

Abstract
Objectives
Isavuconazole, a novel triazole antifungal agent, has broad-spectrum activity against Aspergillus spp. and other pathogenic fungi. The isavuconazole exposure–response relationship in experimental invasive pulmonary aspergillosis using galactomannan index (GMI) suppression as a marker of disease clearance was explored.
Methods
The impact of exposure on GMI suppression in persistently neutropenic rabbits treated with isavuconazonium sulphate (isavuconazole-equivalent dosages of 20, 40 or 60 mg/kg every 24 h, after a 90 mg/kg loading dose) for 12 days was linked using mathematical modelling. Bridging to humans using population pharmacokinetic (PK) data from a clinical trial in invasive aspergillosis was performed using Monte Carlo simulations.
Results
Mean plasma isavuconazole AUC/MIC (EC50) of 79.65 (95% CI 32.2, 127.1) produced a half-maximal effect in GMI suppression. The inhibitory sigmoid Emax curve dropped sharply after an AUC/MIC of ≥30 and was near maximum (EC80) at ∼130. Bridging the experimental PK/pharmacodynamic (PD) target to human population PK data was then used to return to the rabbit model to determine a clinically relevant PD endpoint. The clinical dosing regimen used in the trial would result in a mean GMI of 4.3 ± 1.8, which is a 50% reduction from the starting GMI in the experiment.
Conclusions
The clinical trial results showing the non-inferiority of isavuconazole to voriconazole for all-cause mortality further support the PK-PD endpoint, thereby demonstrating the usefulness of the rabbit model and endpoint for isavuconazole and implications on interpretive breakpoints. Importantly, the analysis supports this model as an important tool for development of antifungal agents.
Introduction
Isavuconazole is a novel broad-spectrum triazole antifungal agent, which is available as the prodrug, isavuconazonium sulphate, in both intravenous and oral formulations, and which was recently approved by the US FDA for the primary treatment of invasive aspergillosis (IA) and mucormycosis in adults.1 IA is a life-threatening disease that causes significant morbidity and mortality in patients who are severely immunocompromised.2,3 Isavuconazole is the first agent that has been developed for IA for 12 years. Despite the recent demonstration of non-inferiority of isavuconazole compared with voriconazole, there is relatively little information regarding exposure–response relationships of isavuconazole against Aspergillus spp., which are required for a complete understanding of the clinical utility of this agent.
Laboratory animal models that mimic human disease have long been used to characterize pharmacological efficacy and safety prior to clinical use. Importantly, well-designed animal models used in antimicrobial development are generally highly predictive of clinical efficacy.4 Over the past 20–30 years these experimental models have enabled an exploration of the relationship between the time-course of drug exposure, penetration of drug to the site of infection and the ultimate antimicrobial effect.5 The treatment of infections caused by Aspergillus spp. and rare filamentous fungi is relatively difficult to study in clinical settings. A detailed understanding of exposure–response relationships underpins the design of antifungal regimens that are safe and effective and enables the right dose to be studied the first time. An understanding of antifungal pharmacokinetic (PK)/pharmacodynamic (PD) properties also provides the foundation for treatment and prevention of antimicrobial resistance, determination of the requirement for therapeutic drug monitoring and decision support for establishing in vitro susceptibility breakpoints.
Here, we describe the use of a well-established experimental rabbit model of invasive pulmonary aspergillosis (IPA) using serum galactomannan (GM) concentrations as the primary model readout to characterize the PK/PD relationships of isavuconazole against Aspergillus fumigatus. We were in a unique position of being able to validate the readout of the experimental PK/PD model with the clinical findings from the recent Phase 3 clinical trial. We defined the decrement in GM in the rabbit that is associated with clinically relevant exposure of isavuconazole and for which clinical efficacy has been established. We then reflected on the PD basis for establishing an in vitro susceptibility interpretive breakpoint of isavuconazole against A. fumigatus.
Materials and methods
Rabbit model of IPA
A previously described persistently neutropenic rabbit model of IPA was used.6,7 Healthy female New Zealand White rabbits (Covance Research Products, Inc., Denver, PA, USA) weighing 2.6–3.5 kg were used in the study. Immunosuppression and profound persistent neutropenia (neutrophil concentration of <100 neutrophils/μL) were established by administration of cytarabine (Ara-C) (Cytarabine injection; Zydus Hospira Oncology Private Ltd, Gujarat, India for Hospira, Inc., Lake Forest, IL, USA) at an initial dose of 525 mg/m2 for 5 consecutive days beginning on day 1 of the experiment and a maintenance dose of 484 mg/m2 on days 8, 9, 12 and 13 of the experiment. Methylprednisolone (Solu-Medrol®, Pfizer for Pharmacia & Upjohn Co., Division of Pfizer Inc., New York, NY, USA) was dosed at 5 mg/kg on days 1 and 2.7 Bacterial infection prophylaxis was maintained with antibiotics (ceftazidime, gentamicin, vancomycin) as previously described.7
Ethics
Rabbits were monitored under humane care and use of standards in facilities accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International, according to the guidelines of the National Research Council for the care and use of laboratory animals, and under the approval of the Animal Care and Use Committee of Weill Cornell Medical Center, New York, NY, USA. Rabbits were housed individually and fed ad libitum.
Organism and MIC
NIH 4215 (ATCC number MYA-1163), which is a well-characterized clinical isolate of A. fumigatus, was used for all experiments.7 The MICs of voriconazole and isavuconazole for A. fumigatus (determined using CLSI standard M38-A2 broth microdilution methodology) were 0.5 and 1 mg/L, respectively.8 The MICs were measured in triplicate.
Treatment
Rabbits received either prodrug, isavuconazonium sulphate, at an isavuconazole (BAL4815; active moiety)-equivalent dose of 20 (ISAV20), 40 (ISAV40) and 60 (ISAV60) mg/kg/day or were untreated controls. A loading dose of the prodrug equivalent to isavuconazole 90 mg/kg was given on the first day of antifungal treatment to all isavuconazole-treated rabbits. Treatment was initiated 24 h after inoculation and administered orally once daily thereafter for up to 12 days. Multiple outcome variables were analysed from the model, including survival, pulmonary infarct score, lung weight, residual fungal burden (log cfu/g), serum GM index (GMI), bronchoalveolar lavage (BAL) fluid GMI and serum (1 → 3)-β-d-glucan levels, as previously described.6
Herein, we report the results of the isavuconazole-treated animals with a particular focus on the serum GM experiments and PK/PD mathematical modelling, with a brief description of the primary efficacy results. The primary efficacy results from the animal model are published elsewhere.6
PD assessment
Blood was collected via the indwelling Silastic catheter at 24 h post-inoculation, just prior to initiation of antifungal treatment and every other day thereafter for determination of the serum GM concentrations. Concentrations were determined by a one-stage immunoenzymatic sandwich microplate assay method [Platelia® Aspergillus Enzyme Immunoassay (EIA), Bio-Rad, Marnes la Coquette, France]9 and according to the manufacturer's instructions.
PK
Single-dose PK analyses were conducted in four non-infected rabbits in each dosage group. Timepoints included pre-dose and 1, 2, 4, 8, 12, 18, 24 and 48 h post-dosing. Multiple-dose PKs were determined in infected animals 7 days post-inoculation. Blood samples were drawn pre-dose and at 1, 4, 8 and 24 h post-dosing. Each blood sample for PK assay was collected in a heparinized syringe. Plasma samples were immediately separated by centrifugation at 400 g and stored at −80°C until assayed.
Measurement
Isavuconazole was measured using LC/MS/MS. Samples were prepared by adding 0.1 M paraoxon (10 μL per 1 mL of plasma) to the blank EDTA/K3 and blank Li-heparin rabbit plasma in order to inhibit esterases. Spiking solutions were prepared by serial dilution in DMSO and acetonitrile (ACN), 0.05% trifluoracetate (TFA) out of a 2 mg/mL stock solution with a range from 0.5 to 500 μg/mL. Calibration samples were performed by spiking 1 μL of each DMSO (or ACN, 0.05% TFA) solution in 99 μL of blank EDTA/K3 plasma. Each sample was then quenched with 300 μL of ACN, 0.05% TFA containing 1 μg/mL BAL0004815-d4, and pyridooxazinone as internal standards.
After centrifugation, 10 μL of the supernatant was injected into the LC/MS/MS device. A similar preparation method was performed for the quality control samples; however, blank Li-heparin plasma was used instead of the EDTA/K3 plasma. For the samples, 50 μL of plasma was mixed with 150 μL of ACN, 0.05% TFA containing 1 μg/mL BAL0004815-d4 and pyridooxazinone as internal standards, then treated like the calibrators. Assay linear range was 0.005–4.45 μg/mL and coefficient of variation ranged from 1.1% to 10.7%. The lower limit of quantification of the plasma assay was 0.005 μg/mL.
PK and PD mathematical modelling
Mathematical modelling was performed in a step-wise manner. First, a population PK (PPK) model was constructed using non-parametric estimation in Pmetrics™ software (v1.3.2, University of Southern CA, Los Angeles, CA, USA)10 and fitted to the rabbit plasma concentration data.10 The weighting functions were estimated using a combination of ADAPT 5 (https://bmsr.usc.edu/software/adapt/; Biomedical Simulations Resource, Los Angeles, CA, USA) and Pmetrics error estimation runs. Specifically, in ADAPT 5, slope and intercept values were estimated using maximum likelihood estimation for each individual rabbit. In Pmetrics, the error estimation run script (i.e. ERRrun) was used to estimate the assay error polynomial coefficients directly from the data. After establishing a model that best described the PK data, the GM concentrations were added to the dataset for each individual rabbit. A linked PK/PD model was built using the following set of differential equations:
| (1) |
| (2) |
| (3) |
| (4) |
The first three equations describe the PK of the drug (compartment 1, theoretical absorptive compartment for oral administration; 2, central compartment; and 3, peripheral compartment). CL is the clearance defined as the amount of drug being cleared from the central compartment over time and V is the volume of the central compartment. Ka is the first-order absorption constant, Kcp is the rate of drug moving from the central to the peripheral compartment and Kpc is the rate of drug moving from the peripheral to the central compartment. Equation 4 describes the rate of change of the serum GM values. Kpmax represents the maximum rate of production, Hp is the slope function for production, C50p is the amount of drug where there is half-maximal production, popmax is the theoretical maximum density of GM, Ksmax is the maximum rate of GM suppression, C50s is the amount of drug where there is half-maximal suppression and Hs is the slope function for the GM suppression.
Acceptance of the final model was evaluated by visual inspection of the observed versus predicted values plotted over time after the Bayesian step and the coefficient of determination (r2) from the linear regression of the observed versus predicted values, as well as evaluation of the estimated bias and precision. After fitting the model to the PK/PD data, the Bayesian posterior estimates for each rabbit were used to estimate the concentration–time profiles for isavuconazole and GMI for each rabbit. Simulations were performed in ADAPT 5. AUCs were calculated by integration from the simulated concentration–time profiles on the last day of dosing (at steady state) in ADAPT 5.
Using the simulated isavuconazole AUC values and the GMI at the end of the dosing period for each rabbit, an inhibitory sigmoid Emax model was constructed to establish the PD relationship between exposure (AUC) divided by MIC and response (GMI). The model used is described below:
| (5) |
E0 is the baseline level of GM prior to exposure to drug, Emax is the maximum GM value, EC50 is a measure of drug potency and H is the slope factor.
Bridging to humans
To bridge the exposure–response results from the rabbits to humans, a PPK model was constructed using the isavuconazole plasma concentration data from the Phase 3 SECURE clinical trial.11 The SECURE (NCT00412893) trial evaluated the efficacy and safety of isavuconazole compared with voriconazole in the treatment of patients with possible, probable or proven IA and other filamentous fungi.11 Patients that were randomized to isavuconazole received the prodrug, isavuconazonium sulphate, at a dose of 372 mg every 8 h for the first 2 days followed by 372 mg once daily thereafter [equivalent to isavuconazole at a dose of 200 mg (intravenous) every 8 h for 2 days, then 200 mg once daily]. The option to receive either intravenous or oral administration was allowed after day 2. The maximum treatment duration was 84 days. Plasma trough samples were collected on days 7, 14 and 42, the end of treatment and at follow-up for some patients. Serial plasma samples (seven in total) from <25 subjects were collected on day 7 (±1 day) or 14 (+1 day) for PK profiling. Overall, 113 patients with isavuconazole concentrations were used for the analysis.
A PPK model using non-parametric estimation was developed in Pmetrics software (v1.3.2, University of Southern California, CA, USA).10 The model-fitting process included evaluating both two- and three-compartment models with and without lag-time and oral bioavailability terms.
Mean Bayesian parameter estimates from the fitted human model were used to perform Monte Carlo simulations of 1000 patients using Pmetrics. The simulations were used to calculate steady-state AUCs within Pmetrics by the trapezoidal rule for each simulated patient. Using Equation (5) above, the effect of AUC/MIC at various MIC values representative of the MIC distribution of isavuconazole for A. fumigatus as described by Espinel-Ingroff et al.12 was calculated.
Total isavuconazole drug levels were used wherever isavuconazole concentration or AUCs are reported unless otherwise specified.
Results
Animal model
Significant reduction of residual fungal burden, lung weights and pulmonary infarct score was demonstrated in the two highest dosage groups (ISAV40 and ISAV60) compared with untreated controls (P < 0.001, ANOVA with Bonferroni's correction for multiple comparisons). The highest dose of isavuconazole significantly prolonged survival compared with untreated controls (P < 0.001, log-rank test). The lowest dosage (ISAV20) also significantly prolonged survival versus untreated controls (P < 0.05, log-rank test).6
Dose-dependent reductions in biomarkers were demonstrated for isavuconazole-treated animals. ISAV40 and ISAV60 dosage groups significantly decreased the GMI during therapy following day 4 compared with increased GMI in the untreated controls (P < 0.01). Figure 1 illustrates GMI versus time for the rabbits treated with isavuconazonium. BAL GMI was also significantly reduced in ISAV40 and ISAV60 dosage groups versus untreated controls (P < 0.001, ANOVA with Bonferroni's correction for multiple comparisons). Serum (1 → 3)-β-d-glucan levels were significantly reduced in ISAV40 and ISAV60 compared with untreated controls (P < 0.05, ANOVA with Bonferroni's correction for multiple comparisons).6
Figure 1.
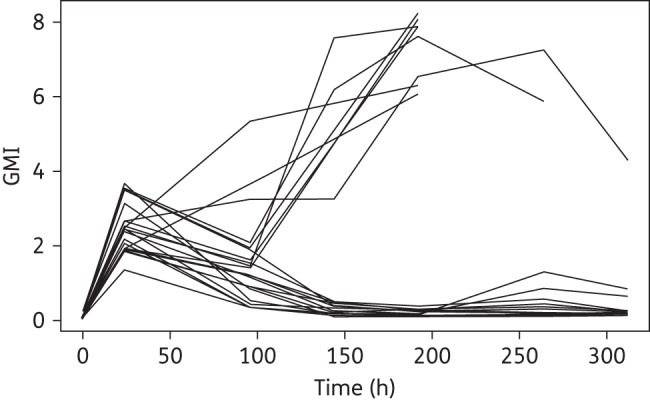
GMI versus time for each rabbit after administration of isavuconazonium sulphate (all dose groups).
PK/PD modelling
Visual inspection of the raw concentration–time profile of the three isavuconazole dosages suggested linear isavuconazole PK in the rabbit. A two-compartment model with first-order absorption fitted the rabbit plasma isavuconazole drug concentration data well. The fit of the linked PK/PD model was also acceptable based on a coefficient of determination for the observed versus predicted values after the Bayesian step of 0.965 and 0.895 for the PK concentrations and GMI, respectively (Figure 2). The estimates of bias and precision for the rabbit PK were −0.14 and 1.05, and −0.288 and 0.892 for GMI, respectively. Parameter estimates from the final model are summarized in Table 1.
Figure 2.

Regression plot of observed (y-axis) versus model predicted (x-axis) plots from rabbit isavuconazole PPK (a) and GMI (b).
Table 1.
Mean and SD values for each parameter estimated from the rabbit PPK/PD-linked model
| Parameter (units) | Mean | SD |
|---|---|---|
| Ka (h−1) | 20.145 | 8.842 |
| CL/F (L/h) | 1.108 | 1.228 |
| V/F (L) | 27.911 | 6.872 |
| Kcp (h−1) | 5.953 | 2.498 |
| Kpc (h−1) | 6.394 | 2.916 |
| Kpmax (GMI/h) | 0.115 | 0.047 |
| Hp | 17.220 | 4.116 |
| C50p (mg/L) | 1.286 | 1.548 |
| IC (GMI) | 0.108 | 0.053 |
| Ksmax (GMI/h) | 0.009 | 0.002 |
| Hs | 12.292 | 5.029 |
| C50s (mg/L) | 0.167 | 0.127 |
| popmax (GMI) | 6.642 | 2.324 |
Ka, first-order rate constant describing the movement of drug from the gut to the central compartment; F, bioavailability; IC, initial condition of GMI prior to drug exposure; Kcp, rate of drug moving from the central to the peripheral compartment; Kpc, rate of drug moving from the peripheral to the central compartment; Kpmax, maximum rate of GM production; Hp, slope function for GM production; C50p, amount of drug where there is half-maximal production; popmax, theoretical maximum density of GM, Ksmax, maximum rate of GM suppression; C50s, amount of drug where there is half-maximal suppression; Hs, slope function for the GM suppression.
The exposure–response relationship showed that the GMI responded sharply for AUC/MIC of ≥30 (Figure 3). The AUC/MIC that induced the half-maximal effect (EC50) was estimated at 79.65 (95% CI 32.2, 127.1), which resulted in a 50% reduction in GMI. Suppression of GMI was a near maximum (EC80) at an AUC/MIC of ∼130.
Figure 3.
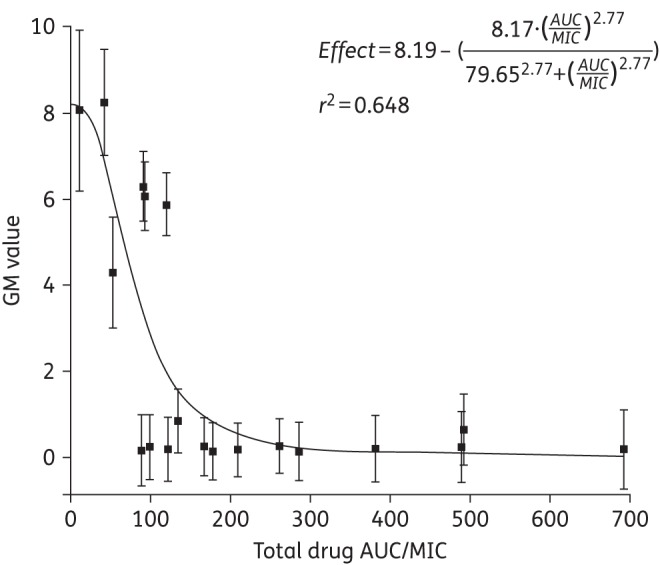
Inhibitory sigmoid Emax curve demonstrating the exposure–response relationship of isavuconazole exposure in terms of plasma AUC/MIC (x-axis) and the associated terminal GM value (y-axis). The black squares represent the observed GMI value for each rabbit (± SEM). The continuous black line represents the fit of the inhibitory sigmoid Emax model to the data.
Bridging to humans
Human isavuconazole plasma drug concentrations from the Phase 3 SECURE clinical trial in patients with IA or other filamentous fungi were used to construct a non-parametric PPK model using Pmetrics software.10 A two-compartment model with first-order absorption fitted the data well, with a coefficient of determination after the Bayesian step of 0.877. Visual inspection of the observed versus predicted values was acceptable, as were measures of bias and precision. The mean parameter estimates from the PPK model for the Phase 3 human plasma concentrations are presented in Table 2.
Table 2.
Mean and SD values for each parameter estimated from the human PPK model
| Parameter (units) | Mean | SD |
|---|---|---|
| Ka (h−1) | 24.3 | 16.7 |
| CL (L/h) | 2.2 | 1.1 |
| V (L) | 398.8 | 225.2 |
| F (%) | 90.2 | 20.9 |
| Tlag (h) | 1.5 | 3.3 |
Ka, first-order rate constant that describes the movement of drug from the gut to the central compartment; F, bioavailability; Tlag, lag time.
Monte Carlo simulations using the mean parameter estimates from the human PPK model were performed. The AUCs for 1000 simulated patients were estimated using the linear trapezoidal rule in the makeAUC function within Pmetrics. The mean steady-state AUC ± SD for the simulated population was 82.6 ± 33.0 mg h/L. The corresponding drop in GMI in rabbits receiving human-like exposures of isavuconazole was 4.3 ± 1.8, which closely approximates the EC50 for GMI reduction (4.1 GMI units) from the start of the experiment in the rabbits (E0 = 8.2).
After Monte Carlo simulations of 1000 patients, the effect of AUC/MIC at different MIC values relevant to the WT MIC distribution of A. fumigatus for isavuconazole as tested by CLSI methodology is displayed in Figure 4. The figure illustrates the predicted GMI at the end of the experiment given the inherent PK variability in humans as a function of each MIC value following the administration of the regimen of isavuconazonium sulphate that was used in the recent clinical trial.
Figure 4.

Frequency distribution of terminal GM values at each MIC value. Monte Carlo simulation of 1000 subjects after exposure to the clinical dosing regimen of isavuconazonium sulphate.
Discussion
Isavuconazole is a novel triazole antifungal agent that is administered as the water-soluble prodrug isavuconazonium sulphate. This novel agent demonstrated non-inferior efficacy (as defined by upper bound of the 95% CI less than the pre-specified 10% non-inferiority margin) compared with voriconazole for all-cause mortality up to day 42 (18.6% versus 20.2%, intent-to-treat population) in the SECURE trial for the treatment of IA and invasive disease caused by other filamentous fungi.11
The clinical trial results enable a retrospective validation of the clinical validity of the endpoints used in the experimental PK/PD model. An isavuconazole AUC that has been associated with clinical efficacy that is at least as good as the current standard of care (i.e. voriconazole) induces a 50% reduction in circulating GM concentrations in the persistently neutropenic rabbit model of IPA. Thus, the rabbit model as currently designed produces an ‘on-scale’ readout that appears clinically relevant and tractable. These analyses provide a unique opportunity to evaluate the predictive nature of the rabbit model endpoints and further facilitate the use of this model for the future development of new antifungal agents.
The findings in this study also have implications for setting in vitro susceptibility breakpoints for isavuconazole against A. fumigatus. The recent clinical study (SECURE) clearly suggests that isavuconazole is an effective agent for the treatment of IA. If an assumption is made that the vast majority of isolates in the clinical trial are WT organisms, then A. fumigatus is considered to be a ‘good target’ for isavuconazole. In the absence of other clinical or PD data, the breakpoint for isavuconazole against A. fumigatus is ordinarily set at the epidemiological cut-off value (ECV or ECOFF), which is ≤1 mg/L.12 There is no evidence from the SECURE study that there is a relationship between MIC and clinical outcome, although the number of patients with a positive culture was too small to enable a definitive analysis. This PD study provides a further perspective on the validity of using 1 mg/L as a breakpoint. Without making any assumptions about a GMI cut-off value that defines therapeutic success and failure, Figure 4 shows the predicted GMI values for patients receiving a standard clinical isavuconazole regimen and infected with isolates with a range of MICs. The histograms in Figure 4 suggest that a ≥50% reduction in GMI is achieved for isavuconazole MICs <1 mg/L. Similarly, GM antigen expression is not suppressed in patients with an MIC ≥2 mg/L. For an MIC of 1 mg/L some patients have suppressed GM antigen expression while others do not. Therefore, an MIC of 1 mg/L may on occasions be difficult to treat and be therefore classified as intermediate to denote the fact that isavuconazole should be used with some caution in this situation.
The PK/PD relationship for isavuconazole against Aspergillus spp. has been described for a variety of other experimental designs; however, given the results of the clinical trial, not all appear to predict the clinical response for isavuconazole well. Therefore, it is important to understand the key differences between the models and how these differences may have an impact on the development of a new antifungal drug. Lepak et al.13 used a neutropenic murine model of IPA. The model used a diverse group of organisms (10 in total) ranging from WT organisms to organisms with well-characterized cyp51a point mutations. In contrast to the current model, primary efficacy was defined by the net stasis in fungal growth as measured by the density of fungal DNA in the lung. The estimated total drug PD target (AUC/MICCLSI) from this experiment was 503. In contrast, Seyedmousavi et al.14 evaluated isavuconazole in a non-neutropenic mouse model of disseminated aspergillosis. Similar to the model of Lepak et al.,13 this model utilized both WT and cyp51a mutants. However, the primary outcome measure was 14-day survival. The total drug AUC/MICCLSI target associated with 50% survival (EC50) was 10-fold lower and estimated to be ∼50.
The estimated half-life of isavuconazole in rabbits (17.5 h) is significantly longer than in mice (1–5 h).13 The difference in half-life between the two animal models could have an impact when estimating the PD target in severe experimental conditions (e.g. profound and prolonged neutropenia). The relative absence of neutrophils combined with long periods of the dosing interval where drug concentrations are beneath a critical threshold may enable fungal regrowth. Progressively higher dosages of isavuconazole are then required to achieve concentrations that prevent fungal regrowth. It is therefore possible that the rapid clearance of drug, the use of dosing intervals (every 12 h) that are many multiples of the half-life and the severe nature of the model conspire to produce an inaccurate estimate of the drug exposure that is ultimately required for therapeutic efficacy.
In addition to the animal models described above, an in vitro human alveolus model of experimental pulmonary aspergillosis was conducted.15 The total drug AUC/MIC estimated for this system was 11. This lower value probably largely reflects the finding that the model best simulates early (first 24 h) versus more established disease (>24 h).16 The in vitro alveolus model also does not reflect the tissue injury, including haemorrhagic infarctions, which may impair penetration of the antifungal agent to the invading hyphae. These factors are important determinants of the drug exposure required to suppress circulating GM antigen concentrations.
When one considers the possible use of these PD targets in drug development, it is clear that even small differences may have a significant impact on the regimen that is ultimately chosen and the establishment of in vitro susceptibility breakpoints. For example, use of the PD target defined in the neutropenic mouse model would have placed the isavuconazole breakpoint in the middle of the WT population (at 0.06 mg/L), which is neither ideal nor consistent with clinical trial findings. A corollary is that a much higher dose would have been predicted to be successful than was ultimately used in the clinical trial. The rabbit model provides ‘on-scale readouts’ for isavuconazole and a clinically relevant model readout of a 50% reduction in GMI. Thus, complete suppression of GM antigen, which may be desirable when treating a patient with IA, is not a relevant consideration for PK/PD bridging studies (in the same way that 100% survival may not be possible or reasonable). Therefore, all the preclinical PK/PD studies for isavuconazole and the subsequent bridging studies provide a salutary lesson in the use of PK/PD models for drug development. Orthogonal reasoning should be applied, models must be stressed, positive controls should be used to benchmark model performance and readouts, and findings should be repeatedly cross-checked.
This study has several limitations. First, only one WT organism of A. fumigatus was used in the experimental model. A more diverse group of organisms with a range of MICs, including organisms with well-characterized resistance mechanisms, would provide a more robust assessment of the exposure–response relationship. Second, the experimental findings as presented are only relevant to CLSI methodology. Third, a small number of terminal GMI values fall in the middle of the curve, suggesting that the results could be driven by the majority of values that occur at the extremes of effect or no effect. Nevertheless, this study provides further evidence that GM is a robust clinically relevant biomarker that can be used to characterize the PD of anti-Aspergillus agents. A 50% reduction in GMI in this persistently neutropenic rabbit model of IPA correlates well with the results of the Phase 3 clinical trial for isavuconazole, which provided a foundation to further characterize the PD of new antifungal agents and the selection of new regimens for study in early-phase clinical trials.
Funding
This work was supported in part by Astellas Pharma Global Development, Inc. T. J. W. receives support as an Investigator of the Save Our Sick Kids Foundation, the Henry Schueler Foundation as a Scholar in Mucormycosis, and the Sharpe Family Foundation as a Scholar in Emerging Infectious Diseases. W. W. H. is supported by a National Institute of Health Research (NIHR) Clinician Scientist Fellowship.
Transparency declarations
L. L. K., A. D. and P. B. are employees of Astellas. T. J. W. and W. W. H. have served as consultants for and received research support from Astellas Pharma, Inc. R. P. and V. P.: none to declare.
References
- 1.Astellas Pharma US Inc. CRESEMBA® (Isavuconazonium Sulfate) Prescribing Information. http://www.astellas.us/docs/cresemba.pdf.
- 2.Garnacho-Montero J, Olaechea P, Alvarez-Lerma F et al. Epidemiology, diagnosis and treatment of fungal respiratory infections in the critically ill patient. Rev Esp Quimioter 2013; 26: 173–88. [PubMed] [Google Scholar]
- 3.Neofytos D, Treadway S, Ostrander D et al. Epidemiology, outcomes, and mortality predictors of invasive mold infections among transplant recipients: a 10-year, single-center experience. Transpl Infect Dis 2013; 15: 233–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rex JH, Eisenstein BI, Alder J et al. A comprehensive regulatory framework to address the unmet need for new antibacterial treatments. Lancet Infect Dis 2013; 13: 269–75. [DOI] [PubMed] [Google Scholar]
- 5.Ambrose PG, Nightingale CH, Murakawa T. Antimicrobial Pharmacodynamics in Theory and Clinical Practice (Infectious Disease and Therapy). New York, NY: Taylor and Francis, 2009. [Google Scholar]
- 6.Petraitis V, Petraitiene R, Moradi PW et al. Pharmacokinetics and concentration-dependent efficacy of isavuconazole for treatment of experimental invasive pulmonary aspergillosis. Antimicrib Agents Chemother 2016; doi:10.1128/AAC.02665-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petraitis V, Petraitiene R, Sarafandi AA et al. Combination therapy in treatment of experimental pulmonary aspergillosis: synergistic interaction between an antifungal triazole and an echinocandin. J Infect Dis 2003; 187: 1834–43. [DOI] [PubMed] [Google Scholar]
- 8.Clinical and Laboratory Standards Institute. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Filamentous Fungi—Approved Standard M38-A2; Second Edition. CLSI, Wayne, PA, USA, 2008. [Google Scholar]
- 9.Stynen D, Sarfati J, Goris A et al. Rat monoclonal antibodies against Aspergillus galactomannan. Infect Immun 1992; 60: 2237–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neely MN, van Guilder MG, Yamada WM et al. Accurate detection of outliers and subpopulations with Pmetrics, a nonparametric and parametric pharmacometric modeling and simulation package for R. Ther Drug Monit 2012; 34: 467–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maertens JA, Raad II, Marr KA et al. Isavuconazole versus voriconazole for primary treatment of invasive mould disease caused by Aspergillus and other filamentous fungi (SECURE): a phase 3, randomised-controlled, non-inferiority trial. Lancet 2016: 387: 760–9. [DOI] [PubMed] [Google Scholar]
- 12.Espinel-Ingroff A, Chowdhary A, Gonzalez GM et al. Multicenter study of isavuconazole MIC distributions and epidemiological cutoff values for Aspergillus spp. for the CLSI M38-A2 broth microdilution method. Antimicrob Agents Chemother 2013; 57: 3823–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lepak AJ, Marchillo K, Vanhecker J et al. Isavuconazole (BAL4815) pharmacodynamic target determination in an in vivo murine model of invasive pulmonary aspergillosis against wild-type and cyp51 mutant isolates of Aspergillus fumigatus. Antimicrob Agents Chemother 2013; 57: 6284–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seyedmousavi S, Bruggemann RJ, Meis JF et al. Pharmacodynamics of isavuconazole in an Aspergillus fumigatus mouse infection model. Antimicrob Agents Chemother 2015; 59: 2855–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Box H, Livermore J, Johnson A et al. Pharmacodynamics of isavuconazole in a dynamic in vitro model of invasive pulmonary aspergillosis. Antimicrob Agents Chemother 2015; 60: 278–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hope WH, Petraitis V, Petraitiene R et al. The initial 96 hours of invasive pulmonary aspergillosis: histopathology, comparative kinetics of galactomannan and (1→3)-β-d-glucan and the consequences of delayed antifungal therapy. Antimicrob Agents Chemother 2010; 54: 4879–86. [DOI] [PMC free article] [PubMed] [Google Scholar]