

Abstract
Catalytic enantioselective diboration of alkenes is accomplished with readily available carbohydrate-derived catalysts. Mechanistic experiments suggest the intermediacy of 1,2-bonded diboronates.
Graphical abstract

Catalytic enantioselective difunctionalization reactions, when they meet the requirement for broad substrate scope, high selectivity, and operationally ease, can be powerful tools for chemical synthesis.1 Enantioselective diboration is one such process that has been developed as an effective reaction that applies to terminal and internal unactivated alkenes.2 Importantly, the 1,2-bis(boronate) products can be converted to a range of useful derivatives.3 While these features are appealing, there are several constraints that currently limit the utility of olefin diboration. First, all currently available asymmetric alkene diboration reactions employ non-earth-abundant precious metal catalysts (Rh4,5, Pt6), and costs associated with these catalysts limit scale-up. Second, to render reactions stereoselective, expensive ligands are required and this similarly limits routine use. Lastly, currently-practiced metal-catalyzed diboration reactions must be conducted with exclusion of air and moisture. In this report, we describe an operationally-simple, enantioselective diboration that is catalyzed by inexpensive carbohydrate-derived glycols. Importantly, this reaction applies to a range of alkenes and is accomplished on useful scales (>10 g).
The reactivity that underlies the glycol-catalyzed diboration is grounded in recent findings that Lewis basic alkoxides can promote reaction of unactivated alkenes,7,8 ostensibly through the intermediacy of an ate complex derived from the diboron reagent (A, Scheme 1). While a study by Fernandez examined stereoinduction with chiral alkoxide promoters, the process was not catalytic (200 mol % chiral alkoxide) and achieved a maximum selectivity of 70:30 er.9 As an alternative, we considered that the boronic ester substituents (OR*) might be used catalytically to control the course of the base-promoted reaction. In our design, we speculated that the 120° O-B-O bond angle of the neopentylglycolate-derived reagent B2(neo)210 might disfavor formation of four-coordinate boron centers, thereby inhibiting tetrahedral ate complex formation and subsequent diboration reaction. Under conditions of boronate ester exchange, we considered that reversible replacement of neopentyl glycol ligands with appropriate chiral alcohols might provide more reactive boronates and would thereby comprise a paradigm for catalytic stereoselective diboration.11
Scheme 1.

Prospective Catalytic Cycle for Alcohol-Catalyzed Alkene Diboration.
Our initial experiments were influenced by Roy and Brown who observed that acyclic and cyclic 1,2-diols undergo exchange with phenyl boronic esters within minutes at room temperature.12 To determine if such exchange reactions might allow chiral alcohols to serve as catalysts for the enantioselective base-promoted diboration, a series of chiral glycols were surveyed in the reaction between 1-tetradecene and B2(neo)2 in the presence of Cs2CO3. Reactions were conducted with 30 mol% of a chiral glycol or related derivative at 60 °C in THF and were subjected to oxidative work-up (Figure 1). Whereas the background reaction in the absence of diol occurs in 47% yield, in the presence of appropriate diols, enhanced reactivity and enantioselectivity can result. Of note, chiral diol 1, a compound known to generate cyclic five-membered boronic esters from diboron precursors13, was found to facilitate a modestly selective catalytic diboration (68:32 er). Similarly, sulfonamide derivative 8, a compound whose parent cis-aminoindanol structure is well known to furnish cyclic five-membered azaborolidines14 was also a competent catalyst for a modestly selective (73:27 er) diboration. Most surprisingly, however, is that the most effective catalysts in the initial survey were found to be trans-1,2-cyclohexanediol derivatives 4 and 7. While cyclic five-membered borates derived from this framework are not unknown15, they are rare and appear to involve significant ring strain.16 Remarkably, even trans-pyrrolidine diol 5 catalyzed the asymmetric diboration reaction even though its derived five-membered cyclic boronate is likely to be prohibitively high in energy. While the features critical to catalysis with trans-1,2-cyclohexanediol (4) were not immediately apparent, this framework was selected as a lead for further development.
Figure 1.
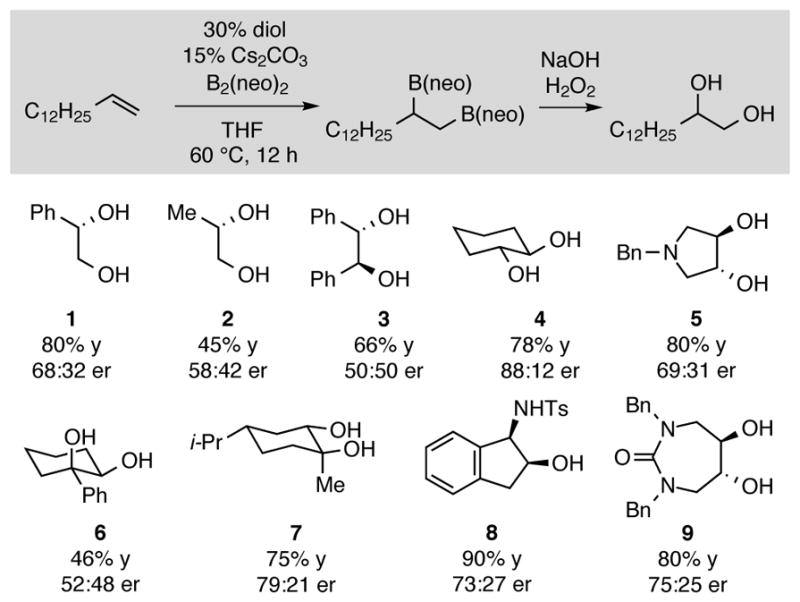
Diboration of 1-tetradecene catalyzed by 1 – 9. Yield and er data are of the diboration product after oxidation to the 1,2-diol.
To furnish catalysts with enhanced levels of stereoselection and improved practicality (diol 4 costs $238/g) carbohydrate derivatives were considered as a pool of readily available, tunable, and non-racemic cyclic 1,2-diols. These studies led to the finding that highly effective enantioselective diboration of alkenes can be accomplished with the pseudoenantiomeric glycols 6-tert-butyldimethylsilyl-1,2-dihydroglucal (TBS-DHG) and dihydrorhamnal (DHR), derived from D-glucal and L-rhamnal as depicted in Scheme 2.17 Of note, the acetate esters of D-glucal and L-rhamnal are inexpensive and they are readily converted to the derived glycol catalysts.
Scheme 2.

Preparation of TBS-DHG and DHR.a
aReagents: (a) H2, Pd/C, EtOAc; (b) lipase from Candida ru-gosa, pH=7 buffer, acetone/iPr2O; (c) TBSCl, imidazole; (d) K2CO3, CH3OH.
Optimal conditions for enantioselective diboration using TBS-DHG and DHR are depicted in Table 1. During optimization, it was found that B2(neo)2 is indeed more effective than B2(pin)2 (60% yield, 69:31 er; conditions of Table 1 with 4 as catalyst), DBU as the base afforded higher selectivity than Cs2CO3, and that reactions were more selective but slower when conducted at room temperature versus 60 °C. With these conditions, reaction of tetradecene with diol 4 (10% catalyst, 48 h) furnished the diboration/oxidation product in 35% yield and 93:7 er. In contrast, with 10% TBS-DHG catalyst, the same reaction occurs in 78% yield and 96:4 er. The pseudo-enantiomeric catalyst DHR is less efficient than TBS-DHG, a problem that was surmounted by conducting the DHR catalyzed reactions with 20 mol % loading at 35 °C for 48 h; under these conditions 1-tetradecene reacts in 70% yield and 93:7 er. As depicted in Table 1, the diboration of a number of other terminal alkenes could be conducted efficiently and with excellent levels of enantiocontrol. While styrenes appear to pose a challenge (product 14), it is notable that other non-activated terminal alkenes, including those with coordinating functional groups (ethers, esters), are processed efficiently and selectively. It is also of note that the reaction of internal alkenes, while still needing improvement, can be accomplished with the glycol-catalyzed diboration (products 20 and 21), a feature not yet observed with chiral platinum catalysts.
Table 1.
Diboration with TBS-DHG and DHR.a

|
With TBS-DHG, reactions conducted at 22 °C for 24 h (products 12, 13, 20) or 48 h (products 10, 11, 14–19, 21); with DHR, reactions conducted for 48 h at 35 °C (products 10, 13, 16, 18) or 60 °C (products 17, 20).
Isolated as the derived acetonide.
NMR analysis indicates >20:1 d.r.
Employed Cs2CO3 as base.
While pinacol boronates have been well studied in organic synthesis, the related neopentylglycolato derivatives have received considerably less attention. To determine if the B(neo) derived products would exhibit similar reactivity as the B(pin) compounds, we examined the glycol-catalyzed diboration when performed as part of a cascade reaction sequence. As shown in Figure 2a, it was found that the components of the glycol-catalyzed enantioselective diboration do not interfere in subsequent Pd-catalyzed cross-coupling reactions and that the B(neo) derivatives serve as competent partners in cascade diboration/cross-coupling sequences18 As shown, this enables single-pot transformation of terminal alkenes into non-racemic secondary alcohols. Also of note, it was determined that the diboration reaction could be conducted easily on larger scale; as depicted in eq. 1 (Figure 2b), the reaction operates effectively on 10 grams of substrate. Moreover, while reactions assembled in a dry box are more efficient, addition of MgSO4 enables reactions assembled in the open atmosphere to be conducted with useful efficiency and selectivity (eq. 2).
Figure 2.

(a) Cascade single-pot diboration/cross-coupling reactions. For 22, 24 and 25 X=Br; for 23 X=Cl. (b) Large scale and glove-box-free diboration reactions.
Several experiments give clues to the inner workings of the glycol-catalyzed diboration reaction:
When B2(neo)2 and one equivalent of styrene diol (1) were mixed in THF, mass spectrometry indicates the presence of both mono and double exchange products (Figure 3a, eq. 3). However, when exchange experiments were conducted with TBS-DHG, the double exchange product was detected but the mono exchange product was not observed (eq. 4).
While NMR analysis of the above exchange experiments indicated <2% exchange between TBS-DHG and B2(neo)2, the complexes B2(4)2 and B2(TBS-DHG)2 could be prepared for study by treatment of B2(OH)4 with the cyclic diol in the presence of 4 Å molecular sieves (i.e. Figure 3b, eq. 5). While physical data (13C NMR, mass spectrometry) of B2(4)2 is consistent with a single symmetric B2(4)2 complex, B2(TBS-DHG)2 appears to exist as more than one complex as determined by 13C NMR spectroscopy.
In connection to the catalytic reaction, addition of isolated B2(4)2 to 1-tetradecene occurs with comparable selectivity to the catalytic reaction, but requires the addition of either Cs2CO3 or DBU/neopentylglycol (eq. 6).
Figure 3.

(a) Exchange experiments with B2neo2 and 1 or TBS-DHG. (b) Preparation and reactions of B2(1)2. (c) 1,1 versus 1,2 bonding modes in neutral diboron reagents.
While Roy and Brown12 observed that a range of acyclic and cyclic cis-1,2-diols undergo rapid exchange with phenyl boronic esters at room temperature, cyclic trans-1,2-diols — presumably because of the high strain energy involved in cyclic five-membered boronates — do not undergo detectable exchange, even after 48 h. This data, in conjunction with the above observations, suggests that the active species in glycol catalyzed diboration is derived from a symmetric B2(trans-diol)2 complex, but that this complex is likely not bonded in the same manner as those derived from acyclic diol ligands. Thus, it was considered the bonding mode for trans-diols with diboron reagents may not be in the common 1,1 bonding motif but instead in the much less common 1,2 bonding arrangement.13,19 As depicted in Figure 3c, DFT calculations (M06-2X/6-31+G*; PCM solvation model with THF) are supportive of this contention: while the calculated low energy bonding mode for B2(neo)2 (eq. 8) and B2(1)2 (eq. 9) is the 1,1-mode as is observed in their X-ray crystal structure, the 1,2-bonded B2(4)2 is calculated to be 8.9 kcal/mol more stable than the 1,1-bonded form (eq. 7).
While more detailed mechanistic experiments are in progress, preliminary computational experiments have probed the ability of 1,2-bonded diboron complexes of cyclic diols to facilitate alkene diboration. With the premise that the reactive species derived from trans-1,2-cyclohexanediol is an ate complex of 1,2-bonded B2(4)2, the reaction pathway for diboration of ethylene was investigated by DFT (Figure 4, M06-2X/6-31+G*; PCM solvation model with THF, Gaussian 09, IRC analysis to confirm transition structures connect with correct ground states). With methoxide as a model Lewis base, the equilibrium between 1,1-B2(neo)2 and 1,2-B2(4)2 (Figure 4a) was investigated and found to favor 1,1-B2(neo)2 by 8.0 kcal/mol in the neutral form (eq. 10), but only 3.2 kcal/mol in the ate complex (eq. 11). Reaction of ethylene with 1,2-B2(4)2•OMe occurs by a two step sequence involving initial rupture of the B-B bond (TS-1, Figure 4b) and formation of an anionic boracycle tethered to a trivalent borate (INT). Mechanistically, this first step appears to be isoelectronic with cyclopropanation involving singlet-carbenes and is related to the mechanism proposed by Gulyás and Fernandez for reaction of 1,1-bonded B2(pin)27; however, it should be noted that Fernandez and Gulyás suggest that the boracycle does not lie on the diboration reaction coordinate. Subsequent to cycloboration, intramolecular reaction between the trivalent borate and the anionic boracycle occurs in a stereoretentive fashion and delivers a macrocyclic vicinal diboronate; ligand exchange between this species and neopentyl glycol seems plausible and would release the 1,2-glycol catalyst from the reaction product. Of note, as required for effective catalysis, reaction through TS-1 is calculated to be favored by 3.8 kcal/mol over the analogous pathway with 1,1-bonded B2(neo)2, such that even though 1,2-B2(4)2•OMe appears to be less stable, it is more reactive than the 1,1-B2(neo)2 compound and a Curtin-Hammett kinetic scenario obtains.20
Figure 4.

(a) Calculated energetics of boronic ester exchange for neutral and ate complexes. Values in kcal/mol; calculations performed by DFT (M06-2X/6-31+G*; PCM solvation model with THF, Gaussian 09). (b) Calculated reaction mechanism for alkene diboration with B2(4)2. (c) Comparison of 1,1- versus 1,2-bonded transition state structures.
While there is still much to learn about the origin of rate acceleration with 1,2-bonded versus 1,1-bonded diboron complexes, comparison of the core transition state structures of the two paths (B and C, Scheme 4c) intimates three features that may be important. First, 1,2-bonded TS-1 (C) is less hindered than the transition structure from the 1,1-bonded reagent (B). With the 1,1-bonded reagent, the methoxide ligand (R) suffers from syn pentane interactions, regardless of its orientation. Second, in 1,2-bonded TS-1 the diolate lone pairs indicated in C are well aligned with the B-B σ* and may assist with B-B bond cleavage (and donate into the developing p orbital on the adjacent boron); geometric constraints preclude this electronic interaction in the 1,1-bonding mode. Third, ring strain that appears to penalize the ground state ate complex of B2(4)2•OMe is lessened as the sp2 hybridized boron becomes pyramidalized in TS-1. Future studies will probe the importance of these effects and examine ways they might be manipulated during the design of more efficient and selective catalysts.
Supplementary Material
Acknowledgments
This work was supported by the NIH (GM-59417).
Footnotes
Supporting Information. Procedures, characterization and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interests.
References
- 1.Busacca CA, Fandrick DR, Song JJ, Senanayake CH. Adv Synth Catal. 2011;353:1825. [Google Scholar]
- 2.Takaya J, Iwasawa N. ACS Catal. 2012;2:1993. [Google Scholar]
- 3.Xu L, Zhang S, Li P. Chem Soc Rev. 2015;44:8848. doi: 10.1039/c5cs00338e. [DOI] [PubMed] [Google Scholar]
- 4.Morgan JB, Miller SP, Morken JP. J Am Chem Soc. 2003;125:8702. doi: 10.1021/ja035851w. [DOI] [PubMed] [Google Scholar]
- 5.Toribatake K, Nishiyama H. Angew Chem Int Ed. 2013;52:11011. doi: 10.1002/anie.201305181. [DOI] [PubMed] [Google Scholar]
- 6.Coombs JR, Haefner F, Kliman LT, Morken JP. J Am Chem Soc. 2013;135:11222. doi: 10.1021/ja4041016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Bonet A, Pubill-Ulldemolins C, Bo C, Gulyás H, Fer-nández E. Angew Chem Int Ed. 2011;50:7158. doi: 10.1002/anie.201101941. [DOI] [PubMed] [Google Scholar]; (b) Blaisdell TP, Caya TC, Zhang L, Sanz-Marco A, Morken JP. J Am Chem Soc. 2014;136:9264. doi: 10.1021/ja504228p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For base promoted diboration of activated alkenes, see: Lee K, Zhugralin AR, Hoveyda AH. J Am Chem Soc. 2009;131:7253. doi: 10.1021/ja902889s.
- 9.Bonet A, Sole C, Gulyás H, Fernández E. Org Biomol Chem. 2012;10:6621. doi: 10.1039/c2ob26079d. [DOI] [PubMed] [Google Scholar]
- 10.Clegg W, Elsegood MRJ, Lawlor FJ, Norman NC, Pickett NL, Robins EG, Scott AJ, Nguyen P, Taylor NJ, Marder TB. Inorg Chem. 1998;37:5289. [Google Scholar]
- 11.Selected studies in catalytic boron activation by boronate exchange: Wu TR, Chong JM. J Am Chem Soc. 2005;127:3244. doi: 10.1021/ja043001q.Pellegrinet SC, Goodman JM. J Am Chem Soc. 2006;128:3116. doi: 10.1021/ja056727a.Lou S, Moquist PN, Schaus SE. J Am Chem Soc. 2006;128:12660. doi: 10.1021/ja0651308.Wu TR, Chong JM. J Am Chem Soc. 2007;129:4908. doi: 10.1021/ja0713734.Luan Yi, Schaus SE. J Am Chem Soc. 2012;134:19965. doi: 10.1021/ja309076g.
- 12.Roy CD, Brown HC. J Organomet Chem. 2007;692:784. [Google Scholar]
- 13.Clegg W, Thorsten J, Marder TB, Norman NC, Orpen AG, Peakman TM, Quayle MJ, Rice CR, Scott AJ. J Chem Soc, Dalton Trans. 1998:1431. [Google Scholar]
- 14.Hong Y, Gao Y, Nie X, Zepp CM. Tetrahedron Lett. 1994;35:6631. [Google Scholar]
- 15.Bishop M, Bott SG, Barron AR. J Chem Soc, Dalton Trans. 2000;3100 [Google Scholar]
- 16.Sugihara JM, Bowman CM. J Am Chem Soc. 1958;80:2443. [Google Scholar]
- 17.For previous synthesis of TBS-DHG: Groaning MD, Rowe BJ, Spilling CD. Tetrahedron Lett. 1998;39:5485.
- 18.Mlynarski SN, Schuster CH, Morken JP. Nature. 2014;505:386. doi: 10.1038/nature12781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.For relevant isomerization of 1,1 and 1,2 B2(cat)2 in the presence of DBN, see: Cade IA, Chau WY, Victorica-Yrezabal I, Ingelson MJ. J Chem Soc, Dalton Trans. 2015;44:7506. doi: 10.1039/c5dt00645g.
- 20.The calculated overall barrier is too high for a reaction that occurs at room temperature. This may arise because of the use of ethylene and methoxide in the calculations, or because the calculations overestimate the decrease in translational (and to a smaller degree rotational) entropy upon complexation of ethene with B2L2OMe anion. For a review of the latter, see: Zhou H-X, Gilson MK. Chem Rev. 2009;109:4092. doi: 10.1021/cr800551w.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.