

Abstract
Retinoic acid (RA) is known to regulate cell growth and differentiation. In HL-60 human myeloblastic leukemia cells, it causes mitogen-activated protein kinase (MAPK) signaling leading to myeloid differentiation and G0 cell cycle arrest. This communication reports that expression of the Cbl adaptor caused enhanced extracellular signal-regulated kinase 2 activation and promoted RA-induced differentiation and G0-arrest. Stable transfectants ectopically expressing c-Cbl underwent myeloid differentiation faster than wild-type (wt) cells when treated with RA. In contrast, c-Cbl knockdown stable transfectants differentiated slower than wt cells when treated with RA. Cells ectopically expressing c-Cbl had enhanced CD38 expression when treated with RA, and cells ectopically expressing CD38 had enhanced c-Cbl expression, even without with RA, suggesting an interaction between c-Cbl and CD38. Fluorescence resource energy transfer and coimmunoprecipitation showed that c-Cbl and CD38 bind each other. RA causes the gradual down-regulation and eventual loss of c-Cbl expression, resulting in loss of the Cbl-CD38 interaction, suggesting that c-Cbl plays a relatively early role in promoting RA-induced differentiation. RA-induced differentiation can thus be propelled by c-Cbl and by CD38, both of which bind together, enhance the expression of each other, and cause MAPK signaling. There thus seems to be a cooperative role for c-Cbl and CD38, reflected in their direct binding, in propulsion of RA-induced differentiation.
Introduction
Retinoic acid (RA) is a form of vitamin A, which is a necessary dietary factor for juvenile development and plays a variety of functions in adults (1). RA induces morphologic and functional terminal differentiation and can be used therapeutically for chemoprevention and treatment of cancer (2), notably acute promyelocytic leukemia, making its mechanism of action of significant interest. The human myeloblastic leukemia cell line (HL-60) has been one of the archetype in vitro models for studying the mechanism of RA action (3–5). HL-60 cells undergo G0 cell cycle arrest and myeloid differentiation in response to RA or monocytic differentiation in response to 1,25-dihydroxyvitamin D3. More interestingly, RA causes activation of mitogen-activated protein kinase (MAPK) signaling, which is necessary to induce differentiation and G0 cell cycle arrest (6, 7).
Cbl is a member of a protein family that is structurally and functionally conserved in multicellular organisms. Several studies have shown the pivotal role of Cbl proteins through adaptor function and E3 ligase activity (8–12). Cbl proteins have a highly conserved NH2-terminal domain, termed the tyrosine kinase–binding domain, which binds to phosphotyrosines on activated receptor tyrosine kinases and other signaling proteins (9, 13, 14), a short linker region, and a Ring finger domain that binds ubiquitin-conjugating enzymes (10, 15, 16). Because of the conserved structure of their NH2 termini, all members of the Cbl family possess the characteristics of recognizing activated target proteins and of mediating their ubiquitination, which is crucial for their function in regulating signaling pathways (15–17). It is well documented that Cbl proteins interact with several intracellular signaling molecules, including kinases, adaptors, and structural proteins, and such an interactome can form signal component networks that control multiple cellular processes (14, 18, 19).
Kontani and colleagues (20) suggested that c-Cbl tyrosine phosphorylation was mediated by the human cell surface antigen CD38, a type II transmembrane glycoprotein originally identified as an activation antigen of T and B cells. CD38 is expressed on several leukocytes and early hematopoietic precursor cells and has a long COOH-terminal extracellular domain and a short NH2-terminal cytoplasmic tail (21). The effects mediated by CD38 include the production of proinflammatory cytokines, proliferation, and protection from apoptosis in lymphocytes (22–24). CD38 can be found in lipid rafts and signals through extracellular signal-regulated kinase (ERK) activation and also causes RAF activation (25, 26). It thus activates MAPK signaling, which regulates cellular processes such as proliferation and differentiation. The studies by Lamkin and colleagues (27) have suggested that RA induces the early expression of CD38, which signals through MAPK to promote RA-induced cell differentiation. In hematologic neoplasias, the role of CD38 has been enigmatic. It has been attributed with potentially both pro-proliferative and antiproliferative effects. It has at times also been considered as a prognostic indicator of better or worse disease (22, 28, 29).
It is becoming evident that multiple direct and indirect interactions between c-Cbl and many signaling proteins have been detected; however, the functional significance of protein complexes assembled around c-Cbl remains unclear. The finding of novel interactions of c-Cbl with CD38 is of significance for understanding the role of c-Cbl and c-Cbl–interacting proteins in the regulation of signaling pathways. In this study, we have succeeded in establishing c-Cbl stable transfectants and knockdown cell lines and clearly indicated the interaction between c-Cbl and CD38 by using independent techniques. We found that high CD38 caused increased c-Cbl expression and overexpression of c-Cbl also enhanced RA-induced CD38 expression. This study also shows the role of c-Cbl in differentiation and cell cycle arrest of HL-60 myeloblastic leukemia cells. We found that stable transfectants ectopically expressing c-Cbl caused enhanced ERK2 activation and promoted RA-induced differentiation and cell cycle arrest. In contrast, knocking down c-Cbl expression by stably transfected small interfering RNA (siRNA) retarded differentiation and arrest. Our study also suggests that CD38/c-Cbl binding together plays a cooperative role in promoting RA-induced differentiation and cell cycle arrest.
Materials and Methods
Cell culture
Human myeloblastic leukemia cells (HL-60) and their derivatives (CD38 stable transfectants, c-Cbl knockdowns, and c-Cbl stable transfectants) were grown and cultured as previously described (30). To maintain CD38 and c-Cbl high expression in stable transfectants or c-Cbl low expression in knockdown cells, the cells were periodically resorted based on high expression of fluorescent enhanced green fluorescent protein (EGFP) using fluorescence-activated cell sorting (FACSAria flow cytometer, BD Biosciences).
Chemicals
RA (Sigma) was used at a final concentration of 1 μmol/L as previously described (30).
siRNA c-Cbl
siRNA c-Cbl (human) was designed and obtained from GenScript and cloned into a commercially available vector, pRNAT-U6.1/Neo, using BamH1 and HindIII sites (GenScript).
Reverse transcription-PCR and plasmid construction
Total RNA of human c-Cbl was isolated from HL-60 cells using the Qiagen RNeasy Mini kit (Qiagen), and the first-strand cDNA was synthesized according to the protocol of the SuperScript First-Strand Synthesis System (Invitrogen). DNA fragments carrying complete human c-Cbl gene were generated by PCR amplication using the oligonucleotides (forward, 5′-CGGGATCCATGGCCGGCAACGTGAAGAA-3′; reverse, 5′-GCGGTACCCTAGGTAGCTACATGGGCA-3′) and cloned into a pRNAT-U6.1/Neo vector using BamH1 and Kpn1 sites. CD38 plasmid construction was described previously (27). All inserts were confirmed by sequencing.
CD11b, CD38, and c-Cbl expression studies by flow cytometry
HL-60 cells (0.5 × 106) were harvested at 1,000 rpm for 5 min. For CD11b and CD38, cells were resuspended in 100 μL PBS containing 5 μL of allophycocyanin (APC)–conjugated anti-CD11b antibody and phycoerythrin (PE)–conjugated anti-CD38 antibody (BD Biosciences). Following incubation for 1 h at 37°C, cells were analyzed by flow cytometry (LSRII flow cytometer, BD Biosciences) using 633-nm red laser and 488-nm blue laser excitations. The threshold to determine percent increase of expression was set at the highest 5% of control cells. To stain for c-Cbl, cells were fixed by resuspension in 100 μL PBS with 2% paraformaldehyde (Alfa Aesar) with 10-min incubation at room temperature and then permeabilized by addition of 900 μL −20°C methanol with 20 min at −20°C. After washing twice with 1 mL PBS, cells were stained with anti-c-Cbl polyclonal antibody (Santa Cruz Biotechnology, Inc.) for 1 h at room temperature. Following incubation and washing, cells were stained with an Alexa Fluor 350–conjugated goat anti-rabbit secondary antibody (Invitrogen) for 1 h and analyzed by flow cytometry. Results given as mean fluorescence intensity were determined by quantifying the fluorescence intensity of the gated entire cell population.
ERK phosphorylation
Cells (0.5 × 106) were fixed and permeabilized as mentioned above. Following incubation and two washes, cells were stained with Alexa Fluor 647–conjugated anti-phospho-p44/42 MAPK (Cell Signaling) antibody for 1 h and analyzed by flow cytometry. The gate to determine percent increase of expression was set at the highest 5% of HL-60 control cells, which represents basal levels of phosphorylated ERK (pERK). Cell populations exceeding basal ERK on RA treatments were detected by their positive shift above the basal levels from control cells.
Measurement of inducible oxidative metabolism
Cells (0.5 × 106) were harvested by centrifugation and resuspended in 200 μL 37°C PBS containing 10 μmol/L 5-(and-6)-chloromethyl-2′,7′-dichlorodihydro-fluorescein diacetate acetyl ester (H2DCF; Molecular Probes) and 0.4 μg/mL 12-O-tetradecanoylphorbol-13-acetate (TPA; Sigma) with incubation for 20 min in a humidified atmosphere of 5% CO2 at 37°C. Flow cytometric analysis was done as previously described (31).
Cell cycle analysis
Cells (0.5 × 106) were collected by centrifugation and resuspended in 200 μL hypotonic staining solution containing 50 μg/mL propidium iodine, 1 μL/mL Triton X-100, and 1 mg/mL sodium citrate. Cells were incubated at room temperature for 1 h and analyzed by flow cytometry (BD LSRII) using 488-nm excitation.
c-Cbl stable transfection
Fifty micrograms of plasmid DNA were transfected into HL-60 cells as previously described (32) and selected with G418 (1 mg/mL) for 2 to 3 wk. Stable transfectants underwent three cycles of cell sorting and amplification to select cells expressing high fluorescent EGFP using fluorescence-activated cell sorting (FACSAria flow cytometer). Western blots probed with anti-c-Cbl antibody (BD Biosciences) were performed to confirm c-Cbl expression.
c-Cbl/CD38 double-stable transfection
Fifty micrograms of CD38 plasmid DNA were transfected into c-Cbl stable transfectant cells as previously described (32) and selected with G418 (1 mg/mL) for 2 wk. Double transfectants were stained with APC-conjugated anti-CD38 antibody, sorted, and amplified three cycles to select cells expressing high CD38 using fluorescence-activated cell sorting. The expression of CD38 was confirmed by flow cytometry using APC-conjugated anti-CD38 antibody.
Immunoprecipitation and Western blot
Cells (2.5 × 107) were lysed using 200 μL lysis buffer (Pierce) and lysates were cleared by centrifugation at 13,000 rpm for 20 min at 4°C. Ten microliters of polyclonal antibody against c-Cbl were added and maintained with gentle shaking at 4°C overnight. Negative control using an equivalent amount of normal rabbit IgG (Santa Cruz Biotechnology) was included. Fifty microliters of protein A/G PLUS agarose beads (Santa Cruz Biotechnology) were then added and the final mixture was incubated at 4°C under rotary agitation overnight. The protein-antibody-protein A/G slurry was centrifuged at 10,000 rpm for 2 min and washed thrice with coimmunoprecipitation buffer (Pierce). The immunoprecipitate was resolved by SDS-PAGE and Western blotted using anti-c-Cbl and anti-CD38 primary antibodies and horseradish peroxidase–linked, anti-rabbit and anti-mouse IgG secondary antibodies (BD Bio-sciences). For Western blot analyses, equal amounts of protein lysates (8 μg) were resolved by SDS-PAGE, transferred to nitrocellulose membranes, and probed with the same antibodies shown in immunoprecipitation. Ponceau S staining (Sigma) and reprobing with β-actin antibody (Cell signaling) were used to check uniform loading.
Fluorescence resonance energy transfer
Flow cytometric fluorescence resonance energy transfer (FRET) experiments were designed and analyzed at the Cornell Biomedical Science Flow Cytometry Core Facility with the assistance and consultation of Dr. James L. Smith. Cells (0.5 × 106) were fixed in paraformaldehyde and permeabilized by methanol as described above. After two washes, cells were resuspended in 200 μL PBS containing 5 μL of primary rabbit anti-c-Cbl (Santa Cruz Biotechnology) and mouse anti-CD38 (BD Biosciences) antibodies and then stained with Alexa Fluor 350–conjugated and Alexa Fluor 430–conjugated goat anti-rabbit and goat anti-mouse secondary antibodies (Invitrogen). The immunocomplexes were analyzed using a BD LSRII flow cytometer. For the HL-60, the FRET signal was measured using 325-nm excitation (from a UV laser) of Alexa Fluor 350 to excite Alexa Fluor 430 measuring the emitted fluorescence through a 505 long-pass dichroic and a 530/30 band-pass filter. For the NB4 FRET measurement, Alexa Fluor 488–conjugated and Alexa Fluor 594–conjugated goat anti-rabbit and goat anti-mouse secondary antibodies were used. The FRET signal was measured using 488-nm excitation (from a blue laser) of Alexa Fluor 488 to excite Alexa Fluor 594 measuring the emitted fluorescence through a 600 long-pass dichroic and a 610/20 band-pass filter. Controls with secondary antibody(s) only or secondary(s) plus c-Cbl or CD38 primary antibody were included. Cells stained with just c-Cbl or CD38 primary antibody and Alexa Fluor 350 or Alexa Fluor 430, respectively, were used for compensation controls for spillover into all fluorescence collection channels. FRET signals were corrected by subtraction of background fluorescence of negative controls with just secondary antibodies and compensation controls.
Statistics
Three independent repeats were conducted in all experiments. Error bars represent SE of three repeats. StatView statistical package (version 5.0.1; SAS Institute, Inc.) was used to analyze the data via ANOVA and Fisher’s protected least-squares difference test.
Results
c-Cbl and CD38 promoted RA-induced cell differentiation
To determine the role of c-Cbl in RA-induced differentiation of myeloid leukemia cell lines, we established c-Cbl stable transfectants (c-Cbl+) and siRNA knockdown (c-Cbl−) stable transfectant cell lines. HL-60 wild-type (wt) and vector control cells showed no differences in proliferation or differentiation and behaved indistinguishably in all experiments reported. Western blots verified that c-Cbl protein expression was appropriately enhanced or diminished in the stable transfectants (Fig. 1). Figure 1 also shows that c-Cbl expression diminished following RA treatment to induce myeloid differentiation. The ability of these different transfectant cell lines (c-Cbl+, c-Cbl−, and vector control) to differentiate in response to RA was measured using a cell surface marker and a functional differentiation marker. First, a cell surface differentiation marker, CD11b, was used to measure cell differentiation by immunofluorescence using APC-conjugated CD11b antibody. We compared CD11b expression in vector control, c-Cbl+, and c-Cbl− cells as well as previously derived (27) CD38 stable transfectants treated with RA for 24, 48, 72, and 96 hours using flow cytometry. Compared with vector controls, c-Cbl+ cells with RA treatment showed enhanced expression of CD11b (P < 0.05, at 24, 48, and 72 hours); however, the loss of c-Cbl led to diminished CD11b expression (P ≤ 0.005, from 24 to 96 hours; Fig. 2A), indicating that c-Cbl promoted differentiation. c-Cbl expression increased the rate of RA-induced CD11b expression from 24 to 96 hours. For example, in c-Cbl+ stable transfectants, CD11b expression was the same as that in vector controls after 96 hours (97.6% in Cbl+, 94.7% in vector), but there was 1.6-fold higher expression after 24 hours (42.7% in Cbl+, 27.0% in vector). This suggested that c-Cbl enhanced the rate of CD11b expression under RA treatment, speeding up induced differentiation. In knockdown transfectant cells, RA induced much less CD11b at an early time point (24 hours, 15% in knockdown, 27% in vector); however, the expression level continued to increase toward that in vector control at a later time point (96 hours, 77.6% in knockdown, 94.7% in vector). This suggested that knockdown cells expressed CD11b more slowly, and c-Cbl was rate limiting for induced differentiation. To compare the CD11b expression in c-Cbl and CD38 stable transfectants, we measured CD11b expression in CD38 stable transfectants too. Figure 2A shows that CD38 enhanced RA-induced CD11b expression and that the enhanced expression levels were not significantly different from that in c-Cbl transfectants.
Figure 1.
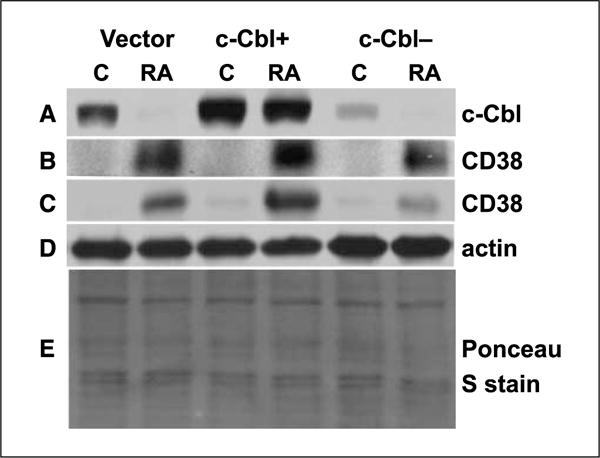
c-Cbl expression in c-Cbl stable transfectants (c-Cbl+) and knockdown (c-Cbl−) stable transfectant cell lines. Western blot of c-Cbl (72 h; A), CD38 (72 h; B), CD38 (10 h; C), and β-actin (D) expression in vector control, c-Cbl+, and c-Cbl siRNA-targeted stable transfectant cells. Protein lysates extracted from different cells were probed with anti-c-Cbl, anti-CD38, and anti-β-actin. β-Actin (D) and Ponceau S stain (E) were used to check uniform loading of protein lysates at 10 and 72 h, respectively. The blots verified c-Cbl overexpression and knockdown, and the Western blot also showed that for RA-treated cells, there was more CD38 in c-Cbl+ and less CD38 in c-Cbl knockdowns compared with vector controls.
Figure 2.
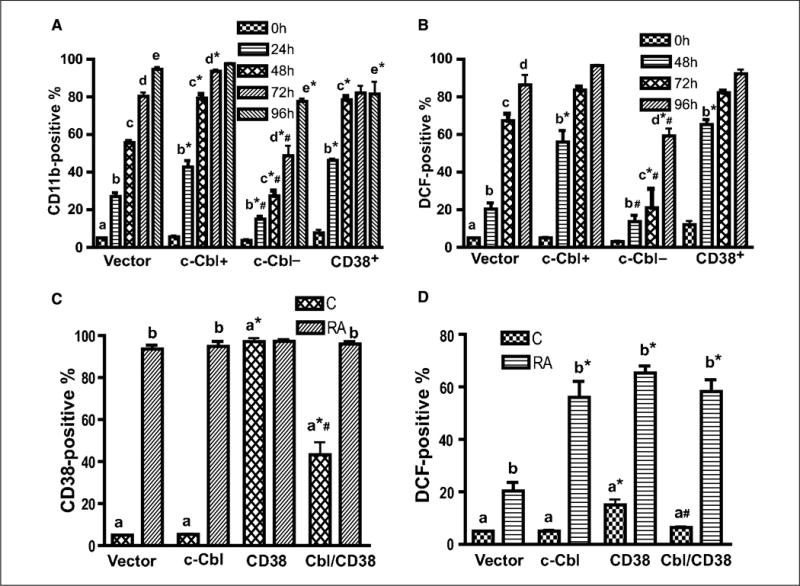
c-Cbl, CD38, and c-Cbl/CD38 double transfectants underwent enhanced myeloid differentiation. A, the expression level of CD11b was higher in c-Cbl stable transfectant cells than that of vector control, and the loss of c-Cbl led to a decreased CD11b expression. Vector controls, c-Cbl transfectant (c-Cbl+), and siRNA targeting c-Cbl transfectant (c-Cbl−) cells were treated with RA for the indicated times and stained with APC-conjugated anti-CD11b antibody, and the percent of cells expressing CD11b was analyzed by flow cytometry. B, c-Cbl enhanced RA-induced expression of a functional differentiation marker, inducible oxidative metabolism. Vector control, c-Cbl+, and c-Cbl− cells were treated with RA for the indicated times, and the percentage of cells capable of inducible oxidation metabolism detected by DCF fluorescence was analyzed by flow cytometry. Cells were incubated in PBS containing DCF and TPA and analyzed by flow cytometry. The threshold to determine percent positive cells was set to exclude 95% of control cells. *, CD11b or DCF expression levels from c-Cbl, c-Cbl−, or CD38 were significantly different from vector controls; #, c-Cbl− transfectants were significantly (P ≤ 0.05) different from c-Cbl+ or CD38 transfectants. The different letters indicate different time points. C, percent of vector control, c-Cbl, CD38, and c-Cbl/CD38 transfectants expressing CD38 before and after 48 h of RA treatment. c-Cbl/CD38 double transfectants had less CD38 expression than wt CD38 stable transfectants. Cells were stained with PE-conjugated anti-CD38 antibody and analyzed by flow cytometry. D, compared with vector controls, c-Cbl/CD38 double transfectants had enhanced expression of a functional differentiation marker, inducible oxidative metabolism, after 48 h of RA treatment similar to c-Cbl or CD38 stable transfectant. *, significant (P ≤ 0.05) difference in same group (untreated or RA-treated); #, c-Cbl/CD38 transfectants were significantly different from CD38 transfectants. The different letters indicate significant difference between untreated and RA-treated in same cell lines.
To confirm the regulation of RA-induced cell differentiation by c-Cbl, a functional differentiation marker, inducible oxidative metabolism was used. The oxidation of the nonfluorescent H2DCFDA to the highly fluorescent 2′,7′-dichlorofluorescein (DCF) was used to detect the generation of reactive oxygen. Inducible oxidative metabolism can be used as a functional marker of mature myeloid and monocytic cells. Figure 2B shows that c-Cbl accelerated HL-60 RA-induced functional differentiation. The difference between c-Cbl transfectants and vector controls was obvious and significant after RA treatment for 48 hours (56% in transfectants, 20% in vector controls; P < 0.001). Furthermore, the c-Cbl knockdown cells showed reduced RA-induced functional differentiation, which was consistent with effects on CD11b expression. We also measured and compared the percentage of DCF-positive cells in RA-treated CD38 transfectants, and the data showed that there was no significant difference between responses achieved by c-Cbl and CD38 stable transfectants. Our data were consistent with studies by Lamkin and colleagues (27), indicating that CD38 enhanced RA-induced myeloid differentiation.
To test how ectopically expressing both c-Cbl and CD38 affects RA-induced cell differentiation, we created c-Cbl/CD38 double-stable transfectants. The double transfectants were done by transfecting a CD38 expression vector into c-Cbl stable transfectant cells. Interestingly, the highest sustainable ectopic expression levels of CD38 in c-Cbl/CD38 double trasfectants were significantly greater than wt cells but significantly lower than that in CD38 stable transfectants, indicating that c-Cbl overexpression diminished achievable CD38 overexpression (Fig. 2C). The results suggest the possibility of an interaction between c-Cbl and CD38. Figure 2D shows that c-Cbl/CD38 double transfectants had enhanced RA-induced functional differentiation, and the response was not different from that in c-Cbl or CD38 transfectants. We also measured CD11b expression and compared c-Cbl, CD38, and c-Cbl/CD38 transfectants. Consistent with the DCF functional differentiation, the induced CD11b expression in c-Cbl/CD38 double transfectants was similar to that in c-Cbl or CD38 single transfectants (data not shown). The lack of enhanced differentiation for c-Cbl/CD38 double transfectants compared with the c-Cbl and CD38 single transfectants may in part reflect the decreased expression of CD38 in c-Cbl/CD38 transfectants.
c-Cbl accelerated G0 arrest and enhanced ERK activation in RA-treated cells
To determine the effects of c-Cbl on RA-induced G1-G0 cell cycle arrest, the percentage of cells (vector, c-Cbl+, c-Cbl−, and CD38) in G1−G0 was measured using flow cytometry (Fig. 3A). G1-G0 arrest would be revealed by an enrichment of cells with G1 DNA. c-Cbl overexpression enhanced G0 arrest on RA treatment, whereas c-Cbl knockdown transfectants diminished induced G0 arrest. This was consistent with the results obtained from the differentiation assays, indicating the role of c-Cbl in promoting RA-induced differentiation and cell cycle arrest.
Figure 3.
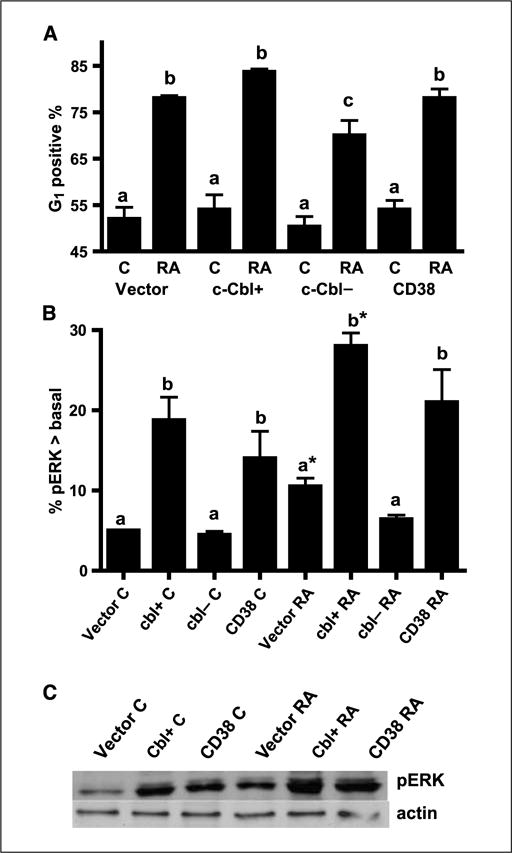
c-Cbl accelerated G0 arrest induced by RA treatment, and both c-Cbl and CD38 enhanced ERK activation. A, percent of vector control, c-Cbl transfectants (c-Cbl+), siRNA-targeted c-Cbl transfectants (c-Cbl−), and CD38 transfectant cells with G1-G0 DNA after 72 h of RA treatment. Cells stained with hypotonic propidium iodine staining solution were analyzed by flow cytometry. The different letters indicate significant difference at the P ≤ 0.05 levels. c, for RA-treated cells, the G1 positive percentage of c-Cbl− transfectant cells is significantly (P ≤ 0.05) lower than that in vector control, c-Cbl+, and CD38 transfectants. B, percentage of cells with pERK exceeding basal levels in untreated controls. Ectopic high expression of c-Cbl and CD38 enhanced activated ERK expression and c-Cbl knockdowns did not activate ERK expression as indicated by flow cytometry. Cells were fixed, permeabilized, and stained with Alexa Fluor 647–conjugated anti-phospho-p44/42 MAPK antibody. Enhancement was measured as a positive shift of the histogram above basal levels in control cells. Induced ERK activation on RA treatment for 15 h was indicated by a shift in percentage of cells with activated ERK above the basal levels of control cells. The threshold for positive shift was set to exclude 95% of control cells with basal level pERK expression. The different letters indicate significant difference at the P ≤ 0.05 levels. *, significant (P ≤ 0.05) difference between untreated and RA-treated in same cell lines. C, Western blot of pERK for vector control, c-Cbl+, and CD38 stable transfectants (top) and β-actin (bottom). C, untreated control; RA, RA-treated for 24 h. The data from the Western blotting of pERK were consistent with flow cytometry, indicating that c-Cbl and CD38 enhanced ERK expression.
It is known that ERK activation is a necessary contributor to propulsion of RA-induced cell differentiation and cell cycle arrest (6). To investigate if RA-induced myeloid differentiation and cell cycle arrest promoted by c-Cbl were related to ERK activation, we compared ERK expression among vector control, c-Cbl+, c-Cbl−, and CD38 stable transfectant cells. Dual pERK [T(203) EY(205)] was measured by flow cytometry. The percentage of cells expressing activated ERK in excess of basal levels is reported as a measure of the shift of the flow cytometrically measured histogram of activated ERK per cell. Figure 3B shows that the amount of activated ERK per cell was increased by ectopic c-Cbl expression and diminished by c-Cbl knockdowns, suggesting that the loss of c-Cbl led to diminished ERK expression and overexpression of c-Cbl caused ERK activation, which accelerates cell differentiation and cell cycle arrest. The amount of activated ERK was increased by RA treatment and by overexpression of CD38 as previously reported (27). To confirm the flow cytometric measurements, Western blotting of pERK was performed for the same cases. The results from Western blots were consistent with flow cytometric data (Fig. 3C), indicating that cells treated with RA and cells ectopically expressing c-Cbl or CD38 had enhanced ERK activation.
c-Cbl and CD38 enhanced the expression of each other
CD38 is among the earliest membrane receptors up-regulated by RA, and its expression causes MAPK signaling and propels RA-induced differentiation and cell cycle arrest in G1−G0. These latter traits are suggestively similar to the capabilities of c-Cbl. To reveal potential interaction between c-Cbl and CD38, the effect of expression of either on expression of the other was determined. To also further characterize the function of c-Cbl in RA-induced differentiation, the effect of c-Cbl expression on RA-induced CD38 expression in c-Cbl transfectants, knockdown, and vector control cells was determined. Figure 4A and B showed that for RA-treated cells, there was more CD38 in c-Cbl+ cells and less CD38 in c-Cbl knockdowns compared with vector controls, indicating that enhanced c-Cbl expression increased the rate of RA-induced CD38 expression, but in knockdowns, this effect was lost. By 24 hours, all cells had attained similar maximal expression. c-Cbl expression thus regulated the rate of RA-induced CD38 expression. Western blots (Fig. 1) confirmed the CD38 expression shown by flow cytometry, indicating that the maximal expression of CD38 per cell was higher in c-Cbl transfectants.
Figure 4.
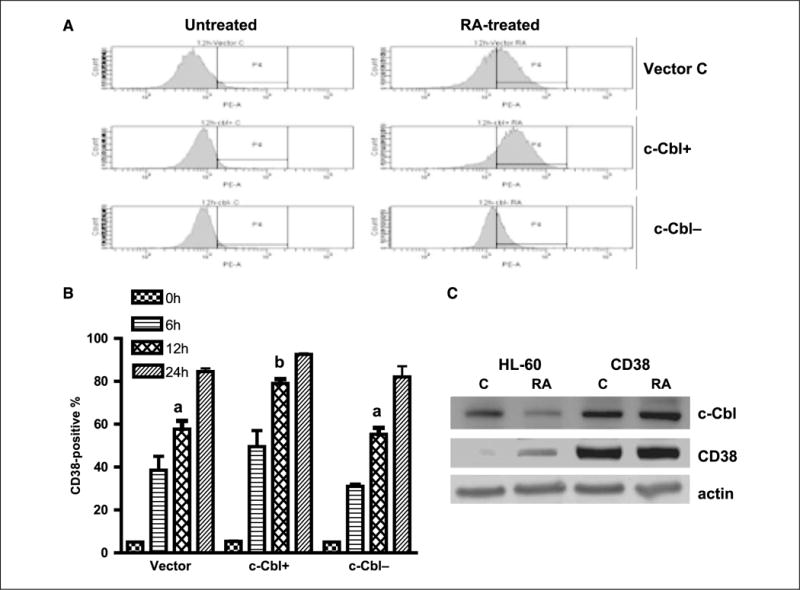
c-Cbl and CD38 enhanced the expression of each other. A, representative CD38 expression histograms of untreated and RA-treated (12 h) vector control, c-Cbl stable transfectants, and knockdown cells. B, percent of vector control, c-Cbl transfectant (c-Cbl+), and siRNA-targeted c-Cbl transfectant (c-Cbl−) cells expressing CD38 after RA treatment for the indicated times. RA-treated c-Cbl+ cells had increased expression of CD38 and c-Cbl− cells had decreased CD38 expression compared with vector control cells. The expression levels were significantly different after 12 h of RA treatment (P ≤ 0.003). Cells were stained with PE-conjugated anti-CD38 antibody and analyzed by flow cytometry. The different letters indicate significant differences between groups. C, Western blots of c-Cbl (top), CD38 (middle), and β-actin (bottom) in RA-treated (48 h) and untreated HL-60 and CD38 transfectant cells. c-Cbl expression in CD38 stable transfectant cells was enhanced. Protein lysates were extracted from wt HL-60 and CD38 overexpression cell lines and probed with anti-c-Cbl, anti-CD38, and anti-β-actin.
To determine if a reciprocal regulation of c-Cbl by CD38 existed, CD38 stable transfectants created as previously described (27) were analyzed. Western blots verified that the CD38 stable transfectant expressed enhanced constitutive and RA-induced CD38 expression (Fig. 4C). The CD38 stable transfectants showed enhanced c-Cbl expression in untreated cells as well as RA-treated (48 hours) cells. CD38 expression thus enhanced c-Cbl expression. There was thus reciprocal enhancement of expression of c-Cbl and CD38, suggesting the possibility of interaction.
Confirmation of the interaction between c-Cbl and CD38
c-Cbl has been shown to form complexes with different proteins. Although Kontani and colleagues (20) showed that CD38 caused tyrosine phosphorylation of target molecules, including the c-Cbl adaptor, there was no demonstration of direct interaction of c-Cbl and CD38. To determine if there is a direct interaction between c-Cbl and CD38, we used immunoprecipitation and FRET to characterize c-Cbl and CD38 interaction.
First, we isolated proteins from HL-60 and CD38 stable transfectant cells to determine if c-Cbl and CD38 coimmunoprecipitated. Coimmunoprecipitation using anti-c-Cbl antibody pulled down c-Cbl protein (Fig. 5A). CD38 protein coimmunoprecipitated with c-Cbl protein in HL-60 cells treated with 1 μmol/L RA to induce CD38 expression and also in HL-60 cells stably transfected with CD38. In both cases, CD38 coimmunoprecipitated with c-Cbl. As expected, no immunoprecipitable CD38 occurred in untreated HL-60 cells, which do not express CD38. Negative controls using IgG as an isotype-specific control were included and did not immunoprecipitate c-Cbl or CD38 (data not shown).
Figure 5.
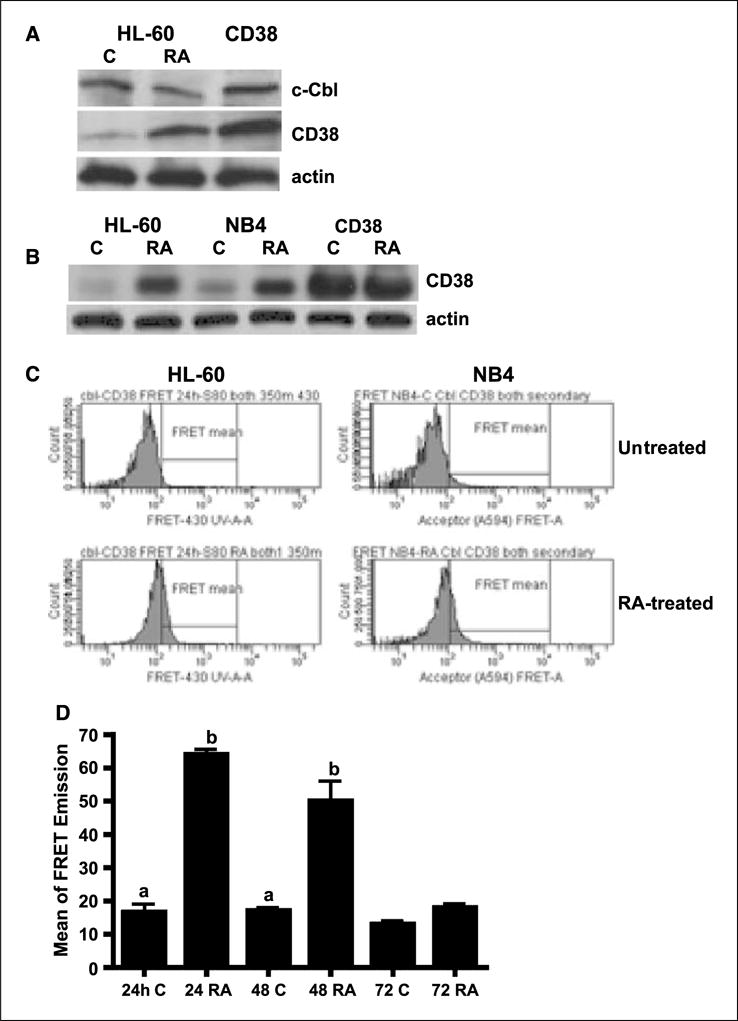
The interaction between c-Cbl and CD38 was confirmed by immunoprecipitation and FRET. A, Western blots of immunoprecipitated c-Cbl (top) and CD38 (middle) in HL-60 cells that were untreated (C) or treated with RA or transfected with CD38. β-Actin (bottom) was used to show input total cellular proteins in the coimmunoprecipitation reactions. Proteins immunoprecipitating with c-Cbl using anti-c-Cbl from protein lysates of wt HL-60 (untreated and RA-treated for 24 h) and CD38 stable transfectant cells were Western blotted using anti-c-Cbl (top) and anti-CD38 (middle) antibodies. Nonspecific immunoglobulin was used as a negative control. B, CD38 was induced by RA in NB4 cells and the inducible CD38 levels were the same compared with HL-60 cells. C, the interaction of c-Cbl and CD38 was observed in both HL-60 and NB4 cells by using FRET techniques. Histograms of FRET signals by flow cytometry showing the difference between 24 h of untreated and RA-treated in HL-60 and NB4 cells. D, fluorescence energy transfer from c-Cbl to CD38 for control and RA-treated HL-60 cells. There was a significant difference in energy transfer between untreated and RA-treated at 24 h (P < 0.0001) and 48 h (P = 0.03), but no difference at 72 h, suggesting an interaction between CD38 and c-Cbl after 24 and 48 h of RA-treatments. The data were analyzed by ANOVA (StatView). The different letters indicate significant difference from control cells. Cells fixed in paraformaldehyde and permeabilized were incubated in PBS containing rabbit anti-c-Cbl and mouse anti-CD38 primary antibodies and then stained with Alexa Fluor 350–conjugated and Alexa Fluor 430–conjugated goat anti-rabbit and goat anti-mouse secondary antibodies.
Second, we also used FRET to corroborate the interaction of CD38 and c-Cbl. FRET is a mechanism by which energy is transferred directly from one molecule to another and can be used to monitor protein-protein interactions in intact cells (33). c-Cbl and CD38 were used as donor and acceptor for measuring FRET emission. HL-60 cells RA untreated (control) or treated were used. RA induced a FRET emission in RA-treated but not untreated cells, which do not express CD38. FRET signals were observed after 24 and 48 hours of RA treatment; however, the FRET signal was lost after 72 hours of RA treatment (Fig. 5C and D). There was thus an interaction between CD38 and Cbl after 24 or 48 hours of RA treatment; however, this interaction was lost after 72 hours. FRET measurements made in reciprocal experiments with a CD38 donor and c-Cbl acceptor gave similar results (data not shown). To show the interaction of c-Cbl and CD38 in another RA-responsive leukemia cell line, we measured CD38 expression and CD38-Cbl FRET signal in NB4 cells. Our data show that CD38 was induced by RA in NB4 cells and that the inducible CD38 levels were the same compared with HL-60 cells (Fig. 5B). Figure 5C shows that the FRET signal was observed in both HL-60 and NB4 cells, indicating an interaction of c-Cbl and CD38.
Loss of the FRET emission at 72 hours suggests possible loss of one of the partners either CD38 or c-Cbl. Previous studies (27) indicated that CD38 expression was induced by RA treatment and persisted in all RA-treated cells. The results thus predict the potential loss of c-Cbl expression in RA-treated cells after 72 hours. To determine the effects of RA on c-Cbl expression in HL-60 cells, we tested c-Cbl expression on treatment with 1 μmol/L RA with Western blots and flow cytometry. Figure 6A shows that RA caused the gradual down-regulation and eventual loss of c-Cbl expression. Flow cytometric data (Fig. 6B and C) were consistent with Western blots, indicating that RA down-regulated c-Cbl expression. The expression level of c-Cbl diminished after 48 hours of RA treatment and was not detected on 72 hours of RA treatment (data not shown). Thus, loss of c-Cbl expression resulted in loss of FRET signal after 72 hours of treatment. This corroborates the FRET conclusions and suggests that c-Cbl–dependent signaling is not needed in cells that are differentiated, as are almost all cells in the population after 72 hours of RA treatment.
Figure 6.
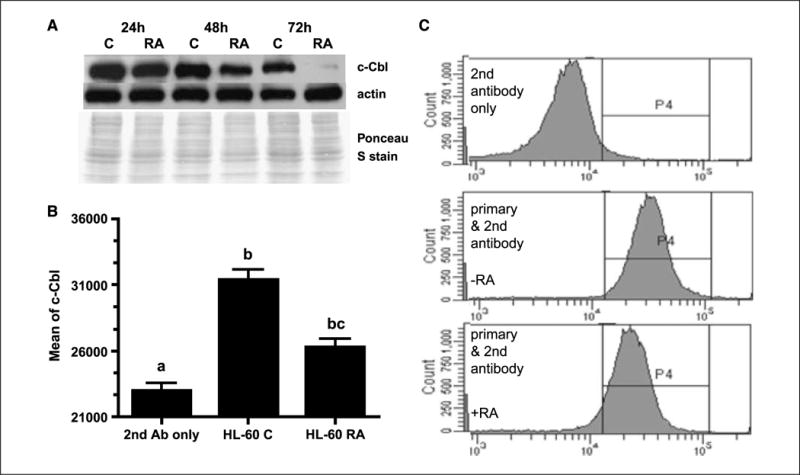
RA reduced c-Cbl expression in HL-60 cell line. A, Western blot of c-Cbl expression in HL-60 cells cultured without (C) or with RA for 24, 48, and 72 h showed that RA down-regulated c-Cbl expression over time. Ponceau S staining and β-actin antibody were used to check uniform loading. B, flow cytometric analysis of c-Cbl expression in untreated (C) and RA-treated (48 h) HL-60 cells. Flow cytometry data were consistent with Western blots, indicating that RA reduced c-Cbl expression. After the cells were fixed and permeabilized, they were stained with a polyclonal primary antibody against c-Cbl and a secondary Alexa Fluor 350–conjugated goat anti-rabbit antibody. Results are given as the mean fluorescence intensity from the entire gated cell population. The different letters indicate significance at the P ≤ 0.05 level. C, representative flow cytometric histograms of c-Cbl expression for untreated (middle) and RA-treated (48 h; bottom) HL-60 cells. Top, secondary antibody only staining shows background signal from the secondary antibody.
Discussion
c-Cbl plays an important role in signal transduction in hematopoietic cells and can drive apoptosis of myeloid leukemia cells (34–36). Consistent with one of the apparent biological roles of Cbl proteins as negative regulators of protein tyrosine kinases (11, 12, 36), their use as anticancer tools has been studied. Although Cbl proteins have known biological roles in terms of their adaptor function and E3 ligase activity, the precise role of c-Cbl in cell differentiation and cell growth still remains to be elucidated. This study is the first to show the role of c-Cbl in regulating differentiation and cell cycle arrest of myeloblastic leukemia cells. We have created HL-60 sublines where c-Cbl was overexpressed or knocked down, and investigated the effect of c-Cbl expression on RA-induced expression of different differentiation markers. Our studies showed that c-Cbl overexpression caused enhanced ERK2 activation. Enhanced c-Cbl expression enhanced RA-induced CD11b and CD38 expression, and diminished c-Cbl expression diminished induced expression of these cellular surface antigens. c-Cbl also accelerated RA-induced functional differentiation using inducible oxidative metabolism as a functional marker. We have found that c-Cbl activated ERK expression, drove MAPK signaling, and promoted cell differentiation. We also found that c-Cbl overexpression enhanced G0 arrest on RA treatment, whereas c-Cbl knockdown cells decreased RA-induced G0 arrest, indicating that c-Cbl regulates cell growth. This, along with the results from the differentiation assays, indicates a role for c-Cbl in promoting RA-induced differentiation and cell cycle arrest.
More interestingly, our results clearly indicated the interaction of c-Cbl and CD38 using independent techniques. Coimmunoprecipitation and Western blots showed that c-Cbl protein associated with CD38 and enhanced CD38 expression, and also that CD38 drove c-Cbl expression by observing CD38 expression in c-Cbl stable transfectants and knockdown cell lines and c-Cbl expression in CD38 stable transfectants. We confirmed the interaction of Cbl and CD38 by using FRET techniques in both HL-60 and NB4 cells. From the FRET studies, we found that the intensity of FRET signals depended on the amount of c-Cbl expressed. The loss of c-Cbl (after 72 hours of RA treatment) resulted in loss of FRET signals, suggesting that the c-Cbl/CD38 interaction was governed by the amount of c-Cbl available. This was consistent with the results from FRET experiments when using high CD38–expressing and low CD38–expressing transfectant cell lines to observe FRET signals (data not shown). From these experiments, we found that FRET signals did not differ between high and low CD38 expression cell lines, suggesting that the interaction depended more on the amount of c-Cbl than on the amount of CD38. There clearly is an interaction between CD38 and c-Cbl; however, it is not yet clear how CD38 and c-Cbl work together in accelerating cell differentiation and cell cycle arrest. One possibility is that their interaction stabilizes each other to promote a signaling complex that uses MAPK signaling, which each has been observed to enhance. CD38 might also sequester c-Cbl and mitigate its reported negative regulatory activity via its E3 ligase activity.
HL-60 cells undergo myeloid differentiation in response to RA (4, 5). In this process, RA induces the early expression of CD38, which activates ERK expression and drives MAPK signaling needed to promote cell differentiation. This study determined that RA gradually down-regulated c-Cbl expression in HL-60 cell lines, with eventual loss of Cbl expression in differentiated cells. Thus far, three mammalian homologues (c-Cbl, Cbl-b, and Cbl-3) have been characterized; all of which are oncoproteins by virtue of their prominent role in regulating signal transduction (37). Studies by Pennock and Wang (38) have indicated that Cbl plays a role in downregulation and trafficking of important mitogenic cell surface receptors, such as epidermal growth factor receptor (EGFR). More interestingly, c-Cbl showed its strongest interaction with EGFR at an early stage of EGFR trafficking, whereas the association of Cbl-b with EGFR occurred at a later stage. Both c-Cbl and Cbl-b binding was associated with EGFR ubiquitination. c-Cbl and Cbl-b may thus have both redundant and differential functions. c-Cbl and Cbl-b are structurally similar, and both were expressed in HL-60 cells (39, 40). In this study, we focused on c-Cbl. It is not known how expression levels of Cbl-b are affected when RA gradually down-regulated c-Cbl expression in HL-60 cell lines, but it is possible that Cbl-b may potentially perform a compensatory role when c-Cbl was lost during the late stages of RA-induced differentiation.
c-Cbl is an adaptor molecule that interacts with a plethora of proteins and has been shown to positively influence certain biological processes (34, 41). The multidomain nature of c-Cbl allows it to interact with many crucial signaling molecules, including kinases, adaptors, and structural proteins, via its various domains (13, 21, 42). It is well documented that c-Cbl is capable of binding to the SH3 domain of Fyn, Lck, and Grb-2 NH2 termini as well as interacting with Src family tyrosine kinases, GTPase-activating protein, and phospholipase Cg via SH2 domains (8, 43). This suggests that c-Cbl protein is a prominent substrate of tyrosine kinases in T cells and may have significant functions in signaling cascades or networks. The discovery of novel partners of c-Cbl is important to an understanding of c-Cbl protein function. In this study, we highlight the interaction of Cbl and CD38. The evidence from differentiation and CD38 interaction experiments showed that c-Cbl enhanced CD38 expression and more CD38 or c-Cbl promoted cell differentiation and cell cycle arrest in HL-60 cell lines. Taken together, the data suggest that the process of RA-induced differentiation can be propelled by both c-Cbl and CD38, both of which bind together, enhance the expression of each other, and cause ERK2 activation and MAPK signaling. This suggests a cooperative role for c-Cbl and CD38 in propulsion of RA-induced differentiation and cell cycle arrest.
Clearly, future work on understanding what domains of c-Cbl are responsible for the interaction and how c-Cbl proteins interact with CD38 or other proteins is now of interest. Studies by Sanjay and colleagues (9) indicated that the Src homology domain 3 domain-binding sites in c-Cbl play a functionally significant role in Src- and Cbl-dependent signaling mechanism, and the c-Cbl RDLPPPP (540–546) sequence contributes to a Src-binding site. More importantly, the Ring finger domain of c-Cbl presents the most conserved region among Cbl family proteins, and it is implicated as an important element in the function of Cbl proteins (8, 11, 44). Mutations spanning the Ring finger abolish c-Cbl–directed polyubiquitination and down-regulation of receptor-associated protein tyrosine kinases (45). The present data motivate future work to identify the important residues of c-Cbl, where mutations disrupt the binding of c-Cbl and CD38. This will permit us to further characterize c-Cbl/CD38 interaction and better understand the role of c-Cbl and c-Cbl–interacting proteins in the regulation of RA-induced differentiation and G0 arrest.
Acknowledgments
Grant support: NIH (USPHS) and New York State Stem Cell Science.
We thank Dr. James L. Smith for expert assistance in flow cytometry, especially in the design and execution of the FRET experiments, and Dr. Michael Spinella (Dartmouth-Hitchcock Medical Center) for cells.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Sklan D. Vitamin A in human nutrition. Prog Food Nut Sci. 1987;11:39–55. [PubMed] [Google Scholar]
- 2.Mann G, Reinhardt D, Ritter J, et al. Treatment with all-trans retinoic acid in acute promyelocytic leukemia reduces early death in children. Ann Hematol. 2001;80:417–22. doi: 10.1007/s002770100304. [DOI] [PubMed] [Google Scholar]
- 3.Collins SJ, Gallo RC, Gallagher RE. Continuous growth and differentiation of human myeloid leukaemic cells in suspension culture. Nature. 1977;270:347–9. doi: 10.1038/270347a0. [DOI] [PubMed] [Google Scholar]
- 4.Bretiman TR, Selonick SE, Collins SJ. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc Natl Acad Sci U S A. 1980;77:2926–40. doi: 10.1073/pnas.77.5.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yen A. HL-60 cells as a model of growth control and differentiation: the significance of variant cells. Hematol Rev. 1990;4:5–46. [Google Scholar]
- 6.Yen A, Roberson MS, Varvayanis S, Lee AT. Retinoic acid induces mitogen-activated protein (MAP)/extracellular signal-regulated kinase (ERK) kinase-dependent MAP kinase activation needed to elicit HL-60 cell differentiation and growth arrest. Cancer Res. 1998;58:3163–72. [PubMed] [Google Scholar]
- 7.Yen A, Roberson MS, Varvayanis S. Retinoic acid selectively activates the ERK2 but not JNK/SAPK or p38 MAP kinases when inducing myeloid differentiation. In Vitro Cell Dev Biol Anim. 1999;35:527–32. doi: 10.1007/s11626-999-0063-z. [DOI] [PubMed] [Google Scholar]
- 8.Swaminathan G, Tsygankov A. The Cbl family proteins: ring leaders in regulation of cell signaling. J Cell Physiol. 2006;209:21–43. doi: 10.1002/jcp.20694. [DOI] [PubMed] [Google Scholar]
- 9.Sanjay A, Houghton A, Neff L, et al. Cbl associates with Pyk2 and Src to regulate Src kinase activity, avh3 integrin-mediated signaling, cell adhesion, and osteoclast motility. J Cell Biol. 2001;152:181–95. doi: 10.1083/jcb.152.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yokouchi M, Kondo T, Houghton A, et al. Ligand-induced ubiquitination of the epidermal growth factor receptor involves the interaction of the c-Cbl RING finger and UbcH7. J Biol Chem. 1999;274:31707–12. doi: 10.1074/jbc.274.44.31707. [DOI] [PubMed] [Google Scholar]
- 11.Ota S, Hazeki K, Rao N, et al. The RING finger domain of Cbl is essential for negative regulation of the Syk tyrosine kinase. J Biol Chem. 2000;275:414–22. doi: 10.1074/jbc.275.1.414. [DOI] [PubMed] [Google Scholar]
- 12.Thien CBF, Langdon WY. c-Cbl and Cbl-b ubiquitin ligases: substrate diversity and the negative regulation of signaling responses. Biochem J. 2005;391:153–66. doi: 10.1042/BJ20050892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonita DP, Miyake S, Lupher ML, Langdon WY, Band H. Phosphotyrosine binding domain-dependent upregulation of the platelet-derived growth factor receptor α signaling cascade by transforming mutants of Cbl: implications for Cbl’s function and oncogenicity. Mol Cell Biol. 1997;17:4597–610. doi: 10.1128/mcb.17.8.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meng W, Sawasdikosol S, Burakoff SJ, Eck MJ. Structure of the aminoterminal domain of Cbl complexed to its binding site on ZAP-70 kinase. Nature. 1999;398:84–90. doi: 10.1038/18050. [DOI] [PubMed] [Google Scholar]
- 15.Joazeiro CA, Wing SS, Huang H, Leverson JD, Hunter T, Liu YC. The tyrosine kinase negative regulator c-Cbl as a RING-type, E2-dependent ubiquitin-protein ligase. Science. 1999;286:309–12. doi: 10.1126/science.286.5438.309. [DOI] [PubMed] [Google Scholar]
- 16.Zheng N, Wang P, Jeffrey PD, Pavletich NP. Structure of a c-Cbl-UbcH7 complex: RING domain function in ubiquitin-protein ligase. Cell. 2000;102:533–9. doi: 10.1016/s0092-8674(00)00057-x. [DOI] [PubMed] [Google Scholar]
- 17.Levkowitz G, Waterman H, Ettenberg SA, et al. Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol Cell. 1999;4:1029–40. doi: 10.1016/s1097-2765(00)80231-2. [DOI] [PubMed] [Google Scholar]
- 18.Szymkiswicz DI, Soubeyran P. Cbl signaling networks in the regulation of cell function. Cell Mol Life Sci. 2003;60:1805–27. doi: 10.1007/s00018-003-3029-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmidt MH, Dikic I. The Cbl interactome and its functions. Nat Rev Mol Cell Biol. 2005;6:907–19. doi: 10.1038/nrm1762. [DOI] [PubMed] [Google Scholar]
- 20.Kontani K, Kikimoto I, Nishina H, et al. Tyrosine phosphorylation of the c-Cbl proto-oncogene product mediated by cell surface antigen CD38 in HL-60 cells. J Biol Chem. 1996;271:1534–7. doi: 10.1074/jbc.271.3.1534. [DOI] [PubMed] [Google Scholar]
- 21.Jackson DG, Bell JI. Isolation of a cDNA encoding the human CD38 (T10) molecule, a cell surface glycoprotein with an unusual discontinuous pattern of expression during lymphocyte differentiation. J Immunol. 1990;144:2811–5. [PubMed] [Google Scholar]
- 22.Albeniz I, Demir O, Turker-Sener L, Yalcintepe L, Nurten R, Bermek E. Erythrocyte CD38 as a prognostic marker in cancer. Hematology. 2007;12:409–14. doi: 10.1080/10245330701383841. [DOI] [PubMed] [Google Scholar]
- 23.Damle RN, Temburni S, Calissano C, et al. CD38 expression labels an activated subset within chronic lymphocytic leukemia clones enriched in proliferating B cells. Blood. 2007;110:3352–9. doi: 10.1182/blood-2007-04-083832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.March S, Graupera M, Rosa Sarrias M, et al. Identification and functional characterization of the hepatic stellate cell CD38 cell surface molecule. Am J Pathol. 2007;170:176–87. doi: 10.2353/ajpath.2007.051212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deaglio S, Mehta K, Malavasi F. Human CD38: a (r)evolutionary story of enzymes and receptors. Leukemia Res. 2001;25:1–12. doi: 10.1016/s0145-2126(00)00093-x. [DOI] [PubMed] [Google Scholar]
- 26.Zubiaur M, Fernandez O, Ferrero E, et al. CD38 is associated with lipid rafts and upon receptor stimulation leads to Akt/Protein kinase B and Erk activation in the absence of the CD3. Immune receptor tyrosine-based activation motifs. J Biol Chem. 2002;277:13–22. doi: 10.1074/jbc.M107474200. [DOI] [PubMed] [Google Scholar]
- 27.Lamkin TJ, Chin V, Varvayanis S, et al. Retinoic acid-induced CD38 expression in HL-60 myeloblastic leukemia cells regulates cell differentiation or viability depending on expression levels. J Cell Biochem. 2006;97:1328–38. doi: 10.1002/jcb.20745. [DOI] [PubMed] [Google Scholar]
- 28.D’Arena G, Musto P, Cascavilla N, et al. CD38 expression correlates with adverse biological features and predicts poor clinical outcome in B-cell chronic lymphocytic leukemia. Leuk Lymphoma. 2001;42:109–14. doi: 10.3109/10428190109097682. [DOI] [PubMed] [Google Scholar]
- 29.Ghia P, Guida G, Stella S, et al. The pattern of CD38 expression defines a distinct sunset of chronic lymphocytic leukemia (CLL) patients at risk of disease progression. Blood. 2003;101:1262–9. doi: 10.1182/blood-2002-06-1801. [DOI] [PubMed] [Google Scholar]
- 30.Brooks SC, III, Kazmer S, Levin AA, Yen A. Myeloid differentiation and RB phosphorylation changes in HL-60 cells induced by RAR- and RXR-selective retinoic acid analogs. Blood. 1996;87:227–37. [PubMed] [Google Scholar]
- 31.Reiterer G, Yen A. Platelet-derived growth factor receptor regulates myeloid and monocytic differentiation of HL-60 cells. Cancer Res. 2007;67:7765–72. doi: 10.1158/0008-5472.CAN-07-0014. [DOI] [PubMed] [Google Scholar]
- 32.Wightman J, Roberson MS, Lamkin TJ, Varvayanis S, Yen A. Retinoic acid-induced growth arrest and differentiation: retinoic acid up-regulates CD32 (FcgRII) RII) expression, the ectopic expression of which retards the cell cycle. Mol Cancer Ther. 2002;1:493–506. [PubMed] [Google Scholar]
- 33.Bhaumik SR. Analysis of in vivo targets of transcriptional activators by fluorescence resonance energy transfer. Methods. 2006;40:353–9. doi: 10.1016/j.ymeth.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 34.Dikic I, Szymkiewicz I, Soubeyran P. Cbl signaling networks in the regulation of cell function. Cell Mol Life Sci. 2003;60:1805–27. doi: 10.1007/s00018-003-3029-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamilton E, Miller KM, Helm KM, Langdon WY, Anderson SM. Suppression of apoptosis induced by growth factor withdrawal by an oncogenic form of c-Cbl. J Biol Chem. 2001;276:9028–37. doi: 10.1074/jbc.M009386200. [DOI] [PubMed] [Google Scholar]
- 36.Dikic I, Giordano S. Negative receptor signaling. Curr Opin Cell Biol. 2003;15:128–35. doi: 10.1016/s0955-0674(03)00004-8. [DOI] [PubMed] [Google Scholar]
- 37.Thien CB, Langdon WY. Cbl: many adaptors to regulate protein tyrosine kinase. Nat Rev Mol Cell Biol. 2001;2:294–307. doi: 10.1038/35067100. [DOI] [PubMed] [Google Scholar]
- 38.Pennock S, Wang Z. A tale of two Cbls: interplay of c-Cbl and Cbl-b in epidermal growth factor receptor downregulation. Mol Cell Biol. 2008;28:3020–37. doi: 10.1128/MCB.01809-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qu X, Liu Y, Ma Y, Zhang Y, Li Y, Hou K. Up-regulation of the Cbl family of ubiquitin ligase is involved in ATRA and bufalin-induced cell adhesion but not cell differentiation. Biochem Biophys Res Commun. 2008;267:183–9. doi: 10.1016/j.bbrc.2007.12.120. [DOI] [PubMed] [Google Scholar]
- 40.Kenae MM, Revero-Lezcano OM, Mitchell JA, Robbins KC, Lipkowitz S. Cloning and characterization of cbl-b: a SH3 binding protein with homology to the c-cbl proto-oncogene. Oncogene. 1995;10:2367–77. [PubMed] [Google Scholar]
- 41.Jacob C, Cottrell GS, Gehringer D, Schmidlin F, Grady EF, Bunnett NW. c-Cbl mediates ubiquitination, degradation, and down-regulation of human protease-activated receptor 2. J Biol Chem. 2005;280:16076–87. doi: 10.1074/jbc.M500109200. [DOI] [PubMed] [Google Scholar]
- 42.Feshchenko EA, Langdon WY, Tsygankov AY. Fyn, Yes, and Syk phosphorylation sites in c-Cbl map to the same tyrosine residues that become phosphorylated in activated T cells. J Biol Chem. 1998;273:8323–31. doi: 10.1074/jbc.273.14.8323. [DOI] [PubMed] [Google Scholar]
- 43.Donovan JA, Wange RL, Langdon WY, Samelson LE. The protein product of the c-cbl protooncogene is the 120-kDa tyrosine-phosphorylated protein in Jurkat cells activated via the T cell antigen receptor. J Biol Chem. 1994;260:22921–4. [PubMed] [Google Scholar]
- 44.Visser GD, Lill NL. The Cbl RING finger C-terminal flank controls epidermal growth factor receptor fate downstream of receptor ubiquitination. Exp Cell Res. 2005;311:281–93. doi: 10.1016/j.yexcr.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 45.Thien CB, Walker F, Langdon W. Ring finger mutations that abolish c-Cbl-directed polyubiquitination and downregulation of the EGF receptor are insufficient for cell transformation. Mol Cell. 2001;7:355–65. doi: 10.1016/s1097-2765(01)00183-6. [DOI] [PubMed] [Google Scholar]