

Abstract
A hydroxyl functional group positioned β to a pinacol boronate can serve to direct palladium-catalyzed cross-coupling reactions, apparently through the agency of a transiently formed palladium alkoxide. This feature can be used to control the reaction site in multiply borylated substrates and can activate boronates for reaction that would otherwise be unreactive.
Graphical abstract
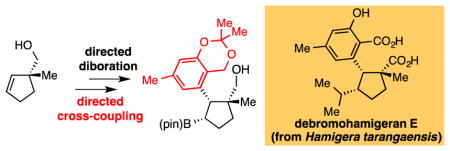
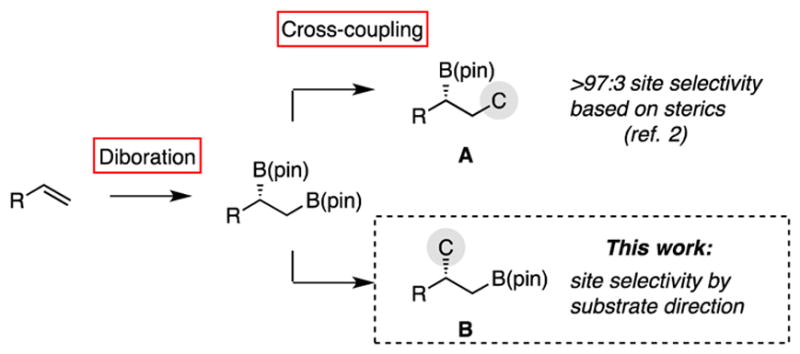
Complexity-generating reactions that apply to alkenes are critical tools for asymmetric synthesis. In this regard, the stereoselective diboration reaction is a versatile approach for the efficient transformation of olefin substrates: the vicinal 1,2-bis(boronate) products possess reactivity that facilitates functionalization of both carbons of the original alkene framework.1 While oxidation of vicinal bis(boronates) to diols is broadly applicable to an array of substrates, differential transformation of the two boronates requires a substrate bias that allows site-selective transformation of one boronate in preference to the other. In this context, the sensitivity of the Suzuki-Miyaura cross-coupling to steric effects allows selective transformation of a primary, terminal boronate without detectable reaction of an adjacent secondary boronate group. Our group has employed this feature in a selective tandem diboration/cross-coupling (DCC) strategy that allows the transformation of terminal alkenes into an array of functionalized non-racemic products via A (Scheme 1).2 It was considered that a different array of target structures would be accessible if the Suzuki-Miyaura cross-coupling could be coaxed to occur preferentially at the secondary boronate in the substrate to give B.3 Importantly, features that overturn the steric bias present in terminal bis(boronates) might also enable the site-selective cross-coupling of bis(boronates) derived from internal alkene substrates. In this Communication, we present a strategy that employs a neighboring hydroxyl group to control the site-selectivity of a Suzuki-Miyaura cross-coupling reaction. A noteworthy feature of this process is that it appears to rely upon a substrate-directed transmetallation that may involve a covalent linkage between the directing hydroxyl group and the Pd center. This reactivity mode is not commonly employed to facilitate catalytic cross-coupling reactions4 and may be a useful element in reaction design.
Scheme 1.
Site Selectivity in Cross-Coupling of Vicinal Bis(boronates).
One strategy for the stereospecific cross-coupling of secondary alkyl boronates employs Lewis basic functional groups to enhance the reactivity of an adjacent boronic acid derivative. In this context, Suginome,5 Molander,6 Hall,7 and Takacs8 have employed adjacent amide or ester functional groups as electron donors to facilitate cross-coupling. Inspired by these studies, we considered that related functional groups positioned proximal to a reacting vicinal bis(boronate) might facilitate site-selective cross-coupling. Considering that protected alcohols are readily accommodated in asymmetric diboration, and since alcohols are an inherent feature in directed metal-free diboration reactions,9 initial experiments probed the ability of a hydroxyl group to control regioselectivity of cross-coupling (Table 1). In these experiments, purified borylated alkyl silyl ethers were first subjected to TBS deprotection with catalytic para-toluenesulfonic acid, then reagents for a Pd/RuPhos10 mediated cross-coupling were introduced. After reaction for 12 hours at 70 °C, the reaction mixture was subjected to oxidative work-up. Preliminary experiments (see Supporting Information) indicated that the reaction solvent plays a critical role in controlling the ratio of elimination products to cross-coupling products with a THF/toluene/H2O combination providing optimal selectivity.
Table 1.
Survey of Substrates for Pd/RuPhos Catalyzed Cross-Coupling of Alkylboronates and Bromobenzene.a

|
Deprotection for 2 h at rt, followed by solvent removal and cross-coupling for 12 h at 70 °C.
Yield determined by 1H NMR versus 1,3,5-trimethoxybenzene as an internal standard.
The substrate was prepared by Cs2CO3-catalyzed diboration in THF/methanol and the solvent evaporated prior to subjection to cross-coupling.
When the above described cross-coupling conditions were applied to non-functionalized bis(boronate) 1, benzylic alcohol 2 was furnished in 71% yield. In contrast, when bis(boronate) 3 was subjected to the deprotection/cross-coupling/oxidation sequence, the presence of the hydroxyl group results in a complete turnover in regioselectivity such that 4 was provided in 72% yield. The remainder of the substrates in Table 1, most notably structures 9, 11 and 12 provide clear evidence that while directing effects from β-position are efficient (entry 5), when the directing group is positioned at the α or γ site (entries 6 and 7) the cross-coupling does not benefit from the presence of a neighboring hydroxyl group.
While the ability of a β hydroxyl group to activate a secondary pinacol boronate for cross-coupling is reminiscent of the activation provided by β-acyl groups, the reactions in Table 1 appear to operate by a distinct mechanistic principle relative to the acyl-promoted cross-couplings developed by Suginome, Molander, and Hall.5–7 This distinction is most clearly indicated by the observation that acyl-promoted cross-couplings proceed with inversion of configuration, ostensibly by generalized chelated “ate” complex C (Scheme 2a), whereas the hydroxyl-directed cross-coupling occurs with complete retention of configuration at carbon (Table 1, entry 8). The stereochemical outcome with hydroxyl direction is reminiscent of Takacs’ secondary-amide-directed stereoretentive cross-coupling, although the mechanistic underpinnings for the this process are unresolved.8 Additional mechanistic information about the hydroxyl-directed reaction was obtained by examining the cross-coupling of bis(boronate) 15 (Scheme 2b). In line with the observations in Table 1, when 15 was subjected to Pd/RuPhos catalyzed coupling with 1.05 equivalents of bromobenzene, the reaction exhibited complete selectivity for the B(pin) group positioned β to the hydroxyl substituent. Of note, in addition to cross-coupling product 16, ketones 17 and 18, and unreacted 15 were isolated. When the reaction was conducted in the presence of excess bromobenzene, complete conversion of 15 occurs and only 16 and 17 were isolated as the reaction products. Considering the known capacity for Pd(II) complexes to oxidize alcohols with aryl halides as terminal oxidant,11 a plausible rationale for formation of 17 and 18 involves β-hydrogen elimination from a substrate-derived L(Ar)Pd(alkoxide). Thus the observation of products 17 and 18 establishes the capacity for substrate-derived Pd(alkoxide) formation during the course of catalytic cross-coupling reactions. A last piece of data pertaining to the origin of hydroxyl-direction arises from the X-ray structure of 15 (Scheme 2c). In the solid state structure, both the β B(pin) and the hydroxyl group occupy axial positions; however, the planar geometry of boron suggests that the two groups are too far removed for internal HO→B coordination. Thus, even in a case where “ate” complex formation appears to be precluded, the cross-coupling is site selective. Taking the above observations into account, a plausible mechanism for the directing effect observed in Table 1 involves binding of the substrate hydroxyl to an LPdAr complex, perhaps by displacement of a halide; subsequent internal delivery of Pd through a complex such as D (Scheme 2d), would generate an organopalladium complex through an inner-sphere stereoretentive transmetallation, and ultimately deliver the corresponding coupling product.
Scheme 2.

Features of Directed Cross-Coupling Reactions.
The hydroxyl-directed transmetallation can merge seamlessly with recently developed hydroxyl-directed metal-free diboration and allows for rapid stereoselective and site-selective functionalization of homoallylic alcohols. As depicted in Scheme 3, sequential directed diboration/cross-coupling, when followed by silylation or acylation, furnishes γ-oxygenated boronates from a range of substrates. Importantly, the reaction can be conducted with aryl, heteroaryl, and alkenyl electrophiles and applies equally well to terminal alkenes, internal olefins, and trisubstituted alkenes. While the terminal and internal olefins furnish 1,3-syn relative stereochemistry, the trisubstituted alkene furnishes modest 1,3-anti induction, an outcome that is inline with the stereoinduction observed in the directed diboration reactions. Lastly, the use of Pd(OAc)2 in reactions of alkenyl electrophiles appeared to minimize alkene-containing side-products derived from β-deborylation of intermediate organopalladium complexes.
Scheme 3. Tandem Directed Diboration/Directed Cross-Coupling.a.

aCross-coupling: aryl electrophiles employed 0.5% Pd2(dba)3, 1% RuPhos and 1.5 equiv ArX; alkenyl electrophiles employed 2.5% Pd(OAc)2, 3% RuPhos and 1.5 equiv alkenyl halide. Yield represents isolated yield of purified material and is an average of two experiments. bCross-coupling employed 10% Pd(OAc)2 and 12% RuPhos.
Tandem diboration/directed cross-coupling reactions can allow alkene functionalization in ways that are not straightforward with current methods. These strategies are illustrated by the examples in Scheme 4. To target CCR1 antagonist 4012, we considered the reaction of homoallylic alcohol 37, a starting material that is readily assembled by allylation of Boc-protected phenylalanine.13 Tandem directed diboration/directed cross-coupling allows stereoselective C-C bond formation at the internal alkene carbon of 37. Upon TEMPO catalyzed oxidation, this reaction sequence delivers lactone 38, which is a precursor to key building block 39.12 In a related fashion, enantioselective double arylation of alkene 41 (Scheme 4b) can be accomplished with Pt-catalyzed enantioselective diboration to initiate the sequence.1c,d With the alcohol protected as a silyl ether, steric effects dominate the cross-coupling of the intermediate bis(boronate) such that bond formation occurs at the terminal carbon. Subsequent TBS deprotection and directed cross-coupling furnishes non-racemic alcohol 43, a plausible intermediate in the synthesis of vitronectin receptor antagonist 44.14
Scheme 4.

Utility of Directed Cross-Coupling.
As a last synthesis example, we considered construction of debromohamigeran E (Scheme 5).15 This structure belongs to a large group of related cyclopentane-containing compounds16, but has not yet been addressed by laboratory synthesis.17 We considered that the target might be accessed by directed cis double alkylation of alkene 46. To effect this transformation, 4618 was subjected to diboration followed by directed cross-coupling with 48. Remarkably, even though the reacting boronate in 47 is at a secondary carbon and resides between quaternary and tertiary carbons, the cross-coupling proceeds stereoselectively and furnishes a single isomer of product. Of note, this reaction was readily accomplished on a preparative scale and was used to furnish 2.7 grams of silyl protected product 49, an intermediate that was characterized by X-ray crystallography. A subsequent Evans-Zweifel olefination19 with 2-lithiopropene furnished 2.1 grams (93% yield) of double alkylation product 50, a versatile intermediate for the synthesis of a number of hamigerans. After C-C bond installation, a portion of 50 was reduced and subjected to silyl deprotection. Subsequent Ru-catalyzed oxidation20 occurred at the primary alcohol site and the benzylic methylene thereby furnishing 52. Saponification afforded the target structure.
Scheme 5.

Synthesis of (±)-Debromohamigeran E.
In summary, the hydroxyl-directed cross-coupling enables regioselective, stereoretentive C-C bond formation in the context of multiply borylated reagents. Moreover, the induced proximity conferred by the putative Pd-O linkage appears to facilitate reaction of boronate groups that are likely to be recalcitrant in the absence of such activating effects.
Supplementary Material
Acknowledgments
We thank Dr. Bo Li for x-ray analysis and Nathan Drake for experimental assistance. The NIH (GM-59417) is acknowledged for financial support; AllyChem is thanked for a donation of B2(pin)2.
Footnotes
Notes
The authors declare no competing financial interest.
Supporting Information. Procedures, characterization and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Reviews: Burks HE, Morken JP. Chem Commun. 2007:4717. doi: 10.1039/b707779c.Suginome M, Ohmura T. Boronic Acids. 2. Vol. 1. Wiley-VCH; New York: 2011. p. 171.Takaya J, Iwasawa N. ACS Catal. 2012;2:1993.
- 2.Mlynarski SN, Schuster CH, Morken JP. Nature. 2014;505:386. doi: 10.1038/nature12781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For important recent advances in the cross-coupling of secondary boronates, see: Li L, Zhao S, Joshi-Pangu A, Diane M, Biscoe MR. J Am Chem Soc. 2014;136:14027. doi: 10.1021/ja508815w.Matthew SC, Glasspoole BW, Eisenberger P, Crudden CM. J Am Chem Soc. 2014;136:5828. doi: 10.1021/ja412159g.Imao D, Glasspoole BW, Laberge VS, Crudden CM. J Am Chem Soc. 2009;131:5024. doi: 10.1021/ja8094075.Dreher SD, Dormer PG, Sandrock DL, Molander GA. J Am Chem Soc. 2008;130:9257. doi: 10.1021/ja8031423.van der Hoogenband A, Lange JHM, Terpstra JW, Koch M, Visser GM, Visser M, Korstanje TJ, Jastrzebski JTBH. Tetrahedron Lett. 2008;49:4122.For a review, see: Leonori D, Aggarwal VK. Angew Chem Int Ed. 2015;54:1082. doi: 10.1002/anie.201407701.
- 4.For directing effects in Stille couplings, see: Crisp GT, Gebauer MG. Tetrahedron Lett. 1995;36:3389.Quayle P, Wang J, Xu J, Urch CJ. Tetrahedron Lett. 1998;39:485.Domínquez B, Pazos Y, de Lera AR. J Org Chem. 2000;65:5917. doi: 10.1021/jo9917588.
- 5.(a) Ohmura T, Awano T, Suginome M. J Am Chem Soc. 2010;132:13191. doi: 10.1021/ja106632j. [DOI] [PubMed] [Google Scholar]; (b) Awano T, Ohmura T, Suginome M. J Am Chem Soc. 2011;133:20738. doi: 10.1021/ja210025q. [DOI] [PubMed] [Google Scholar]
- 6.Sandrock DL, Jean-Gérard L, Chen C, Dreher SD, Molander GAS. J Am Chem Soc. 2010;132:17108. doi: 10.1021/ja108949w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee JCH, McDonald R, Hall DG. Nature Chem. 2011;3:894. doi: 10.1038/nchem.1150. [DOI] [PubMed] [Google Scholar]
- 8.Hoang GL, Yang Z-D, Smith SM, Pal R, Miska JL, Pérez DE, Pelter LSW, Zeng XC, Takacs JM. Org Lett. 2015;17:940. doi: 10.1021/ol503764d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blaisdell TP, Caya TC, Zhang L, Sanz-Marco A, Morken JP. J Am Chem Soc. 2014;136:9264. doi: 10.1021/ja504228p.For non-directed metal-free diboration, see: Bonet A, Pubill-Ulldemolins C, Bo C, Gulyás H, Fernández E. Angew Chem, Int Ed. 2011;50:7158. doi: 10.1002/anie.201101941.
- 10.Charles MD, Schultz P, Buchwald SL. Org Lett. 2005;7:3965. doi: 10.1021/ol0514754. [DOI] [PubMed] [Google Scholar]
- 11.Tamaru Y, Yamamoto Y, Yamada Y, Yoshida Z. Tetrahedron Lett. 1979;20:1401.Tamaru Y, Yamada Y, Inoue K, Yamamoto Y, Yoshida Z. J Org Chem. 1983;48:1286.Bei X, Hagemeyer A, Volpe A, Saxton R, Turner H, Guram A. J Org Chem. 2004;69:8626. doi: 10.1021/jo048715y.For a review, see: Muzart J. Tetrahedron. 2003;59:5789.
- 12.Kath JC, Brissette WH, Brown MF, Conklyn M, DiRico AP, Dorff P, Gladue RP, Lillie BM, Lira PD, Mairs EN, Martin WH, McElroy EB, McGlynn MA, Paradis TJ, Poss CS, Stock IA, Tylaska LA, Zheng D. Bioorg Med Chem Lett. 2004;14:2169. doi: 10.1016/j.bmcl.2004.02.021. [DOI] [PubMed] [Google Scholar]
- 13.Diaz LC, Diaz G, Ferreira AA, Meira PRR, Ferreira E. Synthesis. 2003;4:603. [Google Scholar]
- 14.Manley PJ, Miller WH, Uzinskas IN SmithKline Beecham Corporation. EP1218005 A2. Patent. 2002 Jul 3;
- 15.Isolation: Wellington KD, Cambie RC, Rutledge PS, Bergquist PR. J Nat Prod. 2000;63:79. doi: 10.1021/np9903494.
- 16.For an overview of the hamigerans, see: Nicolaou KC, Gray DLF, Tae J. J Am Chem Soc. 2004;126:613. doi: 10.1021/ja030498f.
- 17.For a review of synthesis efforts towards hamigeran B see: Miesch M, Welsch T, Rietsch V, Miesch L. Strategies and Tactics in Organic Synthesis. 2013;9:203.
- 18.Pichlmair S, de Lera Ruiz M, Basu K, Paquette LA. Tetrahedron. 2006;62:5178. [Google Scholar]
- 19.(a) Zweifel G, Arzoumanian H, Whitney CC. J Am Chem Soc. 1967;89:3652. [Google Scholar]; (b) Evans DA, Crawford TC, Thomas RC, Walker JA. J Org Chem. 1976;41:3947. doi: 10.1021/jo00887a003. [DOI] [PubMed] [Google Scholar]
- 20.Bunch L Kobernhavns Universitet, DK. WO 2014/124651 A1. International Patent. 2014 Aug 21;
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.