

Abstract
Arsenic trioxide (ATO) synergistically promotes retinoic acid (RA)-induced differentiation of HL-60 myeloblastic leukemia cells, a PML-RARα negative cell line. In PML-RARα positive myeloid leukemia cells, ATO is known to cause degradation of PML-RARα with subsequent induced myeloid differentiation. We find now that ATO by itself does not cause differentiation of the PML-RARα negative HL-60 cells, but enhances RA’s capability to cause differentiation. RA-induced differentiation of HL-60 cells is known to be propelled by an induced hyperactive/persistent MAPK signal. ATO augmented RA induced RAF/MEK/ERK axis signaling and expression of CD11b, an integrin receptor that is a myeloid differentiation marker. p47PHOX, a component of the respiratory burst machinery and inducible oxidative metabolism, functional differentiation marker were also enhanced. However, ATO did not enhance RA-induced CD38 expression, an early cell surface differentiation marker. ATO enhanced RA-induced population growth retardation without evidence of apoptosis or an enhanced G1/0 growth arrest. But compared to RA, ATO plus RA showed reduced pAKT, suggesting that an overall biosynthetic/metabolic retardation was seminal to the apparent enhanced growth retardation due to ATO. In sum, our results indicate that ATO can augment action of RA in causing differentiation of myeloid leukemia cells through promoting MAPK signaling and independent of PML-RARα.
1. Introduction
Arsenic has historically been used as a medicinal agent for a variety of diseases such as psoriasis, eczema, asthma, malaria, ulcers, syphillis and leukemia. Arsenic and its derivatives have been used as antiseptics, antispasmodics, antipyretics, sedatives and tonics etc., especially in traditional Chinese medicine (1). The most common compound of arsenic used in medicine is arsenic trioxide. Arsenic Trioxide (As2O3) or ATO is the active ingredient in the Fowler’s solution developed in the 18th century. It has been used to treat different malignancies for over 100 years. In the 1930s, before the introduction of chemotherapy and radiation therapy, arsenic was used in one of the standard treatments for chronic myeloid leukemia and other leukemias (1–2). The modern use of arsenic trioxide as a therapy for Acute Promyelocytic Leukemia (APL) originated in China in the 1990s (3). In a study at the Shanghai Institute of Hematology (SIH) in 1999, patients with relapsed APL were treated with ATO. Complete remission (CR) was achieved in 9/10 of the cases treated with ATO alone and 5/5 cases treated with ATO plus chemotherapy (CT) or Retinoic Acid (3–5). This suggests that ATO is potent as a single agent in APL, a disease cytogentically characterized by a t(15,17) translocation that gives rise to the PML-RARα fusion protein which is a rogue transcription factor. This was further proved by relatively high 2- and 3-year Disease Free Survival (DFS) rates seen recently in newly diagnosed leukemias also (6).
All Trans Retinoic Acid (ATRA) has been used in differentiation induction therapy for APL since the early 1980s (7–8). A recent clinical study of APL treatment with ATRA differentiation therapy in Japan resulted in a complete remission (CR) rate of 94%, a 6-year Disease Free Survival rate of 68.5%, and a 6-year overall survival (OS) rate of 83.9% (9). Clinical studies using a combination of ATRA-ATO showed that the time required to achieve complete remission (CR) with ATRA-ATO was statistically significantly different (25.5 days) from ATRA alone (40.5 days) or ATO alone (31 days). The median disease free survival (DFS) rate had also increased to 20 months for the combination treatment as compared to ATRA alone (13 months) and ATO alone (16 months) (10). Furthermore, the disease burden, determined by the fold change in PML-RARα was much shorter in the combination treatment compared to monotherapy. Clinical studies thus show a synergistic effect of ATRA and ATO treatments, motivating interest in the biochemical cause of this synergy. Both ATRA and ATO have been found to cause degradation of the PML-RARα fusion protein, suggesting a mechanism of action whereby degradation of the transforming protein relieves the block in differentiation and permits progression of differentiation along the myeloid lineage. However, ATO may have many other actions, as is the case for ATRA, and their collaboration may be attributable to other biochemical mechanisms they share as targets. It is thus of interest to determine if they collaborate in a setting with no PML-RARα.
HL-60 is a human myeloblastic leukemia cell line that has been extensively used as a model for pharmacologically induced differentiation. HL-60 cells undergo either myeloid or monocytic differentiation, and G0/G1 growth arrest when treated with All Trans Retinoic Acid (ATRA) or 1–25-dihydroxyvitamin D3 (Vitamin D3) respectively. Previous studies in our laboratory have shown that ATRA or vitamin D3-induced differentiation and G0/G1 cell arrest require sustained activation of MAPK signaling along the RAF/MEK/ERK axis (11–12). ATRA induces the MAPK dependent expression of cell surface differentiation markers such as CD38, CD11b as well as functional differentiation markers such as inducible oxidative metabolism that become progressively expressed as the cells undergo myeloid differentiation.
HL-60 cells are negative for the t(15:17) chromosomal translocation that defines APL and gives rise to the PML-RARα fusion protein. If the ability of ATO to promote differentiation is solely through the degradation of PML-RARα fusion protein, then we should not see any enhanced differentiation for the combination treatment of ATRA and ATO. However, if ATO action collaborates with the mechanism of ATRA induced differentiation, eg. by promoting sustained activation of MAPK, then addition of ATO to ATRA treatment should augment differentiation of HL-60 cells.
The present studies showed that compared to cells treated with ATRA alone, HL-60 cells treated with a combination of ATRA and ATO showed augmented differentiation propelled by an enhanced activation of MAPK signaling. While addition of ATO to ATRA treatment enhanced population growth retardation, there was no corresponding increase seen in the percentage of cells undergoing G0 specific cell-cycle arrest, but rather an apparent overall retardation of cell cycle progression. Unlike PML-RARα positive cells, ATO alone did not cause differentiation in HL-60 cells, as indicated before (13). The results show that ATO can enhance the differentiation-inducing effects of ATRA in PML-RARα negative cells consistent with a paradigm whereby the ATO enhances the ATRA-induced MAPK signaling that propels induced differentiation. Since ATO is already FDA approved (Trisenox), along with ATRA differentiation therapy for APL, a more thorough understanding of the mechanism of action of ATO, especially on the MAPK and related pathways, may be of therapeutic value in treating other leukemias, or even non-hematologic cancers.
2. Materials and Methods
2.1 Cell Culture
HL-60 human myeloblastic leukemia cells were cultured in RPMI 1640 media supplemented with 5% heat-inactivated fetal bovine serum (Invitrogen, Carlsbad, CA, USA) in a 5% CO2 humidified environment at 37° C and were maintained in an exponential growth phase. All the experimental cultures were initiated at a density of 0.2 × 106 cells/mL (14). Cell viability was monitored by Trypan Blue Stain (Invitrogen) and consistently exceeded 95%.
2.2 Reagents Used
All Trans Retinoic Acid (ATRA) (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in 100% ethanol to make a stock solution concentration of 5 mM. The concentration of ATRA used in cultures was 1 μM. Arsenic Trioxide (ATO) (Sigma-Adlrich) was diluted in water to make a stock concentration of 5 mM. ATO was used at a final concentration of 2 μM, and added at the same time as ATRA to the cultures. Antibodies for Western analysis against ERK1/2, phosphorylated ERK-1/2 (Thr-202/Tyr-204), MEK1/2, phosphorylated MEK1/2 (Ser-217/221), RAF1, phosphorylated Akt (Ser-473), phosphorylated Akt (Thr-308), PARP, p47PHOX and GAPDH were obtained from Cell Signaling (Beverly, MA, USA). Antibody against phosphorylated c-RAF1(Ser-621) was from BIOSOURCE/Invitrogen. Western analysis of Akt phosphorylation was done on the nuclear fraction. The nuclear fraction was prepared by re-suspending the cells after centrifugation for 10 min in 100 μL of hypotonic propidium Iodide, sodium citrate (50 μg/mL propidium iodide (PI), 1 μL Triton-X100 and 1 mg/ml sodium citrate) solution. The cells were vortexed and kept on ice for 15 min, and the procedure was repeated 3 times. Nuclei were collected by centrifugation for 10 min, resuspended in 100 μL/sample RIPA buffer (Thermo Scientific, Rockford, IL USA) and freeze-thawed 3 times. Finally, the lysate was centrifuged (16060 × g) for 30 min and the supernatant was collected.
2.3 CD38 and CD11b Expression Studies by Flow Cytometry
At the 24h and 48h time points, 0.5 × 106 cells were collected, centrifuged to A pellet and resuspended in 100 μL PBS containing 5μL allophycocyanin (APC)-conjugated CD11b and 5μL of phycoerythrin(PE)-conjugated CD38 antibodies. The cells were then incubated for 1 hr in a humidified atmosphere of 5% CO2 at 37 °C and expression levels were measured by flow cytometry (BD LSR II flow cytometer, Becton Dickinson, Mountain View, CA). CD11b expression was measured by 633 nm red laser excitation and emission collected through a 735 long-pass dichroic and a 660/20 band-pass filter. CD38 expression was measured with 488 nm laser excitation and emission through a dichroic 550 long-pass and 576/26 band-pass filter. Gates to determine the shift in cellular fluorescence intensity were set to exclude 95% of the control (untreated) cells.
2.4 Measurement of Inducible Oxidative Metabolism
Approximately 0.5 × 106 cells were collected at 48h, 72h time points, centrifuged to a pellet and resuspended in 200 μL PBS at 37 °C containing 5 μmol/L 5- (and 6)-chloromethyl-2′,7′-dichlorodihydro-fluorescence diacetate acetyl ester (H2DCF, Molecular Probes) without or with 0.2 μg/mL 12-O-tetradecanoylphorbol-13-acetate (TPA, Sigma). H2DCF and TPA stock solutions were made in DMSO at concentrations of 0.2 mg/mL and 0.5 mg/mL respectively. Cells were incubated for 20 min in a humidified atmosphere of 5% CO2 at 37 °C. Flow cytometric analysis was done (BD LSR II flow cytometer) using 488 nm excitation and emission from oxidized DCF was collected through a 505-nm long pass dichroic mirror and 530/30 nm band pass filter. The shift in fluorescence intensity in response to TPA was used to determine the percentage of cells capable of inducible oxidative metabolism. Gates to determine percentage of positive cells were set to exclude 95% of the same cells not treated with TPA.
2.5 Cell cycle analysis
About 0.5 × 106 cells were harvested at 72h by centrifugation and resuspended in 200 μL hypotonic staining solution (at 4 °C) containing 50 μg/mL propidium iodide (PI), 1 μL Triton-X100 and 1 mg/ml sodium citrate. Cells were incubated at room temperature for 1 h and analyzed using flow cytometry (BD LSR II). Excitation was provided by a 488 nm laser and the emission was collected using 550 long-pass dichroic and a 576/26 band-pass filter.
2.6 Statistical Analysis
Statistical Analysis was done using MATLAB’s statistical toolbox. Three independent repeats were performed in each case. Means of treatment cases were compared using paired-samples t-test, using the ttest2 function in MATLAB’s statistical toolbox. The bar graphs show means of three independent repeats along with standard errors.
3. Results
3.1 Arsenic Trioxide (ATO) leads to enhanced sustained activation of ATRA induced MAPK proteins
It is well known that transient activation of MAPK leads to proliferation, whereas a sustained activation of MAPK, especially ERK1/2 has been thought to drive differentiation in a variety of cell lines including HL-60 cells (11–12). It is also known that ATO can activate MAPK pathways as part of a stress response. We studied the effect of ATO on the RAF/MEK/ERK axis of MAPK signaling, in particular the potential synergistic effects of ATRA and ATO on these three central kinases. Treating cells with ATO plus ATRA enhanced the increase in phosphorylation at Thr-202/Tyr-204 and activation of ERK1/2 elicited by ATRA alone (Fig 1 (C)). This is seen in the comparison of Western blots of pERK1/2(Thr-202/Tyr-204) after 48 hrs of combination ATRA-ATO) treatment versus ATRA alone treatment. This enhanced activation was sustained and observed at both 24h and 48h. Interestingly, there was also increased activation of ERK1/2(Thr-202/Tyr-204) in the ATO alone treatment case compared to the control (untreated) case. Western analysis of ERK1/2 expression showed that there was no increase in the total expression of ERK1/2 in any of the treatment cases. The expression levels of ERK1/2 were thus unaffected by treatment, although their activation increased.
Figure 1.
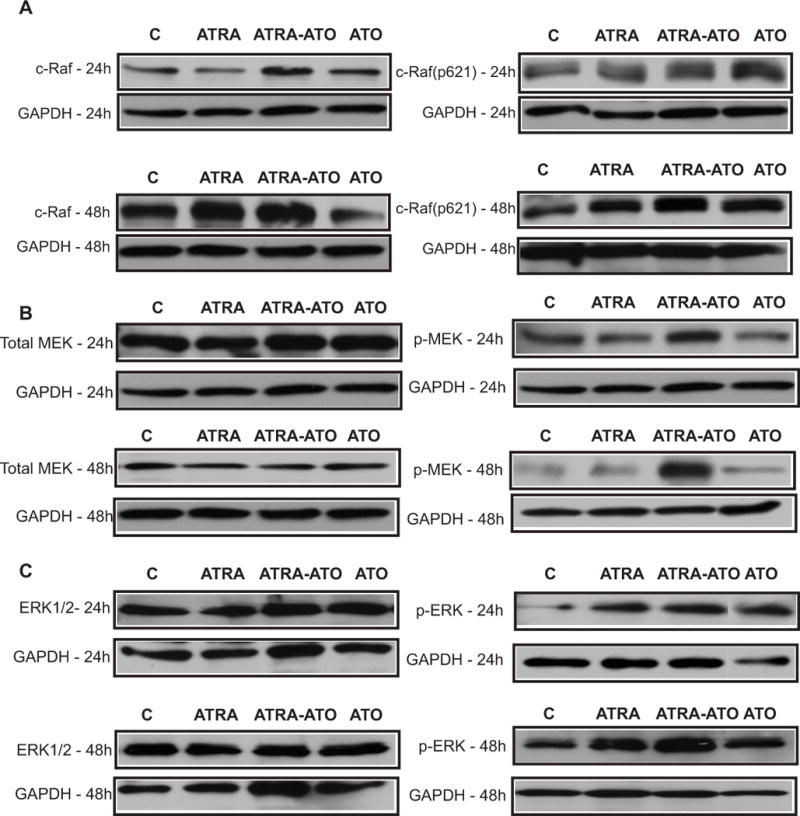
Western Blot analysis of MAPK proteins showed enhanced activation in the combination treatment of All Trans Retinoic Acid (ATRA) and Arsenic Trioxide (ATO) (ATRA-ATO) compared to ATRA treatment. Panels A, B and C show the effect of the different treatments on expression and activation levels of RAF1, MEK1/2 and ERK1/2 respectively. The left columns show the effect on expression levels at 24 h and 48h, whereas the right columns show the effect on activation of MAPK proteins. The lanes show control (untreated) cells, cells treated with ATRA, ATRA plus ATO, and ATO only. ATO leads to an increase in ATRA induced sustained activation of MAPK proteins. This increase was seen in p-ERK at 24h and 48h (see part (c)). Increased activation was also seen for MEK as well as RAF1 at both 24h and 48h. There was thus an increase in activation of p-ERK and c-RAF(Ser621) for the ATO alone treatment case, when compared to untreated (control) case. We did not see any increase in the total expression of MAPK proteins for any of the treatment cases (see left column) at 24h or 48h. This suggests that ATO (in combination or alone) only affects activation (via phosphorylation) of MAPK proteins, without affecting their total expression levels.
Consistent with the results for ERK, Western blot analysis of other MAPK proteins showed similar ATO enhancement of ATRA induced activation of MEK1/2 and RAF1. As seen in Fig. 1(B)., treatment with ATO plus ATRA lead to increased phosphorylation and activation of MEK (Ser-217/221), compared to treatment with ATRA alone. Unlike ERK1/2 phosphorylation however, ATO treatment alone did not lead to an increase in phosphorylated MEK1/2 (Ser-217/221). Western analysis of total MEK1/2 showed no effect of either ATRA or ATO treatment on MEK1/2 expression levels. As for ERK, the increased phosphorylated MEK1/2 in the combination treatment is thus attributable to increased activation and not to increased expression. Further upstream along the MAPK axis, ATO enhanced the ATRA-induced phosphorylation of RAF1(Ser-621 residue), consistent with the results for MEK and ERK. As seen from Fig. 1(A), the combination treatment of ATRA plus ATO leads to more activation of RAF1(Ser-621 phosphorylation) than treatment with ATRA alone. The increased activation of RAF1(Ser-621) is more prominent at 48h, than at 24h. This is consistent with the activation patterns of pMEK(Ser-217/221) and pERK(Thr-202/Tyr-204), which also show more ATO enhancement of activation at 48h than at 24h. It is interesting to note the ATO alone treatment did cause increased p-RAF1(Ser621) as it did for pERK1/2(Thr-202/Tyr-204), especially at 48h, compared with the untreated case. The apparent extent of activation was comparable to that of ATRA alone.
3.2 Arsenic Trioxide (ATO) enhances ATRA induced CD11b but not CD38 expression
Having established enhanced activation of MAPK signaling in the combination treatment, we then proceeded to see if this augmentation translated into enhanced differentiation in HL-60 cells. A visual inspection of different treatments suggested that there was a synergy in the combination treatment which lead to enhanced differentiation. Figure 2 shows that there was an increase in the percentage of potentially differentiated irregularly shaped cells after the combination treatment compared to treatment with ATRA alone. Cells treated with ATO alone did not cause any apparent differentiation, but did exhibit a decrease in growth rate. To investigate if combination treatment lead to enhanced differentiation, we started by looking at the cell surface markers of differentiation, CD38 and CD11b.
Figure 2.
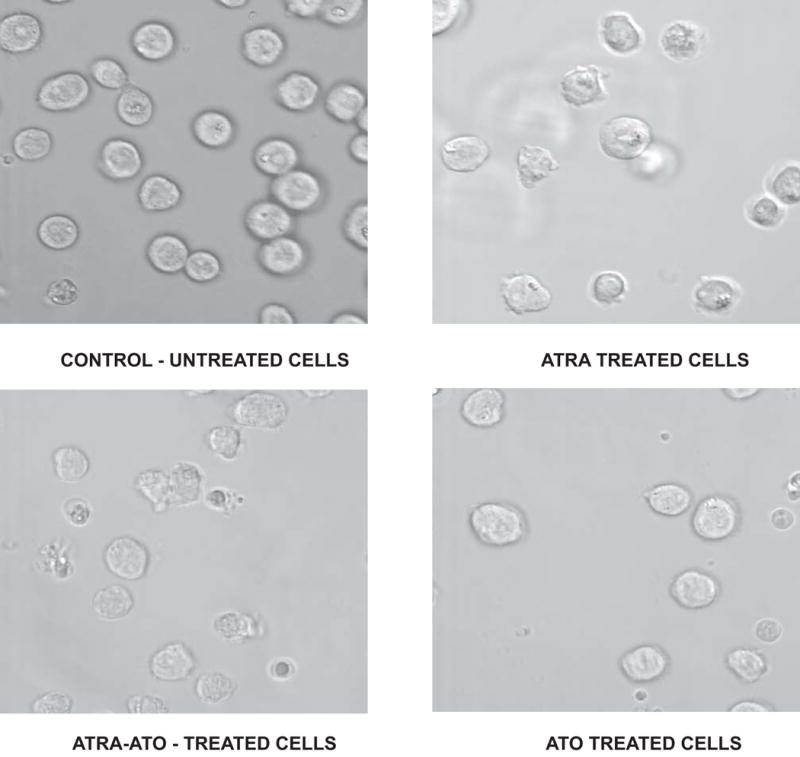
Change in cellular morphology induced by different treatments. Microscopic images showing morphological changes 72h after different treatments are shown here. In the control (untreated) case, mostly round cells were observed. Addition of All Trans Retinoic Acid (ATRA) lead to differentiated cells, seen here as the distorted cells. Differentiation was more apparent in the combination treatment of ATRA and Arsenic Trioxide (ATO), whereas ATO alone treatment case shows a morphology similar to the control case, although a smaller number of cells are seen in this case.
CD38 is 45 kDa ectoenzyme type II transmembrane receptor. The gene is transcriptionally regulated by a Retinoic Acid Response Element (RARE) in its first intron, which confers response to ATRA (15). CD38 can MAPK signal through activation of RAF. One component of the signaling machinery is the c-Cbl adaptor (16). To determine the effects of ATO and ATRA on CD38 expression, we measured expression of CD38 at an early time point (6 h) and at 24 h after various treatments using a PE-conjugated CD38 antibody and flow cytometry. As seen in Figure 3, treatment with ATRA induced progressively increasing CD38 expression that was indistinguishable from treatment with ATRA plus ATO. There was no increase in CD38 expression in the cells treated with ATO alone. These results indicated that ATO had no effect on CD38 expression, either in combination with ATRA or by itself.
Figure 3.
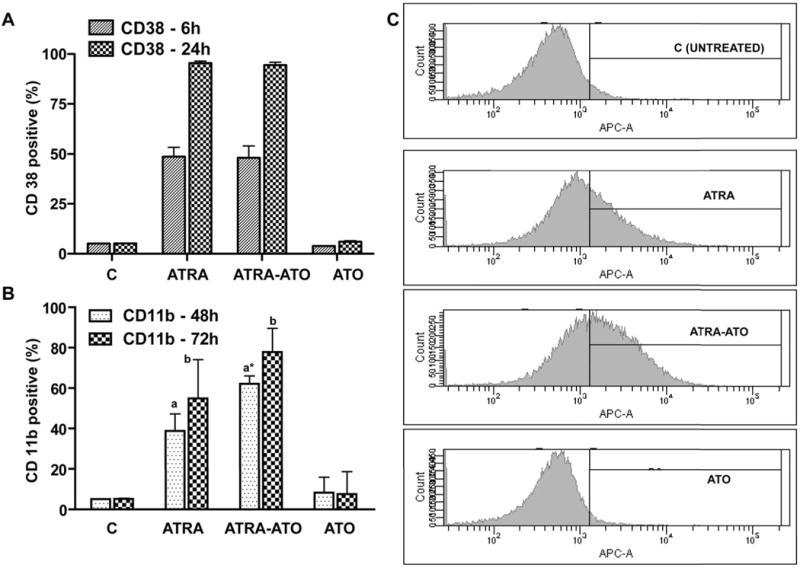
Arsenic Trioxide (ATO) enhanced ATRA-induced CD11b expression, but not CD38 expression. Panel A shows that there was no enhancement in CD38 expression in the combination treatment, but CD11b (Panel B) expression showed a synergistic increase in the ATRA-ATO combined treatment case. CD38 and CD11b expression was analyzed with PE-conjugated CD38 and APC-conjugated CD11b antibodies at 24, 48 and 72 h by flow cytometry. The y-axis shows percentage of CD38 positive cells (Panel A) and CD11b positive cells (Panel B) at indicated time points when compared with untreated cells with gates set to exclude 95% of the cells in the control case. The data show mean values ± standard errors a: CD11b expression in the cells treated with ATRA-ATO was statistically significantly different from that in cells treated with ATRA or ATO alone with p ≤ 0.05. b: CD 11b expression in cells with ATRA treatment was statistically less that that in cells treated with both ATRA and ATO at 90% confidence interval. Panel C shows the representative CD11b histograms of untreated (control) and cells treated with ATRA, ATRA-ATO combination and ATO alone at 48h.
Another cell surface marker, CD11b, an integrin receptor, was used to characterize the effect of Arsenic Trioxide (ATO) on ATRA induced cell differentiation. Expression of CD11b typically marks progression of ATRA-induced differentiation beyond the stage of CD38 expression, which is an early marker. CD11b expression was measured by flow cytometry using an APC-conjugated CD11b antibody (17–18). The effect of ATO and ATRA, alone and in combination, on CD11b expression was measured at 48 and 72 hrs. Compared to untreated control cells, ATRA-treated cells showed an increase in CD11b expression, as expected. However, CD11b expression was higher in the case of ATRA-ATO combination when compared with the ATRA alone case (Fig 3). The difference between CD11b expression was statistically significant at 48h, but not at 72h, indicating that the differentiation machinery was more affected by ATO during its early stages. In contrast, the cells treated with ATO alone did not show an increase when compared with the untreated control case, indicating that ATO alone does not affect CD11b expression.
3.3 Arsenic Trioxide enhances ATRA induced differentiation by upregulating p47PHOX
Inducible oxidative metabolism marked by an increase in Reactive Oxygen Species (ROS) is one of the functional markers of differentiation into mature myelo-monocytic cells. p47PHOX is one of the essential components of the NADPH-oxidase complex which generates ROS when activated in differentiated cells. We measured the effect of ATO and ATRA separately and in combination on p47PHOX expression. The untreated control cells and ATO treated cells did not show p47PHOX expression at 24h or 48h (See Fig. 4). Consistent with the trend seen in MAPK activation, enhanced expression was seen in the ATRA-ATO combination treatment compared to treatment with ATRA alone. Also consistent with the MAPK results, the difference in p47PHOX expression between ATRA and ATRA-ATO treatments was more prominent at 48h than at 24h. This result suggests that ATO synergizes with ATRA to induce expression of p47PHOX although ATO does not induce p47PHOX. Expression by itself.
Figure 4.
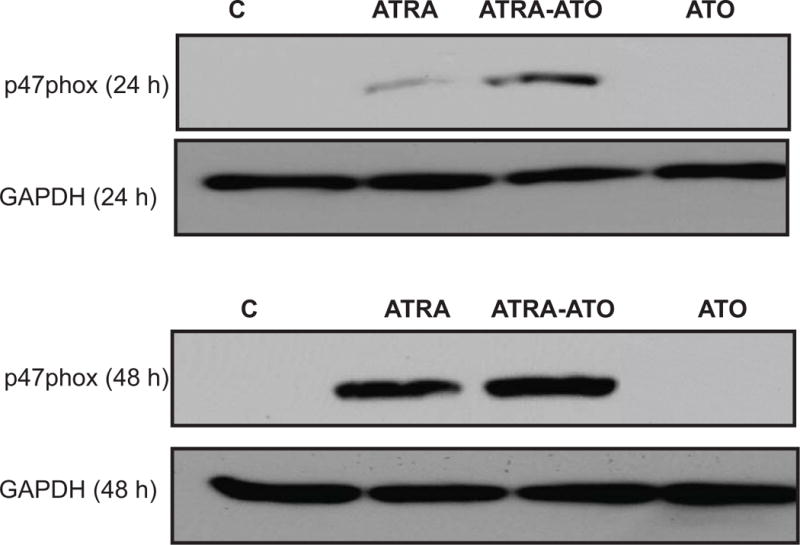
Arsenic Trioxide (ATO) leads to an increase in ATRA induced expression in p47PHOX. Western analysis of total expression of p47PHOX is shown at 24h and 48h time points. p47PHOX is a key member of the NADPH oxidase complex which generates Reactive Oxygen Species (ROS). An upregulation in the expression of p47PHOX is seen at both 24h and 48h, with the difference being more apparent at 48h. Protein lysates were extracted from untreated cells, and cells treated with ATRA, ATRA-ATO combination, and ATO alone. Loading control is shown by GAPDH levels. This suggests that ATO directly affects the ROS part of differentiation machinery also, apart from increase in MAPK proteins.
3.4 Arsenic Trioxide (ATO) enhances ATRA induced functional differentiation
To confirm the functional significance of the induced p47PHOX expression, TPA inducible oxidative metabolism, a late functional differentiation marker that characterizes mature myelo-monocytic cells, was measured. When treated with ATRA, HL-60 cells typically exhibit a functional inducible Reactive Oxygen Species (ROS) response. As before, cells were treated with ATO or ATRA alone or in combination. Inducible oxidative metabolism was measured by flow cytometry using 2′,7′-dichloroduhydrofluorescein diacetate (H2DCFDA), a cell-permeant indicator of reactive oxygen species that is nonfluorescent untill the acetate groups are removed by intercellular esterases when oxidation takes place within the cell. As shown by Fig. 5 (A), HL-60 cells treated with a combination of ATRA-ATO showed increased functional differentiation at both 48h and 72h time-points when compared with cells treated with ATRA alone. For example, at 72h, 88.5 % of cells treated with ATO plus ATRA were able to produce ROS induced by TPA, compared to 66.1 % of cells treated with ATRA alone. The effect of ATO-ATRA was greater than that of ATRA alone with 90% confidence (P = 0.0953). In the case of cells treated with ATO alone, there was no significant increase in ROS induction upon TPA treatment, indicating that ATO alone was unable to induce functional differentiation of HL-60 cells. This is consistent with the inability of ATO to induce the other differentiation markers measured. These results indicate that even though ATO by itself is unable to induce differentiation of HL-60 cells, it works synergistically to enhance ATRA induced differentiation.
Figure 5.
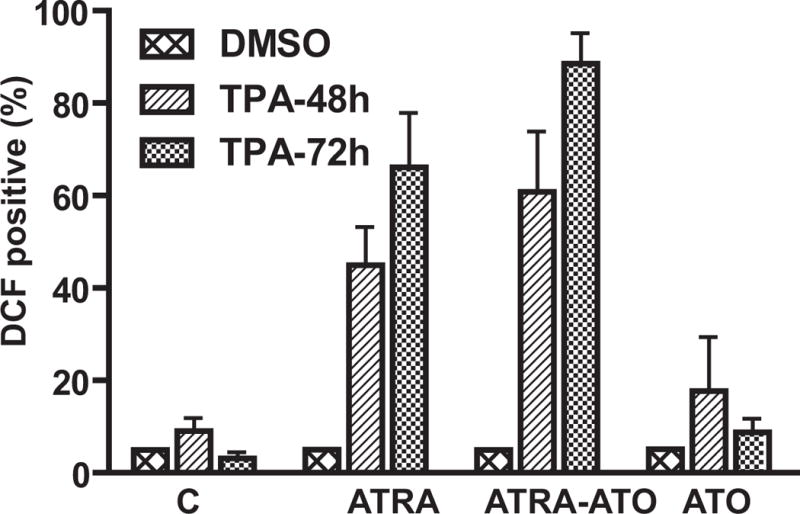
Arsenic Trioxide enhances ATRA induced differentiation of HL-60 cells. Panel A shows the percentage of cells (y-axis) with capability for inducible oxidative metabolism at the indicated time points. Cells were treated with TPA to induce oxidative metabolism. Using a functional marker of differentiation, i.e. generation of Reactive Oxygen Species (ROS) we measured terminal differentiation in untreated cells, cells treated with ATRA, a combination of ATRA-ATO, and ATO alone. The percentage of positive cells with TPA, when gates were set to exclude 95% of control cells (without TPA), is shown. There was no statistically significant difference in the percentage of differentiated cells in the ATRA alone versus ATRA-ATO combination treatments at 48h. At 72h, using a one-tailed test we concluded that percentage of DCF positive cells was less in the ATRA case when compared with the ATRA-ATO combination case with a 90% confidence level (p=0.0953). The background ROS activity in all the samples was measured by cells treated with DMSO.
3.5 Arsenic Trioxide (ATO) leads to reduced growth in HL-60 cells
HL-60 cells treated with Arsenic Trioxide (ATO) had slower population growth rates compared to untreated cells as well as ATRA treated cells, as seen in Fig. 6. Addition of ATRA alone decreased the population growth rate compared to untreated cells. However, addition of ATO had a more drastic effect on the growth rate of cells than ATRA. Cultures with ATO alone or the combination treatment of ATRA-ATO grew much slower than the cultures with ATRA alone. Of note, growth of ATO plus ATRA treated populations was slower than ATRA treated populations. Microscopic images of the cultures at 72h showed morphologically differentiated cells in the ATRA and ATRA-ATO (Fig. 2) combination treated cultures; whereas cells in the ATO alone cultures were not obviously different from the untreated cells morphologically. Taken with the differentiation data, the population growth results suggest that treatment with ATO alone slows down population growth without inducing differentiation.
Figure 6.
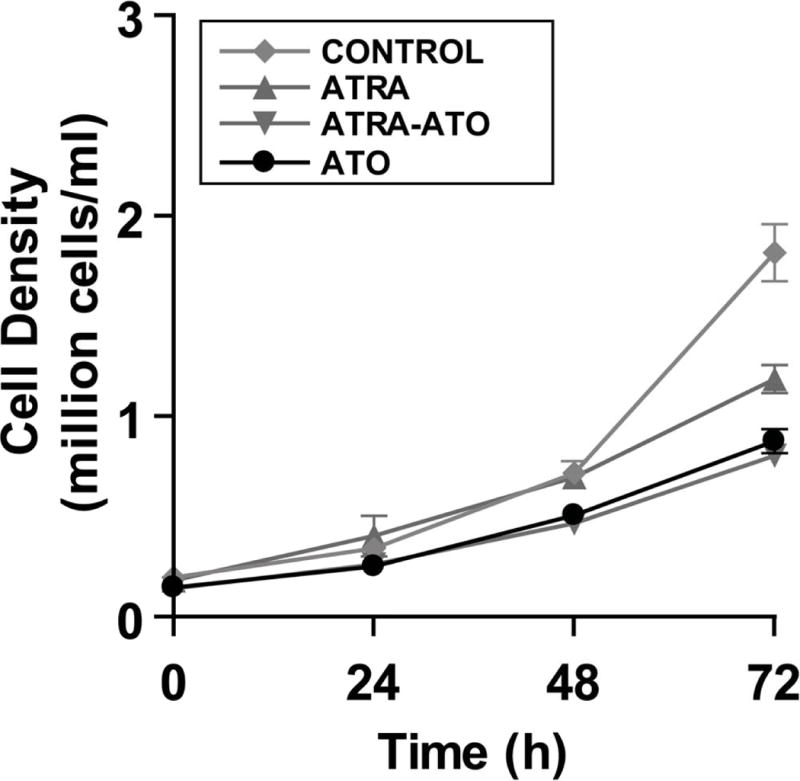
There is a decrease in cell growth rate upon addition of ATO. Cells growth rates were measured over a period of 72h after treatments. The cells were seeded at an initial concentration of 0.2 million cells/mL. Y-axis shows the cell density in the culture in million cells/ml whereas the x-axis shows time points at which the cells were counted. Addition of ATRA also leads to decreased growth rate, however ATO addition has a more significant effect by itself or in combination with ATRA. The ATO cultures did not show an increase in dead cells or debris. This suggests three possibilities, we hypothesized that the cells maybe undergoing apoptosis under Arsenic Trioxide (ATO) treatment, or a higher percentage of cells were getting arrested in the G0/G1 phase or there was a general decrease in the stages of cell cycle. All of these possible reasons were investigated further and the results are shown.
Three possible causes for the decreased growth were hypothesized and tested. A decrease in population growth rate could be due to apoptosis upon addition of ATO. Alternatively, an increase in cells arresting in the G0/G1 phase could lead to an overall decrease in population growth rates due to a diminishing growth fraction. Finally, an overall decrease in biosynthetic activity could cause a general dilation of the cell cycle and decreased cell doubling rates. We looked at the cleavage of PARP for evidence of apoptosis in various treatment cases, cell-cycle distribution by flow cytometry for evidence of G0 cell cycle arrest, and phosphorylation of the Akt kinase as a marker of biosynthetic metabolism for evidence of metabolic slow down.
3.6 Arsenic Trioxide dependent cleavage of PARP is abrogated by ATRA
To test the first hypothesis, namely that reduced growth rates are due to increased apoptosis, we looked at Poly (ADP)-ribose polymerase (PARP) cleavage in the treated cells. PARP acts as a molecular nick sensor and marks sections of damaged DNA for repair. PARP is a substrate for caspase-3 and caspase-7 which cleave it into two fragments, a major fragment of 89 kDa and a minor fragment of 24 kDa. Cleavage of PARP is a marker for the proteolytic activity of caspase and indicates progression of apoptosis. It has been shown that a high dose of ATO (10μM) leads to cleavage of PARP after 12h (19) which continues up to 24h in HL-60 cells and NB4 cells. In NB4 cells, a low dose (2 μM) ATO can lead to cleavage of PARP at 72h. However, it was also seen that chemical agents such as lipoic acid and N-acetylcysteine could block ATO-induced cleavage of PARP and keep the cells from apoptosis without affecting degradation of PML-RARα protein (20). Figure 8 shows the Western analysis of the uncleaved and cleaved PARP. There was no evidence of apoptosis indicated by PARP cleavage in untreated control, ATRA or ATO plus ATRA treated populations. The results suggest that there was some transient apoptosis in the ATO alone treatment that was transiently evident at 24 h and no longer evident by 48h. More importantly, we did not detect apoptosis in the case of ATRA-ATO combination treatment. This result suggests that ATRA protected cells from apoptosis caused by ATO. Consistent with these results trypan blue exclusion also showed no evidence of induced cell death. Thus we conclude that the decrease in population growth rates in the ATRA-ATO combination treatment case compared to ATRA alone is not due to apoptosis. We next tested if the decrease in growth rates was attributable to an increase in the percentage of cells arresting in the G1/G0 phase.
Figure 8.
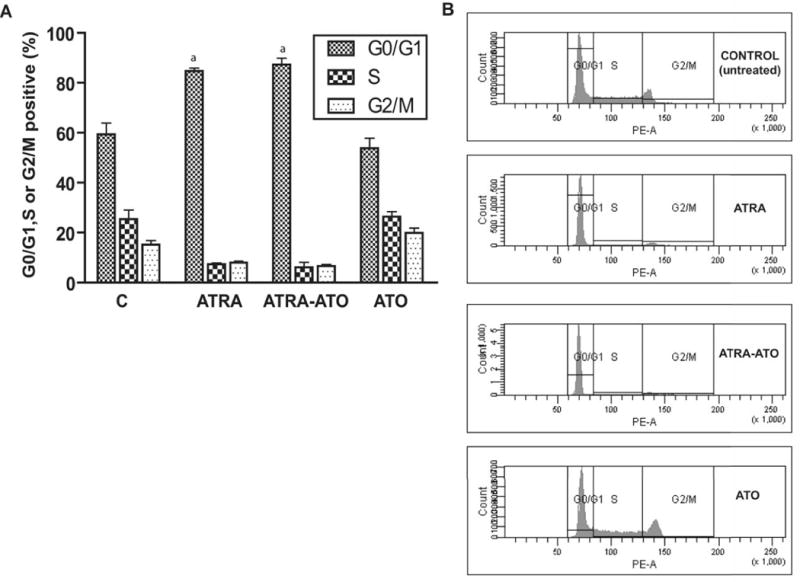
(A) Arsenic Trioxide did not accelerate ATRA induced G0 arrest. The percentage of G1/G0, S and G2/M cells at 72 h after the treatment is shown for control (untreated), ATRA, ATRA-ATO combination treated and ATO alone treated cells. Y-axis here shows the percentage of cells in the different cell-cycle phases at 72h. The X-axis shows the different treatment groups. Cells stained with hypotonic Propidium Iodide (PI) solution were incubated for 1h and analyzed by flow cytometry. Similar DNA histograms were prepared for earlier time points also and we did not see a significant difference in the percentage of cells arrested in G1/G0 arrest between the ATRA-ATO case and ATRA alone case. (B) Representative DNA histograms for G1/G0 arrest in the untreated (control) cells, cells treated with ATRA, cells treated with ATRA-ATO combination and cells treated ATO alone are shown at 72 h.
3.7 Arsenic Trioxide (ATO) did not accelerate ATRA induced cell-cycle arrest in HL-60 cells
ATRA treatment of HL-60 cells leads to myeloid differentiation as well as G1/G0 arrest. To determine if the ATO-induced growth retardation is attributable to G1/0 arrest, the effect of ATO on the percentage of G1/0 cells was measured by flow cytometry for untreated cells and cells treated with ATRA, ATRA-ATO combined, and ATO alone. Treatment with ATRA caused G1/0 arrest that was evidenced by an increase in the percentage of cells in G0/G1 phase, as seen before (18). Treatment with ATO alone however, did not show an increase in the percentage of cells arrested as compared to the control case at 72h (Fig. 7 (A)). Treatment with ATO plus ATRA resulted in an increase in the percentage of G1/0 cells which was indistinguishable from that caused by ATRA alone. The effect of ATO treatment on cell cycle arrest was also measured at earlier time points (24h and 48h, results not shown), and similar results were obtained. Thus, although ATO enhanced ATRA-induced enhanced differentiation, it does not enhance ATRA-induced growth inhibition through enhancing G1/0 arrest. Finally, we tested our hypothesis that the reduced growth rate in ATO cells could reflect a general, overall slowing of cellular biosynthesis needed to support proliferation, thereby slowing population growth without apoptosis or G1/0 specific regulated arrest. To test this possibility the levels of activated PKB/Akt was measured.
Figure 7.
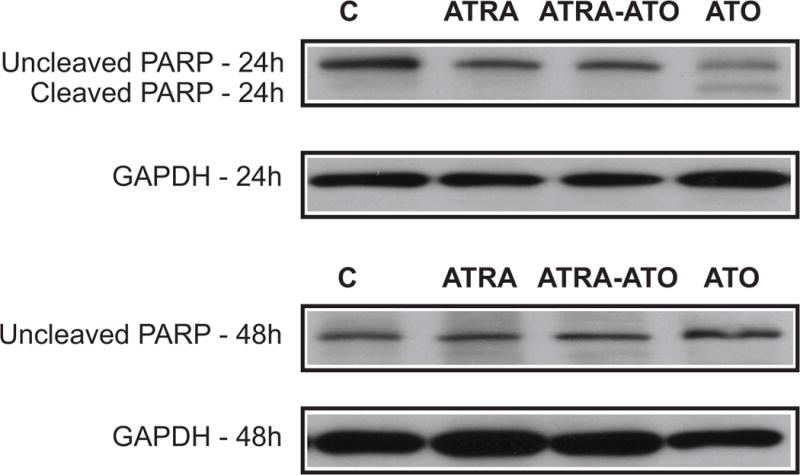
ATRA abrogates the apoptotic effect of ATO in HL-60 cells. Western analysis of cleaved and uncleaved PARP at 24 and 48 h is shown here. The different lanes are control (untreated) cells, and cells treated with ATRA, combination of ATRA-ATO, and ATO alone. Cleavage of PARP is an indicator of apoptosis. The figure shows that we saw some progress towards apoptosis in the ATO alone treatment. However, the combination treatment of ATRA-ATO or the ATRA alone treatment did not show cleavage of PARP. This result suggests that ATRA rescues the cells from apoptotic effect of ATO, and that there is no increase in apoptotic percentage of cells in the combination treatment. Loading control is shown by levels of GAPDH at 24h and 48h.
3.8 Arsenic Trioxide (ATO) in combination with ATRA leads to decreased activation of Akt
The role of Akt phosphorylation (Ser-473/Thr-308) in cellular proliferation has been well documented (21). The Akt/mTOR/ribosomal S6 kinase axis supports formation of the 80S translation initiation complex and thereby protein biosynthesis in general. In HL-60 cells, it has been shown that a constitutively active PI3K/Akt axis was responsible for resistance to ATO-induced apoptosis. ATRA induced differentiation in HL-60 cells was sensitive to pharmacological inhibition of Akt activation (22). To determine if ATO affected Akt, the levels of Akt phosphorylation (Ser-473/Thr-308) in untreated control, ATRA, ATO plus ATRA, and ATO treated cultures were measured by Western analysis. Previous analysis of sub-cellular distribution of p-Akt has showed that the changes in the level of phosphorylated Akt is mostly due to changes in the level of the nuclear enzyme (23–24). As seen from Fig. 9, the combination treatment of ATRA plus ATO causes a decrease in Akt phosphorylation at both 24h and 48h compared to treatment with ATRA alone. There was decreased phosphorylation of residues Thr-308 and Ser-473, which was more prominent at 48h than at 24h. Since, we used the nuclear fraction in measuring Akt phosphorylation, Histone 3 was used as a loading control. The results suggest that the greater growth inhibition of ATO plus ATRA treated cells compared to cells treated with only ATRA reflects a general biosynthetic slow down in ATRA plus ATO-treated cells. The effect appears to depend on both ATO and ATRA since ATO alone did not cause any apparent decrease in Akt phosphorylation.
Figure 9.
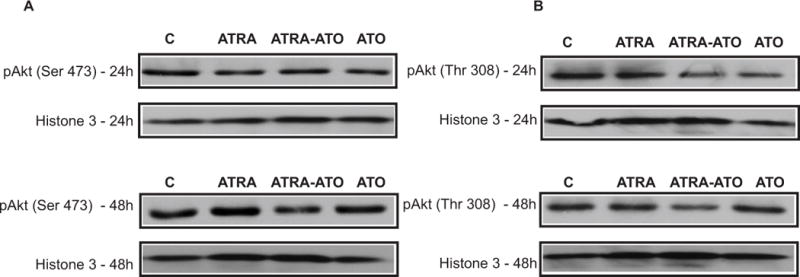
Arsenic Trioxide (ATO) in combination with ATRA leads to a decrease in the phosphorylation of Akt at the Ser-473 and Thr-308 residues. Western analysis of activation of Akt is shown here. Panel A shows the activation at the Ser-473 residue whereas Panel B shows the activation at the Thr-308 residues at 24h and 48h. Protein lysates were extracted from control cells, and cells treated with ATRA, ATRA-ATO combination, and ATO alone. The decrease in phosphorylation in the ATRA-ATO case, compared with the ATRA alone case, is more evident at 48h than at 24h. Since phosphorylation of Akt at these two residues is correlated with growth and proliferation, this result suggests that ATO combined with ATRA affects proliferation negatively. Since the nuclear fraction was used here, histone-3 is the loading control.
4. Discussion
There are a number of potential mechanisms by which ATO may cause or facilitate leukemic cell differentiation. A number of regulatory molecules, pathways and their downstream cellular outcomes have been studied. One of the potentially most significant is the capability of ATO, like ATRA, to cause degradation of PML-RARα, the fusion protein resulting from the t(15,17) translocation that is putatively the seminal cytogenetic cause of APL (25). In the NB4 APL cell line, ATO caused degradation of PML/RARα fusion protein, but not the mRNA, in both ATRA sensitive and resistant variant lines (25). ATO also affects the spatial localization and organization of PML and PML/RARα, altering PML bodies within the nucleus of NB4 cells (13, 25). Clinical correlates have also been reported (4). Degradation of PML-RARα is thus a prominent potential mechanism of action of ATO.
ATO has also been found to affect signaling pathways that have been implicated in ATRA-induced differentiation, as well as growth regulation and apoptosis. ATO can evoke a cellular oxidative stress response (26–27). The glutathione (GSH) redox system is a critical modulator of cellular response in oxidative stress. As such it can regulate apoptosis, a possible sequela of ATO treatment. A function of the GSH redox system is to scavenge free radicals. The GSH redox system is targeted by ATO in various APL cell lines. NB4 cells have one of the lowest levels of basal GSH in leukemic cell lines and were found to be most sensitive to arsenic treatment (20). In NB4 cells, ATO treatment modulated the GSH levels, which sensitized the cells towards apoptosis (20, 28). There were dose dependent growth inhibitory, differentiation enhancing or apoptosis enhancing effects of ATO (13, 29). Previous studies have shown that ATO causes activation of stress-activated protein kinase, c-jun N-terminal kinase (JNK) (27). This activation is directly related to apoptosis as inhibition of p38/JNK was seen to reduce ATO induced apoptosis in NB4 cells. In the case of MAPK signaling, the effect of ATO on p38 has been widely studied, too. Verma et al., showed that p38 is phosphorylated and activated by ATO in a sustained manner and that this activation is downstream of redox events activated by ATO (30) in NB4 cells. Activation of other MAPK proteins, such as Mkk 3 and Mkk 6, was also seen in NB4 cells and in CML-derived KT-1 cells, and found to be essential for p38MAPK activation. Signaling molecules and pathways that are in common downstream of ATO and ATRA may provide a basis for their potential synergistic interaction. Clinically, a synergistic effect has been reported for ATRA-ATO combination treatment, when compared with either ATRA or ATO monotherapies (10). Other studies of the biochemical effects of ATRA-ATO combination treatment have revealed additive effects in JNK/p38, TNF and cAMP/PKA pathways in APL cell lines (31–32). For example, in the cAMP/PKA study, it was found that cAMP, which can induce monocytic differentiation in HL-60 cells, was synergistically induced by ATRA and low doses of ATO (31, 33).
The results of the present study showed that for the PML-RARα negative HL-60 cells, ATO did not induce differentiation, although it did slow cell proliferation. It did enhance the differentiation induction capability of ATRA. Although ATO did not augment ATRA-induced CD38 expression, a marker of the earliest stages of the ATRA-induced progression along the myeloid lineage, it did enhance ATRA-induced expression of CD11b, an integrin receptor that marks further progression of differentiation, as well as p47PHOX, which is a component of the NAPDH oxidase complex used for the inducible oxidative metabolism characteristic of mature myeloid cells. ATO also enhanced the ATRA-induced functional differentiation marker, inducible oxidative metabolism. While ATO by itself thus could not cause differentiation, it enhanced ATRA-induced differentiation, indicating that it affected pathways of importance to propelling differentiation. One such pathway is activation of the RAF/MEK/ERK axis in MAPK signaling (11–12). Activation of this axis was enhanced in cells treated with the combination of ATRA plus ATO compared to ATRA alone. Although phosphorylation of RAF, MEK, and ERK betraying activation was enhanced, expression levels were not detectably affected. ATO enhanced activation of the RAF/MEK/ERK axis was more apparent as differentiation progressed, namely more apparent at 48h than at 24h. The activation and expression patterns of MAPK proteins suggest that the synergy between ATRA and ATO is at the protein-protein interaction level, rather than at the transcriptional level. This agrees with a recent systems level study which indicated that while the effects of ATRA involved transcriptional remodeling, ATO effects were seen at a proteome level, although the study was done for NB4 cells (32).
The present results also showed that ATO retarded population growth, and that populations treated with ATO plus ATRA were more growth inhibited than those treated with ATRA alone. The viability of cultures detected by trypan blue exclusion routinely exceeded 95%, and there was no evidence of PARP cleavage, all indicating a lack of apoptosis. There was also no extra accumulation of cells in G0 in ATO plus ATRA treated cells compared to ATRA treated cells. There was an ATO attributable reduction in Akt phosphorylation, suggestive of an over all slowing of biosynthetic activity causing retardation of cell growth. This effect is ATRA dependent since ATO by itself did not reduce Akt phosphorylation, although it did retard cell growth.
In sum, in PML-RARα negative HL-60 human myeloblastic leukemia cells, ATO causes MAPK signaling and augments ATRA-induced differentiation. The effects are more prominent as the MAPK signal augmentation increases, enhancing expression of later differentiation markers, but not the earliest, detected. A further potential contributor to the enhanced differentiation by ATO is the retardation of growth, possibly slowing down the self-renewal capacity of these progenitor cells and favoring differentiation. The results show that ATO may be of therapeutic use to augment the effects of ATRA.
Acknowledgments
Supported in part by grants from the NIH(USPHS), NYSTEM and NSF CAREER Award #CBET-0846876
References
- 1.Waxman S, Anderson KC. History of the development of arsenic derivatives in cancer therapy. The oncologist. 2001;6(90002):3–10. doi: 10.1634/theoncologist.6-suppl_2-3. [DOI] [PubMed] [Google Scholar]
- 2.Evens AM, Tallman MS, Gartenhaus RB. The potential of arsenic trioxide in the treatment of malignant disease: past, present, and future. Leukemia research. 2004;28(9):891–900. doi: 10.1016/j.leukres.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 3.Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood. 2008;111(5):2505. doi: 10.1182/blood-2007-07-102798. [DOI] [PubMed] [Google Scholar]
- 4.Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood. 1997;89(9):3354. [PubMed] [Google Scholar]
- 5.Niu C, Yan H, Yu T, Sun HP, Liu JX, Li XS, et al. Studies on treatment of acute promyelocytic leukemia with arsenic trioxide: remission induction, follow-up, and molecular monitoring in 11 newly diagnosed and 47 relapsed acute promyelocytic leukemia patients. Blood. 1999;94(10):3315. [PubMed] [Google Scholar]
- 6.Mathews V, George B, Lakshmi KM, Viswabandya A, Bajel A, Balasubramanian P, et al. Single-agent arsenic trioxide in the treatment of newly diagnosed acute promyelocytic leukemia: durable remissions with minimal toxicity. Blood. 2006;107(7):2627–2632. doi: 10.1182/blood-2005-08-3532. [DOI] [PubMed] [Google Scholar]
- 7.Breitman T, Collins S, Keene B. Terminal differentiation of human promyelocytic leukemic cells in primary culture in response to retinoic acid. Blood. 1981;57(6):1000. [PubMed] [Google Scholar]
- 8.Huang M, Ye Y, Chen S, Chai J, Lu J, Zhoa L, et al. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood. 1988;72(2):567. [PubMed] [Google Scholar]
- 9.Asou N, Kishimoto Y, Kiyoi H, Okada M, Kawai Y, Tsuzuki M, et al. A randomized study with or without intensified maintenance chemotherapy in patients with acute promyelocytic leukemia who have become negative for PML-RARalpha transcript after consolidation therapy: The Japan Adult Leukemia Study Group (JALSG) APL97 study. Blood. 2007;110(1):59–66. doi: 10.1182/blood-2006-08-043992. [DOI] [PubMed] [Google Scholar]
- 10.Shen ZX, Shi ZZ, Fang J, Gu BW, Li JM, Zhu YM, et al. All-trans retinoic acid/As2O3 combination yields a high quality remission and survival in newly diagnosed acute promyelocytic leukemia. Proceedings of the National Academy of Sciences. 2004;101(15):5328–5335. doi: 10.1073/pnas.0400053101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J, Yen A. A MAPK-positive Feedback Mechanism for BLR1 Signaling Propels Retinoic Acid-triggered Differentiation and Cell Cycle Arrest. Journal of Biological Chemistry. 2008;283(7):4375. doi: 10.1074/jbc.M708471200. [DOI] [PubMed] [Google Scholar]
- 12.Yen A, Roberson MS, Varvayanis S, Lee AT. Retinoic acid induced mitogen-activated protein (MAP)/extracellular signal-regulated kinase (ERK) kinase-dependent MAP kinase activation needed to elicit HL-60 cell differentiation and growth arrest. Cancer research. 1998;58(14):3163–3172. [PubMed] [Google Scholar]
- 13.Chen G, Zhu J, Shi X, Ni J, Zhong H, Si G, et al. In vitro studies on cellular and molecular mechanisms of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia: As2O3 induces NB4 cell apoptosis with downregulation of Bcl-2 expression and modulation of PML-RAR alpha/PML proteins. Blood. 1996;88(3):1052. [PubMed] [Google Scholar]
- 14.Brooks S, 3rd, Kazmer S, Levin A, Yen A. Myeloid differentiation and retinoblastoma phosphorylation changes in HL-60 cells induced by retinoic acid receptor-and retinoid X receptor-selective retinoic acid analogs. Blood. 1996;87(1):227. [PubMed] [Google Scholar]
- 15.Kishimoto H, Hoshino S, Ohori M, Kontani K, Nishina H, Suzawa M, et al. Molecular mechanism of human CD38 gene expression by retinoic acid Identification of retinoic acid response element in the first intron. Journal of Biological Chemistry. 1998;273(25):15429–15434. doi: 10.1074/jbc.273.25.15429. [DOI] [PubMed] [Google Scholar]
- 16.Shen M, Yen A. c-Cbl Interacts with CD38 and Promotes Retinoic Acid-Induced Differentiation and G0 Arrest of Human Myeloblastic Leukemia Cells. Cancer Research. 2008;68(21):8761. doi: 10.1158/0008-5472.CAN-08-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reiterer G, Bunaciu RP, Smith JL, Yen A. Inhibiting the platelet derived growth factor receptor increases signs of retinoic acid syndrome in myeloid differentiated HL-60 cells. FEBS Letters. 2008;582(17):2508–2514. doi: 10.1016/j.febslet.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen M, Yen A. Nicotinamide Cooperates with Retinoic Acid and 1, 25-Dihydroxyvitamin D 3 to Regulate Cell Differentiation and Cell Cycle Arrest of Human Myeloblastic Leukemia Cells. Oncology. 2009;76:91–100. doi: 10.1159/000188664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang XJ, Wiernik PH, Klein RS, Gallagher RE. Arsenic trioxide induces apoptosis of myeloid leukemia cells by activation of caspases. Cancer Immunology, Immunotherapy. 1999;16(1):58–64. doi: 10.1007/BF02787360. [DOI] [PubMed] [Google Scholar]
- 20.Dai J, Weinberg RS, Waxman S, Jing Y. Malignant cells can be sensitized to undergo growth inhibition and apoptosis by arsenic trioxide through modulation of the glutathione redox system. Blood. 1999;93(1):268. [PubMed] [Google Scholar]
- 21.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase-AKT pathway in human cancer. Nature Reviews Cancer. 2002;2(7):489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 22.Tabellini G, Cappellini A, Tazzari PL, Fala F, Billi AM, Manzoli L, et al. Phosphoinositide 3-kinase/Akt involvement in arsenic trioxide resistance of human leukemia cells Giovanna Tabellini and Alessandra Cappellini equally contributed to this work. Journal of cellular physiology. 2005;202(2) doi: 10.1002/jcp.20153. [DOI] [PubMed] [Google Scholar]
- 23.Matkovic K, Brugnoli F, Bertagnolo V, Banfic H, Visnjic D. The role of the nuclear Akt activation and Akt inhibitors in all-trans-retinoic acid-differentiated HL-60 cells. Leukemia. 2006;20(6):941–951. doi: 10.1038/sj.leu.2404204. [DOI] [PubMed] [Google Scholar]
- 24.Cappellini A, Tabellini G, Zweyer M, Bortul R, Tazzari PL, Billi AM, et al. The phosphoinositide 3-kinase//Akt pathway regulates cell cycle progression of HL60 human leukemia cells through cytoplasmic relocalization of the cyclin-dependent kinase inhibitor p27Kip1 and control of cyclin D1 expression. Leukemia. 2003;17(11):2157–2167. doi: 10.1038/sj.leu.2403111. [DOI] [PubMed] [Google Scholar]
- 25.Shao W. Arsenic trioxide as an inducer of apoptosis and loss of PML/RAR alpha protein in acute promyelocytic leukemia cells. J Natl Cancer I. 1998;90(2):124–133. doi: 10.1093/jnci/90.2.124. [DOI] [PubMed] [Google Scholar]
- 26.Jing Y, Dai J, Chalmers-Redman RME, Tatton WG, Waxman S. Arsenic Trioxide Selectively Induces Acute Promyelocytic Leukemia Cell Apoptosis Via a Hydrogen Peroxide-Dependent Pathway. Blood. 1999 Sep 15;94(6):2102–2111. 1999. [PubMed] [Google Scholar]
- 27.Davison K, Mann KK, Waxman S, Miller WH. JNK activation is a mediator of arsenic trioxide-induced apoptosis in acute promyelocytic leukemia cells. Blood. 2004;103(9):3496–3502. doi: 10.1182/blood-2003-05-1412. [DOI] [PubMed] [Google Scholar]
- 28.Ramos AM, Fernandez C, Amran D, Sancho P, de Blas E, Aller P. Pharmacologic inhibitors of PI3K/Akt potentiate the apoptotic action of the antileukemic drug arsenic trioxide via glutathione depletion and increased peroxide accumulation in myeloid leukemia cells. Blood. 2005;105(10):4013–4020. doi: 10.1182/blood-2004-07-2802. [DOI] [PubMed] [Google Scholar]
- 29.Chen GQ, Shi XG, Tang W, Xiong SM, Zhu J, Cai X, et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): I. As2O3 exerts dose-dependent dual effects on APL cells. Blood. 1997;89(9):3345. [PubMed] [Google Scholar]
- 30.Verma A, Mohindru M, Deb DK, Sassano A, Kambhampati S, Ravandi F, et al. Activation of Rac1 and the p38 mitogen-activated protein kinase pathway in response to arsenic trioxide. Journal of Biological Chemistry. 2002;277(47):44988–44995. doi: 10.1074/jbc.M207176200. [DOI] [PubMed] [Google Scholar]
- 31.Zhao Q, Tao J, Zhu Q, Jia P, Dou A, Li X, et al. Rapid induction of cAMP/PKA pathway during retinoic acid-induced acute promyelocytic leukemia cell differentiation. Leukemia. 2003;18(2):285–292. doi: 10.1038/sj.leu.2403226. [DOI] [PubMed] [Google Scholar]
- 32.Zheng PZ, Wang KK, Zhang QY, Huang QH, Du YZ, Zhang QH, et al. Systems analysis of transcriptome and proteome in retinoic acid/arsenic trioxide-induced cell differentiation/apoptosis of promyelocytic leukemia. Proceedings of the National Academy of Sciences. 2005;102(21):7653–7658. doi: 10.1073/pnas.0502825102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu Q, Zhang JW, Zhu HQ, Shen YL, Flexor M, Jia PM, et al. Synergic effects of arsenic trioxide and cAMP during acute promyelocytic leukemia cell maturation subtends a novel signaling cross-talk. Blood. 2002;99(3):1014. [PubMed] [Google Scholar]