

Abstract
Alzheimer disease is a devastating chronic disease without adequate therapy. More than 10 years ago, it was demonstrated in transgenic mouse models that vaccination may be a novel, disease-modifying therapy for Alzheimer. Subsequent clinical development has been a roller-coaster with some positive and many negative news. Here, we would like to summarize evidence that next generation vaccines optimized for old people and focusing on patients with mild disease stand a good chance to proof efficacious for the treatment of Alzheimer.
Keywords: Abeta, Alzheimer, antibody, third generation, vaccine
Introduction
Alzheimer vaccines
Alzheimer Disease (AD) is an important progressive neurodegenerative illness, and is a major form of dementia. With increasing age, the probability to develop AD steeply increases and currently affects 26 million people worldwide.1 As the age of the human population is globally growing, the increased life expectancy and the consequent increased frequency and number of patients afflicted with this disease is becoming a large social and economic burden.2,3 Only symptomatic and not very effective treatments are currently available and in view of the alarming epidemiological data, novel therapeutic strategies to prevent, mitigate or delay the onset of AD are pursued extensively.4 The hallmark of the pathology observed in AD is plaques in the brain of the afflicted patients. Active or passive vaccination against components of these plaques is currently one of the most promising therapies with disease-modifying potential.5
The 2 major components of the plaques are extracellular aggregates of amyloid-β (Aβ) peptides, which are cleaved out of the amyloid precursor protein (APP) by the β- and γ-secretase, and intracellular neurofibrillary tangles (NFT) consisting of hyperphosphorylated tau proteins.6 Under healthy conditions Aβ peptides as well as tau proteins are constantly expressed but do not show aggregate formation. The normal function of Aβ remains controversial but seems to have e.g., an anti-microbial activity.7 In contrast, tau proteins stabilize microtubules and ensure neuronal integrity and stimulus transfer.
Familial AD is associated with mutations in the APP protein as well as presenilin 1 and 2 as these mutations are known to enhance production of Aβ-peptides and increase their deposition in plaques. Transgenic mice expressing APP associated with familial AD also show plaque deposition and signs of the illness. Furthermore, Aβ peptides are toxic to cells under conditions of oxidative stress, and inflammation8 and APO ϵ4, which is a genetic risk factor for AD, causes delayed and decreased degradation of the peptide.9 Intracellular neurofibriallary tangles (NFT) are also considered valuable targets for treatment of Alzheimer. Progress is, however, much less advanced compared with Aβ1-42 and is therefore not subject of this review.
In 1999, Elan pharmaceuticals reported that active immunization against Aβ1-42 could reduce Aβ pathology in APP transgenic mice.10 Immunization with aggregated Aβ1-42 formulated in strong adjuvants caused reduced plaque burden10 and subsequent studies also demonstrated improved mental performance in immunized mice.11 These findings were not restricted to the particular transgenic mouse strain, as similar results were obtained in additional transgenic mouse models of AD using active and passive immunization (for review, see ref. 12). As passive immunization with mAbs was able to clear the plaques, it was clear that antibodies were the relevant effector molecules induced by vaccination. The exact mechanism of action is, however, still under debate. There is the possibility that Aβ1-42 specific antibodies may act as a sink in the blood and “extract” Aβ1-42 peptide from the brain. Even though Aβ1-42 deposition in plaques seems to be a rather dynamic process, it remains difficult to explain how individual Aβ1-42 peptides are released from the plaques and reach the blood to be captured by antibodies. It may also be possible that specific antibodies, which are known to penetrate the blood brain barrier to some extent, are transporting the peptides in a reverse manner from the brain into the blood. Both hypotheses seem to be supported by the fact that Aβ1-42 levels increase in serum upon immunization as they are bound by antibodies. On the other hand, Aβ1-42 peptide found in blood may not be from the brain but from alternative sources, such as Aβ1-42 depositions found in blood vessels. Yet an alternative possibility is that Aβ1-42 specific-antibodies bind to plaques and promote their removal by microglia cells. As this hypothesis does not call for large amounts of Aβ1-42 to migrate from the brain to the blood, and since aggregates would allow for efficient high-avidity binding of antibodies, this last hypothesis may be the most plausible one. In addition to reducing the plaque-burden, specific antibodies may also mitigate some toxic effects attributed to Aβ1-42.
First generation vaccine
In light of these promising preclinical results a number of clinical studies were initiated to assess safety and potential efficacy of vaccines against AD. The initial vaccine developed by Elan and Wyeth was based on aggregated human Aβ1-42 (AN1792). In first phase I trials, AD patients were immunized with AN1792 formulated in the adjuvant QS21 (AN1792). Aβ specific antibodies could be found in more than half of the immunized AD patients in phase I trials without notable adverse events.13 Encouraged by the positive safety data, AN1792 formulated in QS21 vaccine was tested in a relatively larger phase II trial with 372 AD patients to assess safety as well as efficacy of the therapy. However, treatment had to be discontinued prematurely in this trial since 6% of the vaccinated patients developed aseptic meningo/-encephalitis.14 Although the patients responded well to treatment with corticosteroids, the risk was deemed too high and further development of the vaccine discontinued. However, detailed analysis of safety and efficacy parameters of the study yielded a wealth of information for next generation vaccine development.
The cause of aseptic meningo-/encephalitis
Immediately after the issues with the Elan/Wyeth trial, it was unclear whether T or B cells were responsible for the side effects. The preferred effector cells were T cells, as antibodies are thought to be the effects or molecules responsible for plaque removal. Indeed, evidence was accumulating that T cells were causing the problems. An important piece of evidence was that subsequent analysis revealed that the side effects failed to correlate with antibody titers.14-16 In addition, infiltrating T-cells were found in the brain of diseased patients which suggested Aβ specific T-cells to be the cause of the side observed effects.17-20 The design of the vaccine may have be a key reason for the immunopathology. As human Aβ1-42 was used for immunization, no foreign Th cell epitope has been introduced to bypass Th cell tolerance. As a consequence, Aβ1-42—specific tolerance had to be broken for the induction of Th cell dependent IgG responses. The combination of strong adjuvants (QS21/saponin) and the fibrillar character of the vaccine causing persistence of the antigen for extended time periods at the injection site were able to overcome Th cell tolerance and caused the generation of Th cell dependent IgG responses. Overcoming Th cell tolerance, however, apparently also caused the T cell mediated immunopathology.
Preliminary evidence for efficacy
Even though dosing of patients was stopped in the middle of the trial, there were a substantial number of patients mounting specific IgG responses. This allowed investigator-driven unblinding of a subcohort of the study. These authors described strongly decreased mental decline and delayed disease progression in antibody-responders.13,14 Later studies assessing all patients in a pre-defined manner found some, but not as robust effects.18,21,22 Thus, despite the serious safety issues observed in the AN1792 trial, the vaccine provided first clinical evidence that active vaccination against Aβ may be beneficial for the treatment of AD.18,21,22 From a mechanistic point of view, it was very interesting that post-mortem analysis demonstrated strong reduction of plaque load in antibody responders. Hence, as in the transgenic mice, specific IgG was able to remove plaques consisting of Aβ1-42 from the brain.23 This was an important finding as many researchers doubted the ability of serum IgG antibodies to interfere with plaque burden in a secluded place such as the brain.
Next generation vaccines
The observed cases of meningo/-encephalitis represent the most significant setback for the development of a vaccine against Alzheimer disease. As discussed above, T cells were soon thought to be the pathological culprit, a hypothesis supported by the fact that mAbs specific for Aβ1-42 have caused no signs of meningo-encephalitis. The two major strategies pursued by the industry has been introduction of Th cell epitopes to be able to bypass Th cell tolerance and shortening of the Aβ1-42—derived peptide. Indeed, peptides of sizes smaller than 8 amino acids cannot induce T cell responses as they cannot bind to MHC molecules for presentation to T cells. Figure 1 summarizes these steps and identifies short Aβ1-42—derived peptides coupled to a protein carrier as the ideal vaccine candidate
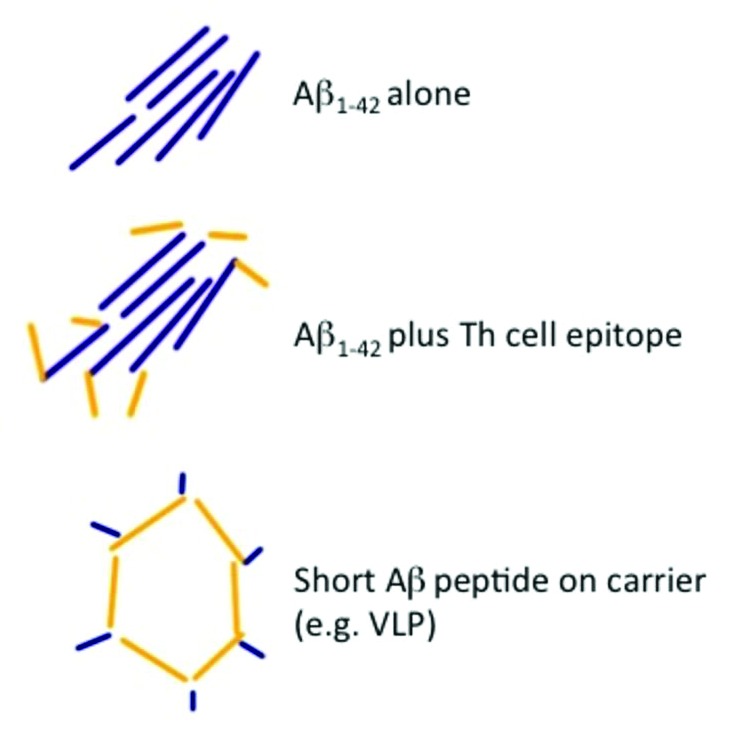
Figure 1. Three versions of vaccines against Aβ1–42. The original version consisting of Aβ1–42 only, an improved version, where a Th cell epitope has been introduced and current versions of the vaccine, where short peptides derived from Aβ1–42 are coupled to protein carriers or virus-like particles.
Indeed, the follow up candidate for AN1792 consists of the N-terminal seven amino acids of Aβ1-42 covalently linked to the diphtheria toxin cross reactive mutant (CRM197) protein carrier and is called ACC-001. As discussed, this peptide is below the minimum length-requirements for any T-cell epitope to bind to MHC molecules. Nevertheless, strong Aβ1-42—specific antibody responses were induced in the transgenic mice and plaque development was efficiently blocked and cognitive functions were improved. This compound is currently subject to intense clinical testing.
Likewise Novartis, using bacteriophage based VLP technology, has embarked on the development of an Alzheimer vaccine targeting amyloid β. This vaccine, which goes by the name of CAD106 consists of the bacteriophage Qβ VLP (Qβ) displaying Aβ1-6 on its surface by means of chemical coupling. As described for the CRM197 vaccine candidate, CAD106 was highly immunogenic for the induction of antibody in transgenic mice as well as rabbits and primates but did not induce measurable Aβ1-42—specific T cells responses.24 As expected, immunization with CAD106 inhibited amyloid cortical plaques deposition in transgenic mouse models of Novartis. Immunization of old mice exhibiting advanced pathology in the brain, resulted in reduced levels of amyloid deposition. However, as expected from immunizing old mice, responses were low compared with immunized young mice. Currently available information from first in man studies with CAD106 suggest that the vaccine is well tolerated and induces Aβ—specific immune response in the majority of the patients in the absence of meningoencephalitis.25-27
Merck and Co., Affirisis/GSK, AC immune and United Biomedical all use a similar approach as described above with small differences in the carrier and/or peptide and adjuvant used for vaccine generation. However, none of the currently tested vaccines has clinical PoC demonstrating delayed disease progression. Another drawback of these second generation vaccines is that they are not optimized for the old and therefore are expected to induce moderate antibody responses. It will be interesting to see whether the attempt to induce anti-inflammatory T cells during vaccination as performed by Mercia Pharma Inc. will have an impact on safety and efficacy of the approach.
What can we learn from mAbs
There are a number of mAbs in development. Most advanced are Bapineuzumab (PFE/JNJ/ELN) and Solanezumab. (LLY). Both antibodies have been in large phase III studies. Even though they failed to show efficacy in the overall disease population, subgroups analysis showed a strong impact of Apo ϵ4, which is involved in degradation of Aβ1-42 and predisposes to poor responsiveness to mAb-treatment. Most remarkable, a subgroup analysis of the LLY trial with Solanezumab demonstrated efficacy in pooled data of mild but not moderate patients (a 34% reduction in cognitive decline, which was significant compared with placebo [P = 0.001]). This may be the most important insight, as such a vaccine moves toward a preventative rather than therapeutic modality. It seems rather obvious that prevention of plaque accumulation is far superior over removal of existing plaques, as the latter process is expected to simply leave “a hole in the brain,” which is not necessarily helpful for mental performance. As PET tomography now can specifically visualize Alzheimer plaques in the living patient, it will be possible to treat patients very early, in fact sometimes before they even show any symptoms.24 In familial Alzheimer, plaque deposition could be visualized a decade before onset of symptoms. As most of the potential side effects will be due to interaction of antibodies with plaques, it is obvious that early immunization will also keep the side-effects low. PET may also be used as a surrogate marker for efficacy as it is now possible to demonstrate reduction in plaque load in situ in patients treated with mAbs.28
There are additional biomarkers that may be used to assist diagnosis and progression of disease. Levels of Aβ peptides and tau in cerebrospinal fluid (CSF) have been shown to be altered in familial Alzheimer disease patients more than 10 y before onset of disease and may therefore represent additional ways to diagnose AD early.29 In addition, levels of tau protein in CSF may also be used to follow disease development under treatment. In contrast, levels of Aβ1-40 and Aβ1-42 -peptides may be less useful biomarkers to monitor therapy success, as antibodies will stabilize those peptides and increase their levels at least in the blood. Indeed, antibody bound Aβ peptides in serum seen after vaccination may be viewed as auspicious sign for potential efficacy. Levels of phospho-tau, N-glycan or lipid profiles in blood may be additional promising markers; they are, however, far from routine clinical application and it is unclear how robust the signals are.30
The way Aβ1-42 deposits are recognized in treated patients may also be an important distinction between mAbs and vaccines in terms of their safety. mAbs are injected at high doses in a very short time period; interaction of antibodies and Aβ1-42 deposits in e.g., blood vessels or the brain will therefore be rapid and strong. In contrast, onset of antibody production upon vaccination will be slow, likely reducing the potential side effects. Indeed Bapineuzumab has a number of side-effects (e.g., vasogenic edema) which do not seem to occur in vaccinated individuals.
Table 1 summarizes the various strategies to treat Alzheimer disease. It also includes classical small molecules. The marketed drugs are all not disease modifying but there is a number of new molecules as well as mAbs and vaccines in development which are expected to be disease-modifying by reducing plaque burden or preventing their build-up.
Table 1. Strategies to treat Alzheimer disease31-35.
| Molecule Type | Target | Phase |
|---|---|---|
| Small Molecules | Acetycholine Esterase | Market |
| NMDA receptor | Market | |
| β-amyloid | Phase II | |
| Phospho-Tau | Phase II | |
| β/γ-secretase | Phase II | |
| mAbs | β-amyloid | Phase III |
| TNF | Phase II | |
| Vaccine | β-amyloid | Phase II |
| Phospho-Tau | Phase I |
The way forward: From therapy to prophylaxis
There are two key changes that need to be implemented in new Alzheimer vaccine programs. (1) The vaccines need to be optimized for old people. This may be achieved by introducing new structural features into the vaccine backbone and/or use of optimized adjuvants as well as regimens. (2) Patients with mild disease will need to be recruited. PET-imaging will be the key to allow early diagnosis and monitoring treatment success. Thus, vaccine-based treatment of Alzheimer disease will move from late therapeutic to early almost prophylactic interventions. This will allow preventing the build-up of plaques, bringing benefit to the patient with minimal damage expected.
Side effects caused by vaccination against Aβ are expected to be minimal in the absence of Aβ1-42 aggregates, i.e., plaques. Thus, the earlier a vaccine may be used, the smaller the expected side effects. Indeed, vaccination before plaque deposition in a prophylactic manner may be possible without causing any undue side effects. Thus, in a way similar to the childhood vaccines that we are all familiar with, a vaccine against Alzheimer disease may become the first prophylactic vaccine specifically used in senior citizens.
Disclosure of Potential Conflicts of interest
All authors are involved in the development of active immunotherapies.
Acknowledgments
We would like to thank Adrian Hill, Thomas Kündig, and Pål Johansen for helpful discussions.
References
- 1.Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. . Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement 2007; 3:186 - 91; http://dx.doi.org/ 10.1016/j.jalz.2007.04.381; PMID: 19595937 [DOI] [PubMed] [Google Scholar]
- 2.Corrada MM, Brookmeyer R, Paganini-Hill A, Berlau D, Kawas CH. . Dementia incidence continues to increase with age in the oldest old: the 90+ study. Ann Neurol 2010; 67:114 - 21; http://dx.doi.org/ 10.1002/ana.21915; PMID: 20186856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wimo A, Winblad B, Jönsson L. . The worldwide societal costs of dementia: Estimates for 2009. Alzheimers Dement 2010; 6:98 - 103; http://dx.doi.org/ 10.1016/j.jalz.2010.01.010; PMID: 20298969 [DOI] [PubMed] [Google Scholar]
- 4.Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M. . Alzheimer’s disease: clinical trials and drug development. Lancet Neurol 2010; 9:702 - 16; http://dx.doi.org/ 10.1016/S1474-4422(10)70119-8; PMID: 20610346 [DOI] [PubMed] [Google Scholar]
- 5.Lemere CA, Masliah E. . Can Alzheimer disease be prevented by amyloid-beta immunotherapy?. Nat Rev Neurol 2010; 6:108 - 19; http://dx.doi.org/ 10.1038/nrneurol.2009.219; PMID: 20140000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. . Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci U S A 1988; 85:4051 - 5; http://dx.doi.org/ 10.1073/pnas.85.11.4051; PMID: 3131773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, Burton MA, Goldstein LE, Duong S, Tanzi RE, et al. . The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One 2010; 5:e9505; http://dx.doi.org/ 10.1371/journal.pone.0009505; PMID: 20209079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mattson MP. . Pathways towards and away from Alzheimer’s disease. Nature 2004; 430:631 - 9; http://dx.doi.org/ 10.1038/nature02621; PMID: 15295589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu CC, Kanekiyo T, Xu H, Bu G. . Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 2013; 9:106 - 18; http://dx.doi.org/ 10.1038/nrneurol.2012.263; PMID: 23296339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, et al. . Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999; 400:173 - 7; http://dx.doi.org/ 10.1038/22124; PMID: 10408445 [DOI] [PubMed] [Google Scholar]
- 11.Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, et al. . A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature 2000; 408:982 - 5; http://dx.doi.org/ 10.1038/35050116; PMID: 11140686 [DOI] [PubMed] [Google Scholar]
- 12.Röskam S, Neff F, Schwarting R, Bacher M, Dodel R. . APP transgenic mice: the effect of active and passive immunotherapy in cognitive tasks. Neurosci Biobehav Rev 2010; 34:487 - 99; http://dx.doi.org/ 10.1016/j.neubiorev.2009.10.006; PMID: 19857518 [DOI] [PubMed] [Google Scholar]
- 13.Bayer AJ, Bullock R, Jones RW, Wilkinson D, Paterson KR, Jenkins L, Millais SB, Donoghue S. . Evaluation of the safety and immunogenicity of synthetic Abeta42 (AN1792) in patients with AD. Neurology 2005; 64:94 - 101; http://dx.doi.org/ 10.1212/01.WNL.0000148604.77591.67; PMID: 15642910 [DOI] [PubMed] [Google Scholar]
- 14.Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, et al. . Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 2003; 61:46 - 54; http://dx.doi.org/ 10.1212/01.WNL.0000073623.84147.A8; PMID: 12847155 [DOI] [PubMed] [Google Scholar]
- 15.Hock C, Konietzko U, Papassotiropoulos A, Wollmer A, Streffer J, von Rotz RC, Davey G, Moritz E, Nitsch RM. . Generation of antibodies specific for beta-amyloid by vaccination of patients with Alzheimer disease. Nat Med 2002; 8:1270 - 5; http://dx.doi.org/ 10.1038/nm783; PMID: 12379846 [DOI] [PubMed] [Google Scholar]
- 16.Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Müller-Tillmanns B, Lemke U, Henke K, Moritz E, Garcia E, et al. . Antibodies against beta-amyloid slow cognitive decline in Alzheimer’s disease. Neuron 2003; 38:547 - 54; http://dx.doi.org/ 10.1016/S0896-6273(03)00294-0; PMID: 12765607 [DOI] [PubMed] [Google Scholar]
- 17.Ferrer I, Boada Rovira M, Sánchez Guerra ML, Rey MJ, Costa-Jussá F. . Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer’s disease. Brain Pathol 2004; 14:11 - 20; http://dx.doi.org/ 10.1111/j.1750-3639.2004.tb00493.x; PMID: 14997933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Rovira MB, Forette F, et al. , AN1792(QS-21)-201 Study Team. . Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 2005; 64:1553 - 62; http://dx.doi.org/ 10.1212/01.WNL.0000159740.16984.3C; PMID: 15883316 [DOI] [PubMed] [Google Scholar]
- 19.Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C, Seubert P, Games D, Kirby L, Schenk D. . Abeta vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology 2005; 64:129 - 31; http://dx.doi.org/ 10.1212/01.WNL.0000148590.39911.DF; PMID: 15642916 [DOI] [PubMed] [Google Scholar]
- 20.Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. . Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med 2003; 9:448 - 52; http://dx.doi.org/ 10.1038/nm840; PMID: 12640446 [DOI] [PubMed] [Google Scholar]
- 21.Fox NC, Black RS, Gilman S, Rossor MN, Griffith SG, Jenkins L, Koller M, AN1792(QS-21)-201 Study. . Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology 2005; 64:1563 - 72; http://dx.doi.org/ 10.1212/01.WNL.0000159743.08996.99; PMID: 15883317 [DOI] [PubMed] [Google Scholar]
- 22.Vellas B, Black R, Thal LJ, Fox NC, Daniels M, McLennan G, Tompkins C, Leibman C, Pomfret M, Grundman M, AN1792 (QS-21)-251 Study Team. . Long-term follow-up of patients immunized with AN1792: reduced functional decline in antibody responders. Curr Alzheimer Res 2009; 6:144 - 51; http://dx.doi.org/ 10.2174/156720509787602852; PMID: 19355849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, et al. . Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet 2008; 372:216 - 23; http://dx.doi.org/ 10.1016/S0140-6736(08)61075-2; PMID: 18640458 [DOI] [PubMed] [Google Scholar]
- 24.Wiessner C, Wiederhold KH, Tissot AC, Frey P, Danner S, Jacobson LH, Jennings GT, Lüönd R, Ortmann R, Reichwald J, et al. . The second-generation active Aβ immunotherapy CAD106 reduces amyloid accumulation in APP transgenic mice while minimizing potential side effects. J Neurosci 2011; 31:9323 - 31; http://dx.doi.org/ 10.1523/JNEUROSCI.0293-11.2011; PMID: 21697382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Graf A, et al. . Optimization of the treatment regimen with active Aβ immunotherapy CAD106 in alzheimer patients. Alzheimers Dement 2010; 6:S532; http://dx.doi.org/ 10.1016/j.jalz.2010.05.1775 [DOI] [Google Scholar]
- 26.Winblad B. . S2-04-06: Safety, tolerability and immunogenicity of the Aβ immunotherapeutic vaccine CAD106 in a first-in-man study in Alzheimer patients. Alzheimers Dement 2008; 4:T128; http://dx.doi.org/ 10.1016/j.jalz.2008.05.295 [DOI] [Google Scholar]
- 27.Winblad BG, et al. . Results of the first-in-man study with the active Aβ Immunotherapy CAD106 in Alzheimer patients. Alzheimers Dement 2009; 5:113 - P114; http://dx.doi.org/ 10.1016/j.jalz.2009.05.356 [DOI] [Google Scholar]
- 28.Rinne JO, Brooks DJ, Rossor MN, Fox NC, Bullock R, Klunk WE, Mathis CA, Blennow K, Barakos J, Okello AA, et al. . 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol 2010; 9:363 - 72; http://dx.doi.org/ 10.1016/S1474-4422(10)70043-0; PMID: 20189881 [DOI] [PubMed] [Google Scholar]
- 29.Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, et al. , Dominantly Inherited Alzheimer Network. . Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med 2012; 367:795 - 804; http://dx.doi.org/ 10.1056/NEJMoa1202753; PMID: 22784036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fletcher LC, Burke KE, Caine PL, Rinne NL, Braniff CA, Davis HR, Miles KA, Packer C. . Diagnosing Alzheimer’s disease: are we any nearer to useful biomarker-based, non-invasive tests?. GMS Health Technol Assess 2013; 9:Doc01; PMID: 23755087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Belluti F, Rampa A, Gobbi S, Bisi A. . Small-molecule inhibitors/modulators of amyloid-β peptide aggregation and toxicity for the treatment of Alzheimer’s disease: a patent review (2010 - 2012). Expert Opin Ther Pat 2013; 23:581 - 96; http://dx.doi.org/ 10.1517/13543776.2013.772983; PMID: 23425062 [DOI] [PubMed] [Google Scholar]
- 32.Himmelstein DS, Ward SM, Lancia JK, Patterson KR, Binder LI. . Tau as a therapeutic target in neurodegenerative disease. Pharmacol Ther 2012; 136:8 - 22; http://dx.doi.org/ 10.1016/j.pharmthera.2012.07.001; PMID: 22790092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tobinick E. . Tumour necrosis factor modulation for treatment of Alzheimer’s disease: rationale and current evidence. CNS Drugs 2009; 23:713 - 25; http://dx.doi.org/ 10.2165/11310810-000000000-00000; PMID: 19689163 [DOI] [PubMed] [Google Scholar]
- 34.Boutajangout A, Sigurdsson EM, Krishnamurthy PK. . Tau as a therapeutic target for Alzheimer’s disease. Curr Alzheimer Res 2011; 8:666 - 77; http://dx.doi.org/ 10.2174/156720511796717195; PMID: 21679154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. www.clinicaltrials.gov