeled [2-13C]-l-tyrosine and [2-13C]-l-leucine and confirmed their incorporation into 4 (Figure S4). Additional feeding studies and/or genetic analyses were performed to conclude the amino acid sources of 4, 5, and 6. These experiments are described in detail in the Supporting Information (see the SI text and Figures S1–S5).
To investigate the non-enzymatic multicomponent formation of the discoipyrrole scaffold, we took advantage of our previously established synthetic model reaction system in organic solvent. This model takes advantage of starting with oxidized 4-methoxysattabacin (7) (Figure 2), the formation of which we have previously found to be the rate-limiting step in both fermentation media and organic solvent. Additionally, the model system removes analytical barriers associated with the complex fermentation medium. Past successes using NMR-active isotope labels to study reactions through the use of kinetic isotopes,9 to elucidate biosynthetic pathways,10 and in the identification of natural products11 led us to utilize 13C- and 15N-labeled substrates to probe the multicomponent reaction by NMR spectroscopy and mass spectrometry.
Figure 2.

Initial studies of the formation of DPA in the fermentation flask were unsuccessful due to low reagent yields, interfering medium components, and complicated analytical sample preparation. To combat these issues, precursors were combined in deuterated organic solvent and observed constantly by NMR spectroscopy. The base 4-(dimethylamino)pyridine (DMAP) was used to simulate the basic environment of the fermentation flask (pH ∼9). The oxidation of the acyloin of 4-hydroxysattabacin to form a diketone is known to be the rate-limiting step in DPA formation, and thus, the diketone was prepared with Dess–Martin periodinane (DMP) beforehand to increase the efficiency of the model system.
To track the formation of reaction intermediates, the chemical shifts and heteronuclear correlations of key carbon and nitrogen atoms around the pyrrole core were observed in real time. Utilizing [1-13C]-p-hydroxybenzaldehyde and 15N-anthranilic acid allowed the observation of heteronuclear NMR correlations from these isotope labels during each of the key bond-forming steps. All of the following experiments, unless otherwise noted, were run using 1.0 equiv of 7, 2.0 equiv of 5 and 6, and 3 equiv of 4-(dimethylamino)pyridine (DMAP). The reactions were carried out in dimethyl sulfoxide (DMSO). Reactions performed in NMR tubes were run in a total volume of 700 μL in DMSO-d6.
Experimentally we began by carrying out the multicomponent reaction with 15N-anthranilic acid and conducted continuous reaction monitoring in 30 min intervals for 36 h using 1H–15N heteronuclear multiple-bond correlation NMR spectroscopy (HMBC). This methodology provided evidence of C–N bond formation through the development of 1H–15N correlations, specifically to upfield 1H signals of the isobutyl chain of 7. Because of the spectral range of 15N NMR spectroscopy, the 15N chemical shift of anthranilate-derived intermediates provides key structural information on the nature of the C–N bond of an intermediate (Figure S6).
The first experiment performed provided a time frame for key 15N-containing intermediates (Figure 3 and video S1). For example, from the time-lapse video it can be seen that the first major intermediate (red box in Figure 3a) with correlations from the 15N to protons on 7 begin appearing at 30 minutes and disappeared by 12 h, whereas the second major intermediate (blue box in Figure 3a) appears at 4 h and disappears at 14 h. From this timeline, we carried out additional two-dimensional NMR experiments, such as 1H–1H correlation spectroscopy (COSY), 1H–13C heteronuclear single-quantum coherence spectroscopy (HSQC), and 1H–13C HMBC to elucidate the structural identities of the time-sensitive intermediates (Figure 3b).
Figure 3.

Reaction time course monitored by 1H–15N NMR spectroscopy. (a) Representative time points from 1H–15N HMBC experiments. Major correlations are highlighted using colored boxes on the spectra. (b) Construction of a reaction timeline on the basis of 1H–15N HMBC experiments allows the design of additional NMR experiments to elucidate the structures of intermediates. (c) Structures of intermediates determined through the use of one- and two-dimensional NMR data.
The starting material 15N-anthranilic acid has a 15N shift of −311.1 ppm and a strong correlation to aromatic protons at 6.71 and 7.67 ppm. Immediately after the three reagents were added to the NMR tube in DMSO-d6, we saw the appearance of 1H–15N HMBC correlations from an 15N shift of −225.0 ppm to protons at 6.96, 2.17, and 2.38 ppm, suggesting bond formation between 7 and 5. This was validated by further COSY and HSQC correlations that established the proton resonances at 2.17 and 2.38 ppm to correspond to the methylene protons of the isobutyl side chain of the discoipyrroles. The key COSY correlations were from 2.17 and 2.38 ppm to a septet at 1.28 ppm that was further coupled to methyl doublets at 0.62 and 0.70 ppm. The slightly downfield chemical shift of the diastereotopic methylene and the 15N chemical shift indicated an electron-withdrawing environment, potentially suggesting a pyrrole ring. This was confirmed by 1H–13C HMBC correlations from methylene protons to carbons at 127.0 and 138.5 ppm, consistent with structure of I. The absence of 1H–13C HMBC correlations between these carbons and exchangeable NH 1H signals further suggested the formation of the pyrrole ring.
A weaker 15N signal at −254.9 ppm appeared with correlations to an aromatic proton at 6.95 ppm and aliphatic signals at 4.40 and 1.34 ppm. By means of similar strategies as described for I, correlations between these signals and to the isobutyl chain led to structure II, the ketone tautomer of I.
After 16 h, the 1H–15N HMBC correlations of I and II decrease, while a third 15N signal at −248.3 ppm appears with correlations to protons at 7.24 and 1.69 ppm, representing compound III. Up to 24 h into the reaction, this signal existed independently and continued to build in intensity. Because of the apparent stability and abundance of the intermediate, an aliquot of the reaction mixture was removed and subjected to LC–MS analysis. The retention time and mass indicated that compound III is the open form of discoipyrrole that exists in pH-dependent equilibrium with 1. This reaction was repeated at a later time to yield larger quantities of III, which was isolated and characterized in full (Table S1).
After 24 h, 1% trifluoroacetic acid (TFA) was added to the NMR tube, and monitoring via 1H–15N HMBC was continued. In the first NMR scan after acid addition, two 15N species at −251.3 ppm, with 1H correlations to 7.29 and 1.67 ppm, and −246.4 ppm, with proton correlations to 7.61, 1.75, and 1.44 ppm, appeared. Within minutes, a major 15N species at −263.8 ppm with correlations to protons at 6.29, 2.06, and 1.91 ppm appeared. This was assigned as the final product, IV.
These experiments demonstrated that the formation of the pyrrole core of the discoipyrroles occurs rapidly to produce the first major intermediate, I, which persists in equilibrium with the tautomer II. The loss of oxygen indicated that formation of the C–N bond in I induces an elimination of water. Subsequent oxidation of the pyrrolone ring yields III, which upon exposure to acid undergoes cyclization to IV. It should be noted that it is also possible that I is directly oxidized to III and that we only observe II as part of the equilibrium with I. To confirm this mechanism, we carried out the formation of III under identical conditions with an atmosphere of 18O2. Monitoring of the reaction by LC–MS led to the observation of a peak with an m/z [M – H]− of 475.2, representing a +2 shift from canonical III, as is evident by the control reaction run with 16O2 (Figure 4). Acid-free isolation and high-resolution mass spectrometry of the 18O-labeled product gave an experimental m/z [M + H]+ of 477.1923 (expected m/z [M + H]+ = 477.1924). The isolated 16O control product gave an experimental m/z [M + H]+ of 475.1881 (expected m/z [M + H]+ = 475.1881) (Figures S7 and S8). This unusual oxidation of the pyrrole moiety is unaffected by substitution of the substitutents on either the benzaldehyde or anthranilic acid moiety.
Figure 4.
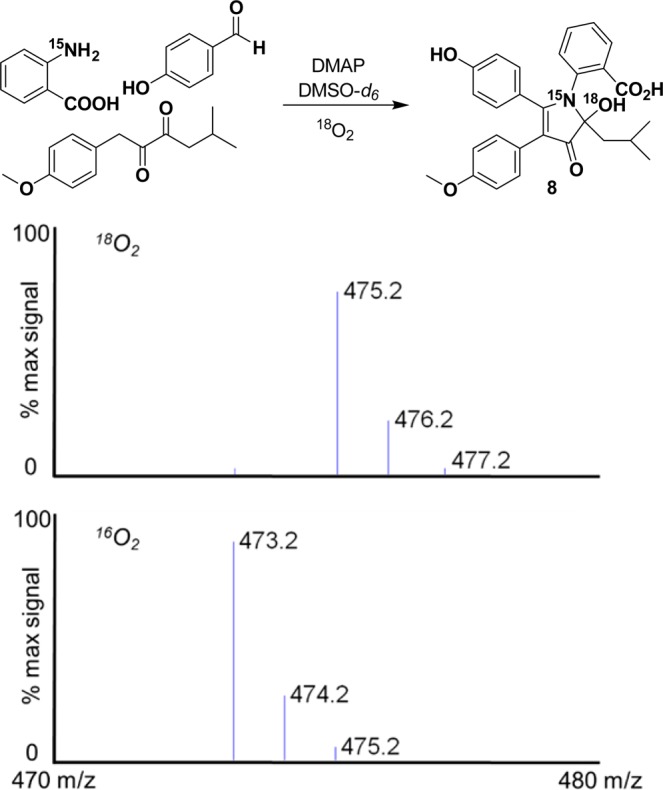
Formation of discoipyrrole analogue 8 under an atmosphere of 18O2 and [M – H]− data confirming the incorporation of 18O with a representative control experiment carried out with 16O2.
Isolation of the 18O-labeled analogue of III allowed us to explore the acid-induced ring closure. Surprisingly, exposure to TFA led to an isolated product with an m/z [M + H]+ of 457.1774 (expected m/z [M + H]+ = 457.1776) that indicated the loss of the 18O label upon ring closure (Figure S9). This suggests that under these anhydrous, acidic conditions the elimination of H2O leads to imine formation followed by rapid cyclization.
While monitoring the 1H–15N HMBC spectrum, we could detect no intermediates prior to pyrrole formation, i.e., no combination of 5 with either 7 or 6 alone. This indicated to us that the formation of intermediate I is irreversible and that the multicomponent reaction occurs too rapidly to be detected by isotope-enhanced NMR spectroscopy. We hypothesized that there are three potential pathways to form I (Figure 5, pathways 1–3). To test these individually, we performed three independent reactions, each with one of the substrates omitted. These reactions were performed using the described model conditions, except as otherwise noted.
Figure 5.

Possible pathways to pyrrole core intermediate I.
Pathway 1 relies on the initial formation of an aldimine between 5 and 6, setting up a Mannich reaction. We monitored for imine formation using 1H–15N HMBC with an expected imine 15N shift in the 0–100 ppm range. Under model conditions, after 24 h there was no formation of 9 (Figure 6). This was further confirmed by LC–MS analysis. As a control reaction, we were readily able to form aldimine 10 between 15N-aniline and 6 under the model conditions to give a product with an 15N resonance at −65.0 ppm (Figure 6). This suggests that the mechanism of discoipyrrole formation does not go through a Mannich reaction. To further test this, we subjected commercially available 9, 7, and DMAP to our model conditions and monitored for the formation of III by LC–MS. No product was detected after 48 h, well within the time frame expected for product formation to occur.
Figure 6.
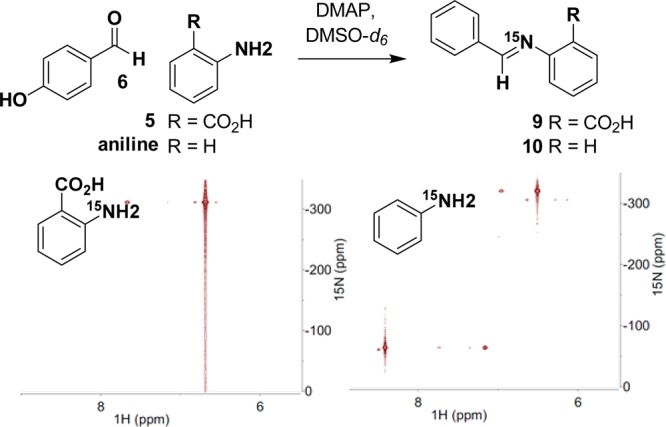
Use of 1H–15N HMBC to monitor the formation of an aldimine between 6 and 5 and between 6 and aniline.
To test pathway 2, [1-13C]-6 and 7 were treated with DMAP, and the reaction mixture was monitored by NMR spectroscopy and LC–MS, looking for the Claisen–Schmidt product. After 48 h we observed only the presence of [1-13C]-6 and 7. Increasing the reaction time, temperature, or strength of the base did not facilitate any condensation. It is possible that anthranilic acid could act as a catalyst for the Claisen–Schmidt reaction, but the complete lack of product formation under all of the conditions attempted is strong evidence against pathway 2.
Pathway 3, in which 7 undergoes imine formation, was tested by omitting 6 in the reaction. However, to our surprise, after 24 h the main product was a dimethoxydiscoipyrrole analogue. Utilizing NMR and LC/MS, we observed 4-methoxybenzaldehyde in the reaction, demonstrating that 7 can be degraded to form 4-methoxybenzaldehyde. Therefore, aldehyde contamination of reactions involving oxidized sattabacin analogues prevented further analysis because of the shuttling of intermediates to pyrrole formation.
To circumvent this issue, we utilized 4-hydroxysattabacin in lieu of 7, removing the possibility for condensation with aldehyde coming from 7. No discernible amount of the desired product was detected, but slightly increased temperatures induced a minor amount of product formation. We reasoned that an equilibrium between the acyloin group and any imine might favor the former compared with the diketone of 7. Therefore, we set out to trap potential amination products using reductive conditions. Indeed, reacting 4-hydroxysattabacin and anthranilate in the presence of sodium triacetoxyborohydride in dichloroethane with acetic acid yielded a reductive amination product (31% yield) within 16 h (Figure S10). Reduction of 4-hydroxysattabacin to the diol side product was prevalent, potentially obscuring the calculated reaction efficiency.
The efficiency of these omission experiments compared with the complete reaction containing all three reactants indicated that the mechanism is enhanced by a concerted flow of reactivity. Our current hypothesis is that pyrrole formation can proceed through amination of 7 by anthranilic acid to form an imine intermediate that can be trapped by a Claisen–Schmidt condensation between C1 of sattabacin and 4-hydroxybenzaldehyde. We believe that the amination increases the acidity of the C1 methylene to cause the “snapping” together of the three substituents. The resulting conjugated olefin can then be attacked by the nitrogen of anthranilic acid to form the five-membered core.
Conclusion
The discoipyrroles present a unique biosynthetic case in that they can be produced by a specific and concerted mechanism in aqueous media from diverse excreted metabolites. To study how anthranilic acid, 4-hydroxybenzadehyde, and 4-hydroxysattabacin were able to undergo such a reaction, we constructed a model system in organic solvent and utilized various analytical techniques, including the novel application of 1H–15N HMBC as a tool for monitoring reactions in situ. Building on the foundation made possible by this technique, we performed various isotope-labeling experiments with 13C, 15N, and 18O to elucidate the mechanism of non-enzymatic discoipyrrole formation. This multicomponent reaction was characterized by initial rapid steps to induce pyrrole formation and the following changes in oxidation state that dictate the final closure of the lactone ring. Particularly useful were the 18O-labeling experiments, which allowed us to clearly demonstrate the atmospheric oxidation of the pyrrolone using careful isolation and high-resolution mass spectrometry. Furthermore, the subsequent loss of this isotope was integral in establishing the dehydration-dependent lactone ring closure.
Importantly, once the details of the mechanism were uncovered, we constructed fermentation media with deuterated water and used our 1H–15N HMBC monitoring method to validate discoipyrrole formation under biologically equivalent conditions without organic solvent or added DMAP (Figure S11). We observed identical intermediates and similar reaction time courses under the two sets of conditions.
Arylamines have been found to participate in many of the recent examples of natural products amenable to non-enzymatic incorporation, including the ammosamides, elansolids, and oxazinin A.5,6 Through our mechanistic studies of the DPs, we have shown that anthranilic acid acts as the initiating factor through imine formation with sattabacin and that the ortho carboxylic acid substitution of anthranilate drives the selectivity, as seen in the aldimine formation experiments (Figure 6). Our work and that of others suggest that arylamines play a significant role in non-enzymatic reactions and that aryl substitution patterns, the nature of the amination sites, and the stability of intermediates all contribute to the complexity of these reactions. Using these types of nucleophiles could be a potentially cost-effective and simple way to screen bacterial libraries for natural products amenable to non-enzymatic perturbations. Concordantly, detection methods using 15N labels, such as 1H–15N HMBC as demonstrated here, could provide quick and high-sensitivity analysis.
Acknowledgments
We acknowledge Jonathan R. Goodman and Dr. Wade C. Winkler (University of Maryland) for their gracious contribution in the sequencing and processing of the B. hunanensis SNA-048 genome. We acknowledge following grants for funding of this project: Welch Foundation I-1689, NIH R01CA1499833, NIH U01CA176284, CPRIT RP140152, the Chilton/Bell Foundation, and the Martha Steiner family.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b13320.
The authors declare no competing financial interest.
Supplementary Material
References
- Hu Y.; Potts M. B.; Colosimo D.; Herrera-Herrera M. L.; Legako A. G.; Yousufuddin M.; White M. A.; MacMillan J. B. J. Am. Chem. Soc. 2013, 135, 13387–92. 10.1021/ja403412y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih J. L.; Nguyen T. S.; May J. A. Angew. Chem., Int. Ed. 2015, 54, 9931–5. 10.1002/anie.201503528. [DOI] [PubMed] [Google Scholar]
- a Clardy J.; Walsh C. Nature 2004, 432, 829–837. 10.1038/nature03194. [DOI] [PubMed] [Google Scholar]; b Fischbach M. A.; Walsh C. T. Chem. Rev. 2006, 106, 3468–3496. 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- a Cottreau K. M.; Spencer C.; Wentzell J. R.; Graham C. L.; Borissow C. N.; Jakeman D. L.; McFarland S. A. Org. Lett. 2010, 12, 1172–5. 10.1021/ol902907r. [DOI] [PubMed] [Google Scholar]; b Zhu X.; Liu J.; Zhang W. Nat. Chem. Biol. 2015, 11, 115–120. 10.1038/nchembio.1718. [DOI] [PubMed] [Google Scholar]; c Asai T.; Tsukada K.; Ise S.; Shirata N.; Hashimoto M.; Fujii I.; Gomi K.; Nakagawara K.; Kodama E. N.; Oshima Y. Nat. Chem. 2015, 7, 737–743. 10.1038/nchem.2308. [DOI] [PubMed] [Google Scholar]
- Pan E.; Oswald N. W.; Legako A. G.; Life J. M.; Posner B. A.; MacMillan J. B. Chem. Sci. 2013, 4, 482–488. 10.1039/C2SC21442C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lin Z.; Koch M.; Abdel Aziz M. H.; Galindo-Murillo R.; Tianero M. D.; Cheatham T. E.; Barrows L. R.; Reilly C. A.; Schmidt E. W. Org. Lett. 2014, 16, 4774–4777. 10.1021/ol502227x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Steinmetz H.; Gerth K.; Jansen R.; Schläger N.; Dehn R.; Reinecke S.; Kirschning A.; Müller R. Angew. Chem., Int. Ed. 2011, 50, 532–536. 10.1002/anie.201005226. [DOI] [PubMed] [Google Scholar]; c Steinmetz H.; Zander W.; Shushni M. A. M.; Jansen R.; Gerth K.; Dehn R.; Dräger G.; Kirschning A.; Müller R. ChemBioChem 2012, 13, 1813–1817. 10.1002/cbic.201200228. [DOI] [PubMed] [Google Scholar]
- a Kanehisa M.; Goto S. Nucleic Acids Res. 2000, 28, 27–30. 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kanehisa M.; Goto S.; Sato Y.; Kawashima M.; Furumichi M.; Tanabe M. Nucleic Acids Res. 2014, 42, D199–205. 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Park J. S.; Kagaya N.; Hashimoto J.; Izumikawa M.; Yabe S.; Shin-Ya K.; Nishiyama M.; Kuzuyama T. ChemBioChem 2014, 15, 527–32. 10.1002/cbic.201300690. [DOI] [PubMed] [Google Scholar]; b Balskus E. P.; Walsh C. T. J. Am. Chem. Soc. 2008, 130, 15260–1. 10.1021/ja807192u. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Gocke D.; Nguyen C. L.; Pohl M.; Stillger T.; Walter L.; Müller M. Adv. Synth. Catal. 2007, 349, 1425–1435. 10.1002/adsc.200700057. [DOI] [Google Scholar]
- a Cleland W. W. Arch. Biochem. Biophys. 2005, 433, 2–12. 10.1016/j.abb.2004.08.027. [DOI] [PubMed] [Google Scholar]; b Gomez-Gallego M.; Sierra M. A. Chem. Rev. 2011, 111, 4857–963. 10.1021/cr100436k. [DOI] [PubMed] [Google Scholar]
- a Mahmud T. J. Labelled Compd. Radiopharm. 2007, 50, 1039–1051. 10.1002/jlcr.1391. [DOI] [Google Scholar]; b Esquenazi E.; Jones A. C.; Byrum T.; Dorrestein P. C.; Gerwick W. H. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 5226–31. 10.1073/pnas.1012813108. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Rinkel J.; Dickschat J. S. Beilstein J. Org. Chem. 2015, 11, 2493–2508. 10.3762/bjoc.11.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Vizcaino M. I.; Crawford J. M. Nat. Chem. 2015, 7, 411–7. 10.1038/nchem.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bode H. B.; Reimer D.; Fuchs S. W.; Kirchner F.; Dauth C.; Kegler C.; Lorenzen W.; Brachmann A. O.; Grun P. Chem. - Eur. J. 2012, 18, 2342–8. 10.1002/chem.201103479. [DOI] [PubMed] [Google Scholar]; c Kwon Y.; Park S.; Shin J.; Oh D. C. Arch. Pharmacal Res. 2014, 37, 967–71. 10.1007/s12272-013-0254-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.