

Abstract
There is great medical need for estrogens with favorable pharmacological profiles, that support desirable activities for menopausal women such as metabolic and vascular protection but that lack stimulatory activities on the breast and uterus. Here, we report the development of structurally novel estrogens that preferentially activate a subset of estrogen receptor (ER) signaling pathways and result in favorable target tissue-selective activity. Through a process of structural alteration of estrogenic ligands that was designed to preserve their essential chemical and physical features but greatly reduced their binding affinity for ERs, we obtained “Pathway Preferential Estrogens” (PaPEs) which interacted with ERs to activate the extranuclear-initiated signaling pathway preferentially over the nuclear-initiated pathway. PaPEs elicited a pattern of gene regulation and cellular and biological processes that did not stimulate reproductive and mammary tissues or breast cancer cells. However, in ovariectomized mice, PaPEs triggered beneficial responses both in metabolic tissues (adipose tissue and liver) that reduced body weight gain and fat accumulation and in the vasculature that accelerated repair of endothelial damage. This process of designed ligand structure alteration represents a novel approach to develop ligands that shift the balance in ER-mediated extranuclear and nuclear pathways to obtain tissue-selective, non-nuclear pathway-preferential estrogens, which may be beneficial for postmenopausal hormone replacement. The approach may also have broad applicability for other members of the nuclear hormone receptor superfamily.
INTRODUCTION
Estrogens regulate many essential physiological processes and are needed for the functional maintenance of many adult target tissues within and outside of the reproductive system. They can, however, have deleterious actions in promoting breast and uterine cancers (1–4). This balance between desirable and undesirable activities in diverse target tissues offers an intriguing opportunity for the development of tissue-selective estrogens that provide a net benefit with minimal risk for menopausal hormone replacement, such as ones affording metabolic and vascular protection without stimulation of the breast or uterus. Estrogens act through estrogen receptors (ERs) by utilizing two distinct signaling pathways, the nuclear-initiated (“genomic”) pathway, wherein ER functions as a chromatin-binding ligand-regulated transcription factor, and the extranuclear-initiated (“non-genomic”) pathway, which involves kinase cascades initiated by ER action from outside the nucleus (5, 6). In this report, we used a process involving structural alteration of steroidal and non-steroidal ER ligands in ways that enhanced their selectivity for the extranuclear-initiated pathway over the nuclear-initiated ER pathway, resulting in ER ligands having effective metabolic and vascular protective activity, but lacking stimulatory activity on reproductive tissues.
The activation of specific kinases by the action of estrogens through extranuclear ER action is generally rapid and often transient (7–12), and its initiation likely requires only the input of a triggering signal by the ER-hormone complex to initiate a kinase cascade and cellular activities through the extranuclear-initiated ER signaling pathway that could collectively have important downstream cellular effects. By contrast, the activation of genes through the nuclear ER signaling pathway appears to require a sustained action of ER-hormone complexes, sufficient to effect dissociation of heat shock proteins, recruit coregulator proteins, stimulate ER binding to chromatin, alter chromatin architecture and modify histones, and activate RNA polymerase (pol) II to initiate gene transcription. ER ligands with potent nuclear ER activity form kinetically stable receptor-cofactor complexes, and coactivator binding can slow ligand dissociation rates by orders of magnitude (13). Thus, it seemed possible that ER ligands preferential for extranuclear over nuclear ER signaling might be obtained by redesigning the structures of certain estrogens in ways that would preserve their essential chemical features, a phenol and often a secondary alcohol, as well as their overall composition and geometry, but would reduce considerably their high affinity ER binding. Our aim was to preserve the ability of such a modified estrogen to have an effective interaction with ER that would be appropriate and sufficient to activate the extranuclear-initiated ER pathway, but insufficient to sustain activity through the nuclear pathway.
In this report, we describe the unique activities of such Pathway Preferential Estrogens (denoted PaPEs) that alter the balance of utilization of extranuclear and nuclear receptor-initiated signaling pathways. By selectively triggering extranuclear-initiated ER signaling pathway activities, PaPEs regulate the transcription of a subset of ER target genes and evoke biological outcomes both in cells in culture and in animals in vivo in ways that provide a favorable balance of desired vs. undesired effects. Thus, PaPEs represent novel tissue-selective estrogens that might have potential as clinically useful pharmaceuticals. The structural alteration approach we have used to design PaPEs could be applied to other nuclear hormone receptors as well.
RESULTS
A pathway preferential estrogen, PaPE-1, has impeded estrogen receptor binding and coactivator interactions
To create the pathway preferential estrogen, PaPE-1, we rearranged key elements of the steroid structure of estradiol that, while preserving its key functional groups, reduced considerably its ER binding affinity (Figure 1A). First, we deleted the B-ring of the steroid, a change that we knew from prior work on A-CD and other B-seco estrogens would cause a drop in ER binding affinity (14). (b) Second, we added the two carbon atoms lost from the B-ring deletion to the A-ring, flanking both sides of the phenol; this type of ortho substitution sterically impedes the important hydrogen bonds between the phenol and the glutamate and arginine residues (Glu353 and Arg394) in the ER ligand-binding pocket and also reduces receptor binding (15). Finally, we converted the C-ring to a benzene, simplifying the structure by eliminating the C-18 methyl group and at the same time creating a biphenyl system, a ligand core element that is present in various non-steroidal estrogens (16, 17). From a comparison of their properties (Figure 1A), it is evident that both E2 and the structurally permuted estrogen, PaPE-1, have closely matched estrogenic functional groups and physical properties (composition, shape, size, volume, lipophilicity, and polar surface area) but very different binding affinities for ERα and ERβ.
Figure 1. Structures and molecular and binding properties of E2 and four PaPEs, and a model of E2 and PaPE-1 binding to ERα ligand-binding domain.
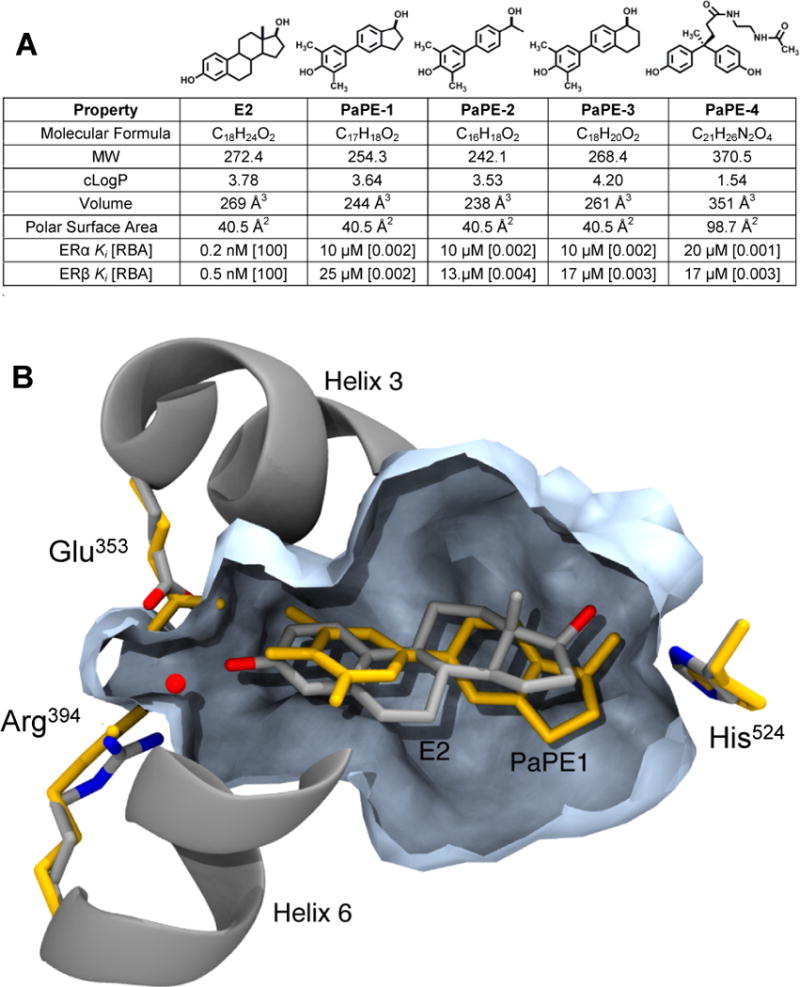
(A) MW is molecular weight; cLogP is Log10 of the calculated octanol-water partition coefficient; Volume is molecular volume; Polar Surface Area is a measure of compound polarity. All values were obtained using ChemBioDraw Ultra (ver. 13.0.0.3015). Relative binding affinity (RBA) values were determined by competitive radiometric binding assays (18). E2 is set at 100 on both ERs. Ki values calculated as Ki = Kd (for E2) × (100/RBA), where Kd of E2 is 0.2 nM (ERα) and 0.5 nM (ERβ). Values are average of 3–4 determinations with coefficients of variation <0.3. (B) Computational model comparing PaPE-1 and E2 in the ligand-binding pocket of ERα. The model of ERα+E2, based on a crystal structure (PDB ID 1GWR), has E2 and helical elements shown in silver/grey and the pocket volume contour in slate blue. The model for PaPE-1 was generated from the ERα+E2 structure by progressive transformation of the ligand structure from E2 to PaPE-1, partnered with progressive minimization of the ligand and the ligand-binding domain. The resulting positions of the PaPE-1 ligand and hydrogen bonding residues are shown in orange.
The relative binding affinity (RBA) values of E2 and PaPE-1 (Figure 1A) were obtained by a competitive radiometric binding assay using purified full-length human ERα and ERβ (18). The RBAs for E2 are defined as 100, and relative to E2, PaPE-1 binds 50,000-fold less well to ERα and ERβ (Supplementary Figure S1A). The equivalent KD values are 10 and 25 µM for PaPE-1 on ERα and ERβ, respectively, compared to the sub-nanomolar KD values for E2. Thus, our design greatly lowered the ER binding affinities of PaPE-1 for both ERs and also greatly increased its dissociation rate from ERα (Supplementary Figure S1B), while preserving as much as possible the physical and functional group attributes of E2.
A computational model showing the structural details of how the ligand-binding pocket of ERα accommodates PaPE-1 in comparison to E2 is presented in Figure 1B. Helices 3 and 6 constrict the A-ring end of the binding pocket, and E2 (silver-gray) forms hydrogen bonds to both Glu353 and Arg394, further stabilized by a crystallographic water (red dot) bridging these two residues. By contrast, the ortho-methyl groups of PaPE-1 (in yellow) introduce steric clashes with H3 and H6, inducing a substantial shift in ligand positioning. While the A-ring OH of PaPE-1 maintains a hydrogen bond to Glu353 and the crystallographic water still bridges between Glu353 and Arg394, direct ligand contact to Arg394 is broken (ER structure in yellow). At the D-ring end of the pocket, there is also a subtle shift in the positioning of His524. Overall, it appears that the reduced volume of the PaPE-1 ligand and the increased planarity due to the aromatic C-ring mimetic result in fewer van der Waals contacts throughout the central region of the pocket. All of these changes with respect to the ER-E2 complex are consistent with the greatly lower binding affinity of PaPE-1. Nevertheless, the overall shape of the ligand-binding pocket is not altered in a major way by the binding of PaPE-1, suggesting that PaPE-1 can still form a structurally competent complex with ER.
Using a time-resolved Förster resonance energy transfer (tr-FRET) assay we have previously developed, we monitored the binding of the steroid receptor coactivator 3 (SRC3) to ERα (19, 20). In coactivator titration assays (Supplementary Figure S1C), SRC3 bound with high affinity to ERα complexes with E2, but showed no binding to ERα complexes with PaPE-1. Notably, however, the antagonist trans-hydroxytamoxifen (OH-Tam) reversed the ER-SRC3 interaction promoted by E2; PaPE-1 also reversed the ER-SRC3 interaction, but only at much higher concentrations, commensurate with its lower ERα binding affinity (Supplementary Figure S1D. The E2-elicited interaction of ERα with SRC3 was also observed by coimmunoprecipitation of the complexes from MCF-7 cells, whereas no coimmunoprecipitated complexes were observed for ERα and SRC3 after exposure to PaPE-1 (Supplementary Figure S1E).
PaPE-1 regulates a subset of estrogen-modulated genes and activates kinases, but does not stimulate breast cancer cell proliferation
In earlier studies of an estrogen-dendrimer conjugate (EDC) that activates ER non-genomic signaling with high selectivity (8, 10, 21, 22), we identified a set of genes in MCF-7 human breast cancer cells whose expression was increased effectively by either extranuclear ER action and by nuclear ER action, as well as ones that required only nuclear action by ER (10). We found that PaPE-1 was selective in activating extranuclear-initiated ER-regulated genes, as shown by LRRC54 stimulation, but was essentially without activity on the nuclear-initiated ER gene target PgR (Figure 2A). Activation of LRRC54 gene expression by PaPE-1 was blocked by treatment with the antiestrogen (Fulvestrant) ICI 182,780 (Figure 2B) and by knockdown of ERα (Figure 2C and Supplementary Figure S2). By contrast, knockdown of GPR30 did not affect gene stimulation (Figure 2C and Supplementary Figure S2). PaPE-1 also did not stimulate proliferation of MCF-7 cells, whereas E2 potently stimulated proliferation (Figure 2D).
Figure 2. Comparison of regulation of gene expression, cell proliferation, and pathway signaling by PaPE-1 and E2.
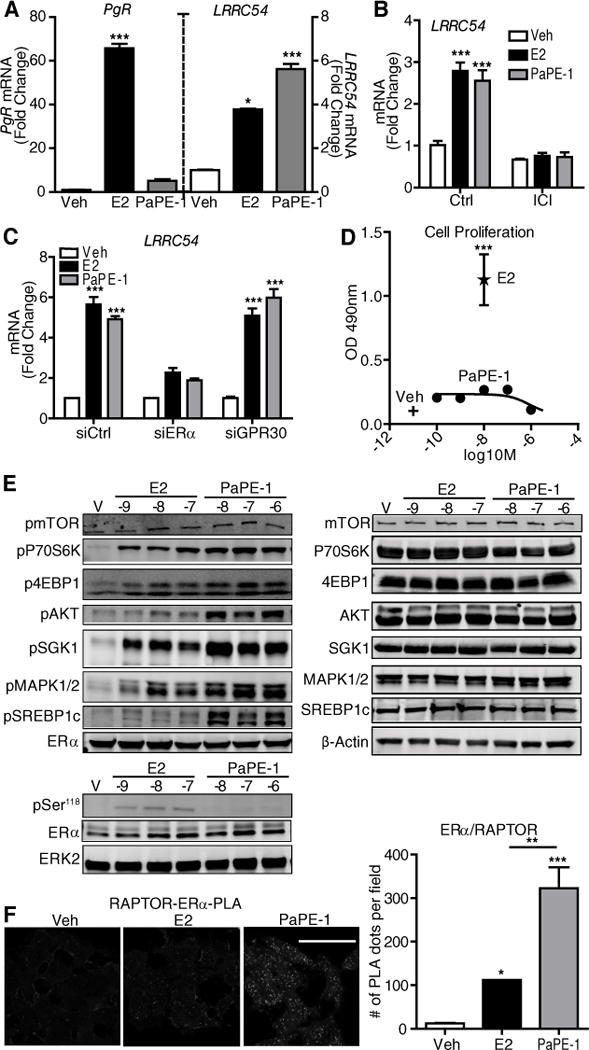
(A) PaPE-1 preferentially activates extranuclear-initiated genes (LRRC54) over nuclear genes (PgR) compared to E2 in MCF-7 cells. Cells were treated with control vehicle (Veh), E2 or PaPE-1 and gene expression was monitored by qPCR. N=3 biological replicates. (B) MCF-7 cells were pretreated with ICI182,780 (ICI) and then treated with Veh, E2 or PaPE-1 in the presence or absence of ICI. RNA was isolated and subjected to qPCR analysis for the indicated genes. N=3 biological replicates. (C) MCF-7 cells transfected with siCtrl, siERα or siGPR30 were treated with Veh, E2 or PaPE-1. RNA was isolated and subjected to qPCR analysis. N=3 biological replicates. (D) MCF-7 cells were treated with Veh, E2 or the indicated concentrations of PaPE-1 for 6 days and proliferation was monitored by WST assay. N=4 biological replicates. (E) PaPE-1 selectively activated mTOR and MAPK signaling in MCF-7 cells. Left: Cells were treated with control Vehicle (V), or the indicated concentrations of E2 or PaPE-1 for 15 min (upper panel) and 45 min (lower panel) and Western blots were done to assess the activation of signaling proteins and phosphorylation (p) of Ser118 in ERα. Total ERα was monitored, and total ERK2 was used as a loading control. N=3 biological replicates. Right: Total amount of these factors in cells receiving the indicated treatments. N=3 biological replicates. (F) PaPE-1 induces interaction between ERα and Raptor. MCF-7 cells were treated with E2 or PaPE-1. Cells were crosslinked and incubated with ERα and Raptor antibodies overnight and PLA was performed. Quantitation of signal intensities are shown in the graph. N=3 biological replicates. Two-way ANOVA, Bonferroni posttest, * p<0.05, ** p<0.01, *** p<0.001. Scale bar, 50μm.
When we monitored activation of major signaling pathways in MCF-7 cells, we observed that PaPE-1 efficiently activated mTOR and MAPK signaling (Figure 2E), as seen by increased phosphorylation of P70S6K and 4EBP1 which are associated with mTORC1 activation, increased phosphorylation SGK1 which is associated with mTORC2 activation, and increased phosphorylation of MAPK. PaPE-1 increased phosphorylation of AKT and SREBP1 to a greater extent than E2. However, PaPE-1 did not detectably induce phosphorylation of Ser118 in ERα, which was observed with E2 (Figure 2E) and is associated with the mitogenic activity of ERα (23).
The mTOR pathway is the major signaling system that senses the nutrient state of the environment and modulates metabolic functions in the cell. mTOR kinase is present in two distinct complexes, mTORC1 and mTORC2, which are distinguished by the presence of RAPTOR and RICTOR scaffolding proteins, respectively. It is thought that mTORC1 primarily modulates cell metabolism, whereas mTORC2 is principally involved in regulating the cytoskeleton and cell proliferation (24, 25). To further characterize the mTOR activation by PaPE-1, we performed proximity ligation assays (PLAs) in MCF-7 cells and found that ERα interacted with RAPTOR to a greater extent in the presence of PaPE-1 than E2 (Figure 2F). We did not detect any interaction between ERα and mTOR, or ERα and proline-rich Akt substrate of 40 kDa (PRAS40), which is important in Akt and mTOR signaling, suggesting that ERα modulated the mTOR pathway through direct interactions with RAPTOR (Supplementary Figure S3).
PaPE-1 regulates metabolism-related genes in an mTOR and MAPK activity-dependent manner, but does not cause recruitment of ERα to chromatin
Next, we performed RNA-Seq analysis to compare genes that were changed in their expression by PaPE-1 and/or E2. We treated MCF-7 cells with 10 nM E2 or 1 μM PaPE-1 for 4h and 24h, and compared genes regulated by each compound at these two time points (Figure 3A). At both times, E2 regulated the expression of more genes than did PaPE-1, and the magnitude of gene regulation by E2 was generally higher than that by PaPE-1. At 4h, E2 regulated nearly 1500 genes, whereas PaPE-1 modulated expression of only 500 genes (Figure 3A upper Venn diagram). At 24h, about 3000 genes were regulated by E2, whereas PaPE-1 regulated ~2200 genes (Figure 3A lower Venn diagram). At both 4h and 24h, approximately three quarters of the genes regulated by PaPE-1 were also targets of E2. Notably, only E2 increased the expression of mitosis genes and decreased the expression of apoptosis genes at 24 h (Supplementary Table 1), consistent with our observation that PaPE-1 did not stimulate MCF-7 cell proliferation.
Figure 3. PaPE-1 and E2 regulate common as well as distinct groups of genes in MCF-7 cells.
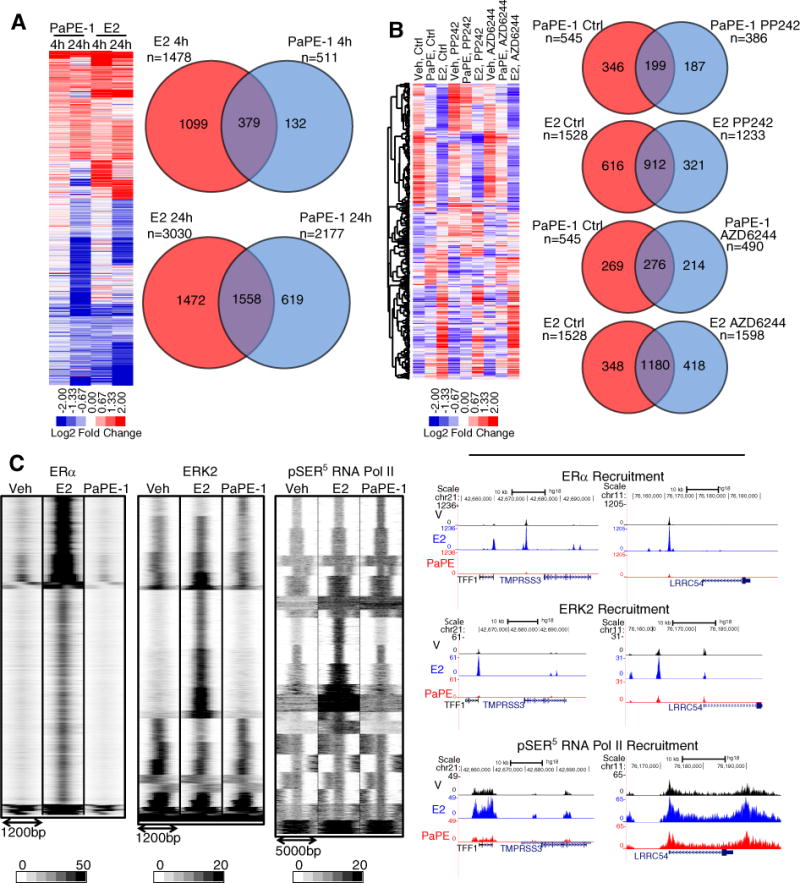
(A) Cells were treated with E2 or PaPE-1 for 4h and 24h. RNA was isolated and RNA-Seq was performed. Regulated genes are considered to be those with P<0.05 and expression fold change >2. N=2 biological replicates. (B) PaPE-1 mediated gene expression changes are sensitive to mTOR and MAPK pathway inhibitors. Effect of mTOR and MAPK inhibitors on gene regulation by E2 and PaPE-1 in MCF-7 cells. Cells were pretreated with PP242 or AZD6244 before treatment with E2 or PaPE-1 in the presence or absence of inhibitors. RNA was isolated and RNA-Seq performed. (P<0.05, fold change >2). N=2 biological replicates. (C) PaPE-1 does not induce recruitment of ERα or ERK2 to chromatin but stimulates recruitment of RNA Pol II. MCF-7 cells were treated with E2 or PaPE-1. ChIP-Seq was performed for the indicated proteins. UCSD genome tracks of cistromes in the presence of E2 or PaPE-1 are shown (right panel). N=3 biological replicates that were pooled.
Next, we used pathway-selective inhibitors to assess the effect of mTOR or MAPK pathway activation on PaPE- and E2-mediated gene regulation. The mTOR pathway inhibitor PP242 blocked the regulation of ~60% of PaPE-1 regulated genes, but only 40% of E2 regulated genes. Similarly, the MAPK inhibitor AZD6244 blocked almost 50% of PaPE-1 regulated genes, but only ~23% of E2 regulated genes (Figure 3B). Examination of enriched gene ontology (GO) functions revealed that these inhibitors blocked E2 regulation of cell migration, immune, cell cycle and angiogenesis related genes, whereas they blocked PaPE-1 regulation of genes involved in nucleotide metabolism, inflammatory response, ncRNA processing, amino acid transport, and glycoprotein metabolism (Supplementary Table 1).
To investigate the roles of ERα, ERK2 and active RNA Pol II recruitment in PaPE-1-mediated transcriptional events, we performed ChIP-Seq analysis which revealed that ERα was recruited to chromatin upon treatment of cells with E2, but not with PaPE-1 (Figure 3C, Supplementary Table 2). PaPE-1 induced recruitment of ERK2 to the proximal promoter regions of genes, whereas E2 caused recruitment of ERK2 to enhancer regions together with ERα. We observed distinct RNA Pol II recruitment sites with PaPE-1, further suggesting that PaPE-1 induces transcriptional events through recruitment of RNA Pol II without affecting ERα or ERK2 recruitment to enhancer regions of target genes. These distinct patterns of ERα, ERK2, and phosphorylation of Ser5 in RNA pol II binding after cell treatment with Veh, E2, and PaPE-1 are shown for the TFF1/pS2 and LRRC54/TSKU genes in Figure 3C, right. The lack of ERα and ERK2 recruitment by PaPE-1 may be associated with the lack of effect of PaPE-1 on the phosphorylation of Ser118 in ERα (Figure 2E).
PaPE-1 is a tissue-specific modulator of ER and mTOR signaling, with preferential estrogen-like activity in non-reproductive (metabolic and vascular) tissues
We next characterized the in vivo biological activities of PaPE-1 in ovariectomized female mice. Similar to E2, PaPE-1 was cleared rapidly after subcutaneous injection (t1/2 ca. 1 h, Supplementary Figure S4A); so, to provide more prolonged exposure for in vivo studies, we used either administration by ALZET minipump or by a pellet containing PaPE-1, both implanted subcutaneously (Supplementary Figure S4B). Mice at 8 weeks of age were ovariectomized, and after 2 weeks, they were treated with control vehicle, E2 or PaPE-1 for 3 weeks. While E2 was effective in increasing growth of the uterus, PaPE-1 did not exhibit any stimulatory effects on uterine weight (Figure 4A). As has been previously demonstrated (26), we observed that E2 induced marked involution of the thymus, but PaPE-1, even at a high dose, had no effect on thymus size (Figure 4A). Likewise, PaPE-1 did not change mammary gland ductal morphology from that of vehicle control mice, whereas E2 elicited marked ductal growth (elongation and branching) (Figure 4B). Of interest, however, both E2 and PaPE-1 reduced the mammary gland adiposity that develops in mice after ovariectomy. This effect was associated with reduced adipocyte area after E2 or PaPE-1 treatment (Figure 4C).
Figure 4. Unlike E2, PaPE-1 does not change uterus or thymus weight and does not induce mammary gland ductal branching but like E2, PaPE-1 reduces mammary gland adipocyte area.
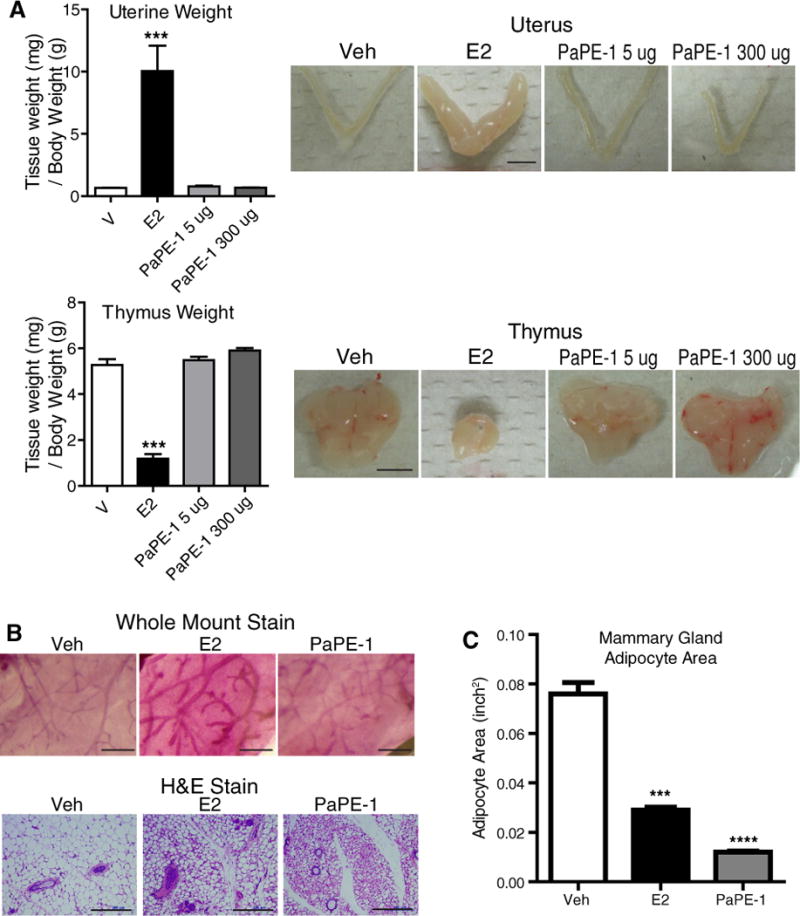
(A) PaPE-1 does not affect uterus or thymus weight. C57BL/6 mice were ovariectomized and then were given daily subcutaneous injections of E2 or were implanted with PaPE-1 pellets for 4 days. Weights of uterus and thymus were monitored. N=8 mice per treatment. Scale bars, 5 mm. (B) PaPE-1 stimulates minimal mammary ductal elongation but it greatly reduces adipocyte size in mammary gland. Ovariectomized C57BL/6 mice were implanted with E2 or PaPE-1 pellets. Whole mount stain (scale bars, 1 mm) and H&E stain (scale bars, 200 μm) of mammary gland are shown. (C) Mammary gland adipocyte area was calculated from the H&E images. N=4 mice per treatment. A one-way analysis of variance (ANOVA) model was fitted to assess the contribution of ligand treatment on uterine weight, thymus weight or mammary gland adipocyte area. When the main effects were statistically significant at α=0.05, pairwise t-tests with a Bonferroni correction were employed to identify the treatments that were significantly different from each other. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
Body weights of the mice increased after ovariectomy, as has been previously reported, and this increase in weight was suppressed by PaPE-1 and by E2 over the 3-week period monitored (Figure 5A). Body weights were statistically lower in E2- and PaPE-1-treated mice than in Veh control treated animals, and the PaPE-1 and E2 groups were not statistically different from one another. The reduction in body weight gain that we observed with PaPE-1 was not due to a change in food consumption (Figure 5B). Because of the anorexigenic effect of E2, there was an early drop in food intake in E2-treated animals, an effect that was not observed in PaPE-1-treated animals, suggesting that this effect did not trigger the differences in body weights and changes in the fat depots.
Figure 5. Like E2, PaPE-1 reduces the increase in body weight after ovariectomy and reduces adipose stores and blood triglyceride concentrations.
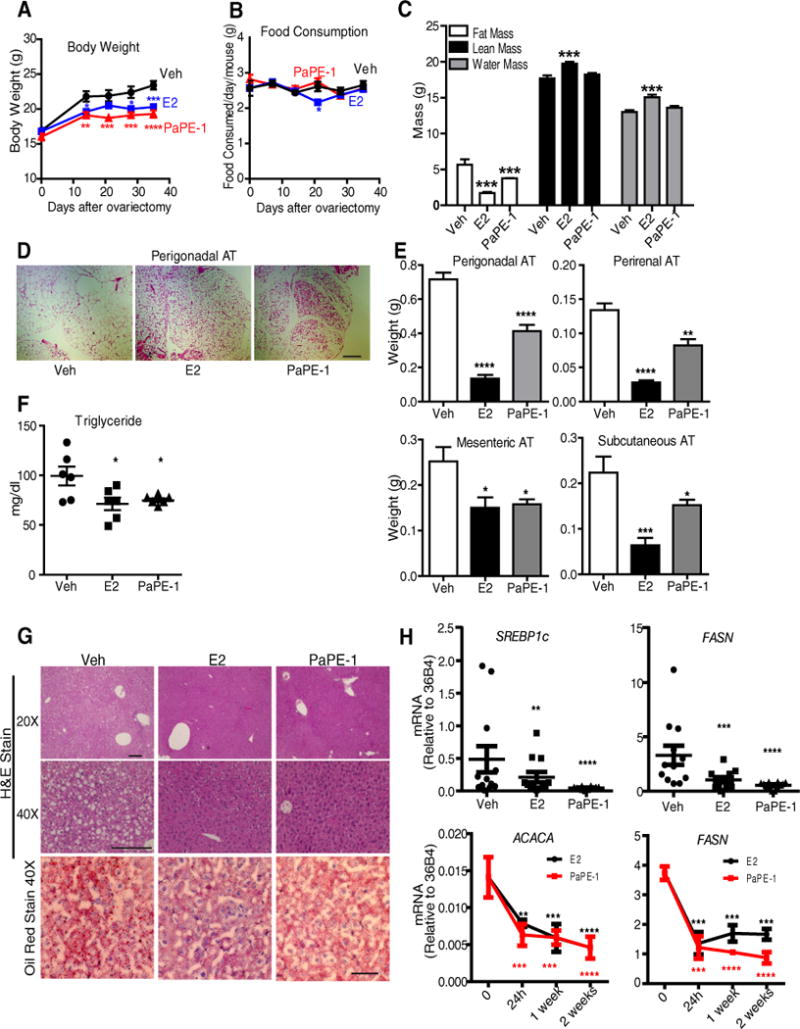
(A) PaPE-1 is effective in normalizing body weight after ovariectomy. Ovariectomized C57BL/6 mice were implanted with pellets containing E2 or PaPE-1 or Veh control for 3 weeks (n = 8 mice/group). Animals were on a normal chow diet. Two-way ANOVA, Bonferroni posttest, * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001, comparing treatments to Veh. (B) Food consumption for each mouse from A was monitored weekly. (C) Body composition for each mouse from A was monitored using EchoMRI at the end of 3 weeks. One-way ANOVA, Newman-Keuls post-test, * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001. (D) H&E staining of perigonadal adipose tissue (AT). Images are representative of 8 mice per group. Scale bar, 500 μm. (E) Weights of various adipose tissue (AT) depots after 3 weeks of control vehicle or ligand exposure. (F) Triglycerides were measured in the blood of animals (n=6 mice/group) at the end of 3 weeks of Veh, E2 or PaPE-1 treatment. (G) H&E staining (upper 2 rows; scale bars, 100 μm) and Oil Red O staining (lower panel; scale bar, 20 μm) of the liver after 3 weeks of treatment. Images are representative of 8 mice per group. (H) Gene expression analysis of SREBP1c and FASN in the liver at 3 weeks (upper panel) (n=12 mice/group), and time course of FASN and ACACA expression in the livers of E2 and PaPE-1 treated mice (n=3 mice/group). A two-way analysis of variance (ANOVA) model was fitted to assess the contribution of ligand treatment and time of ligand treatment on body weight, food consumption or gene expression. A one-way analysis of variance (ANOVA) model was fitted to assess the contribution of ligand treatment on fat mass, lean mass, water mass, triglyceride concentrations, weight of different fat depots or gene expression. When the main effects were statistically significant at α=0.05, pairwise t-tests with a Bonferroni correction were employed to identify the treatments that were significantly different from each other. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
A major impact of PaPE-1 was on fat depots of the animals, as observed in EchoMRI measurements. Both E2 and PaPE-1 greatly reduced fat mass, but total lean mass and water mass were increased only by E2 and were not affected by PaPE-1, further indicating that PaPE-1 worked by changing overall adiposity rather than lean mass of the animals (Figure 5C). H&E and Oil Red O staining of perigonadal adipose tissue (Figure 5D) highlighted the similar and marked estrogenic effects of both PaPE-1 and E2 on this tissue. PaPE-1 and E2 decreased the weight of all of the fat depots examined (Figure 5E). The weights of the perigonadal, perirenal, and subcutaneous adipose tissues were reduced by both ligands, but to a larger extent by E2 at the dosages tested. Mesenteric adipose tissue weight was similarly reduced by both ligands, and both ligands also decreased triglyceride concentrations in blood (Figure 5F).
Ovariectomy resulted in increased lipid droplet accumulation in the liver, and E2 or PaPE-1 decreased hepatocyte lipid content, as observed in Oil Red O staining of liver sections (Figure 5G). We verified changes in fatty acid synthesis pathways by monitoring two biomarkers of hepatic steatosis, FASN (27, 28) and SREBP-1c (29, 30), and found that expression of these genes was reduced following 3 weeks of E2 or PaPE-1 treatment (Figure 5H). Expression of SREBP-1 transcriptional targets, FASN and the gene encoding the enzyme acetyl-CoA carboxylase alpha (ACACA), which catalyzes the carboxylation of acetyl-CoA to malonyl-CoA, the rate-limiting step in fatty acid synthesis, was decreased by 24h of PaPE-1 or E2 treatment, and remained low over the 2 week monitoring period (Figure 5H).
PaPE-1 and E2 show similarities and differences in gene expression and signaling pathway activations in multiple tissues in vivo
PaPE-1 induced tissue-specific molecular changes in gene expression and signaling pathway activation patterns. To characterize these changes in the various tissues harvested from animals in long-term studies, we performed analysis of the expression of genes reported in the literature to be altered in liver, skeletal muscle, perigonadal fat, pancreas, and uterus. PaPE-1 and E2 generated quite similar profiles in metabolic tissues, namely in liver and skeletal muscle; in contrast, only E2 induced gene expression changes in uterus (Figure 6A), consistent with the selective stimulatory actions of PaPE-1 in non-reproductive tissues.
Figure 6. Gene regulation and signaling pathway activations by PaPE-1 and E2 in tissues in vivo.
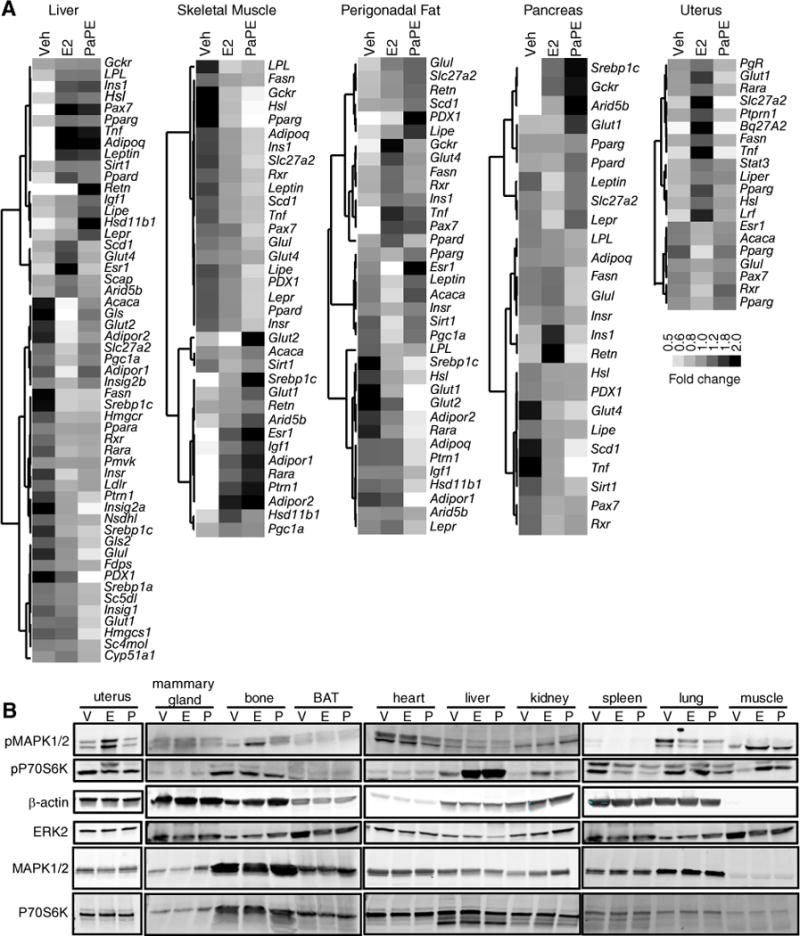
(A) Ovariectomized C57BL/6 mice were implanted with pellets containing E2 and PaPE-1. Liver, skeletal muscle, perigonadal fat, pancreas, and uterus were harvested. RNA was isolated and qPCR performed for the indicated genes. N=8 mice/treatment. (B) PaPE-1 activates mTOR signaling (as monitored by increased phosphorylation of S6) in liver and skeletal muscle. Ovariectomized C57BL/6 mice were injected with E2 (E) or PaPE-1 (P) for 2h. The indicated tissues were collected and subjected to Western blot analysis for phosphorylated (p) S6 and MAPK1/2. β-Actin and total ERK2 were used as loading controls. Total MAPK1/2 and total P70S6K are also shown. N=3 mice/treatment.
To gain more mechanistic insight, we profiled the signaling pathways activated by E2 and PaPE-1 in different tissues (Figure 6B). E2 and PaPE-1 had similar pathway effects in non-reproductive tissues, whereas E2, but not PaPE-1, stimulated reproductive tissues (uterus and mammary gland). Furthermore, in metabolic tissues such as liver and skeletal muscle, both E2 and PaPE-1 were effective in inducing mTOR signaling, as observed by S6 phosphorylation.
PaPE-1 activity is lost in ERα-knockout mice
A select number of effects of E2 and PaPE-1 on physiologic processes and gene expression were compared in wild-type and ERα-knockout mice (ERKO), in which the complete ERα gene was deleted (Supplementary Figure S5). OVX mice were treated with E2 or PaPE-1 for 3 weeks, and the ability of E2 or PaPE-1 to diminish the body weight gain after ovariectomy was lost in the ERKO mice (Figure S5A). As expected, E2 did not stimulate uterine growth in ERKO mice (Supplementary Figure S5B), and E2 or PaPE-1 did not decrease blood triglycerides in these mice (Supplementary Figure S5C). Furthermore, ERα mediated the suppression of FASN and SREBP1c gene expression by PaPE-1 or E2 in the liver of wild-type mice, an effect that was lost in ERKO mice (Supplementary Figure S5D).
PaPE-1 and E2 show beneficial vascular effects
Estrogens have potential beneficial actions on vascular cells, as exemplified by in vivo studies of carotid artery reendothelialization following perivascular electric injury in female mice. Following ovariectomy, mice were treated with vehicle, E2 or PaPE-1 for 18 days, at which time E2 and PaPE-1-treated mice were also administered a single dose of vehicle or the antiestrogen ICI 182,780. Three days later, carotid artery denudation was performed, and the mice received a second dose of vehicle or ICI while E2 or PaPE-1 was continued, and 72h after denudation Evans blue dye was administered systemically to assess the remaining area of denudation in the intimal surface of the carotid artery (Figure 7A, upper panel). E2 and PaPE-1 caused similar marked endothelial repair, as indicated by the minimal area of remaining denudation, and the responses elicited by E2 and PaPE-1 were fully prevented by the antiestrogen ICI 182,780 (ICI, Fulvestrant) (Figure 7A, lower panel). In the same animals, uterine weight, which was greatly elevated by E2, was unaffected by PaPE-1, and as expected, the antiestrogen ICI blocked uterine stimulation by E2 (Figure 7B).
Figure 7. Like E2, PaPE-1 elicits repair of the vascular endothelium after injury, an effect that is prevented by the antiestrogen ICI.
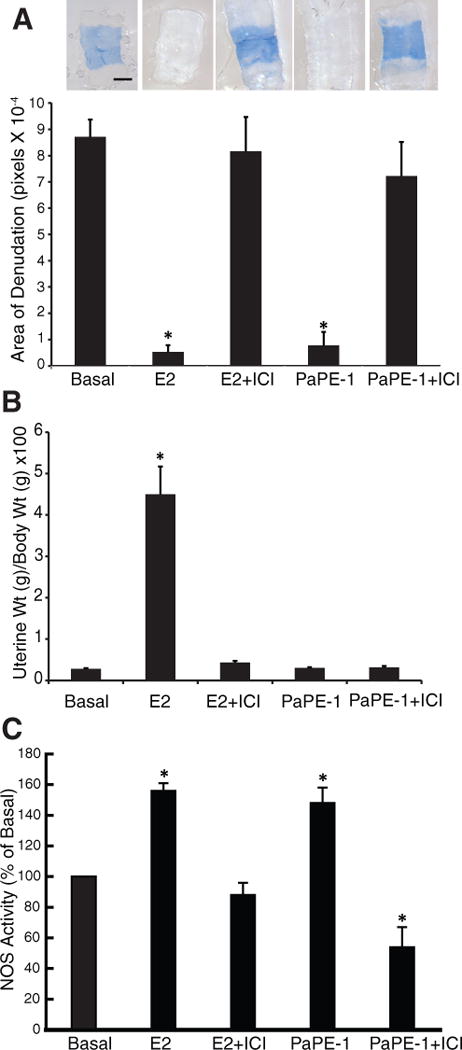
(A) Carotid artery reendothelialization after an injury that denudes the endothelial layer in ovariectomized mice treated with PaPE-1 or E2 in the absence or presence of the antiestrogen ICI 182,780 (ICI) (N=6–9 mice/group). *, p<0.05 compared to basal control. Scale bar, 400 μm. (B) E2, but not PaPE-1, increases uterine weight, an effect that is blocked by ICI. N=7 mice/treatment. (C) eNOS stimulation by E2 and PaPE-1 in the presence and absence of the antiestrogen ICI 182,780 in BAECs. N= 4 biological replicates. *, p<0.05 compared to control.
To evaluate direct effects on endothelium, endothelial NO synthase (eNOS) activation was assessed by measuring the conversion of 14C-L-arginine to 14C-L-citrulline by intact primary bovine endothelial cells in culture. eNOS was activated by E2, as previously observed (22), and there was a comparable response to PaPE-1; the effects of both E2 and PaPE-1 were fully attenuated by ICI (Figure 7C).
Additional PaPEs show pathway- and tissue-selective activities similar to those of PaPE-1
To further exemplify compounds that preferentially activate ER non-genomic signaling, we present results from PaPE-2 and PaPE-3, two ligands that are structurally related to PaPE-1 by being altered forms of the steroidal ligand, estradiol, but having variations in the structure of what was originally the estradiol D-ring, namely ring cleavage in PaPE-2 and ring enlargement in PaPE-3 (Figure 1A). To further diversify the structures of PaPEs, we also studied an additional ligand, PaPE-4; this PaPE is derived from a non-steroidal estrogen, bisphenol A (BPA), and it retains the core bisphenol structure of BPA but has been modified to have reduced ER binding affinity by replacing one of the methyl groups with a polar bis-amide substituent. The binding and physical properties of PaPE-2, -3, and -4 are given in Figure 1A.
All of the PaPEs showed similar biological activities in vitro and in vivo. They all caused preferential stimulation of extranuclear-initiated over nuclear gene (LRRC54 > PgR) compared to E2 (10), which efficiently stimulated expression of both genes (Figure 8A). In contrast to E2, none of the PaPEs stimulated proliferation of MCF-7 cells over a broad concentration range tested (Figure 8B). All of the PaPEs increased activation of MAPK, mTOR and AKT signaling pathways, as monitored by the phosphorylation of MAPK, P70S6 and SREBP1, and AKT in these cells (Figure 8C and 8D). In endothelial cells, PaPE-2, -3, and -4 also increased NOS activity (Figure 8E), similar to E2 and PaPE-1 (Figure 7C), and the NOS stimulation was fully blocked with the antiestrogen ICI. In vivo, the four PaPEs and E2 reduced body weight gain after ovariectomy (Figure 8F), without altering food consumption (Figure 8G). In contrast, unlike E2, the four PAPEs did not elicit increases in uterine weight (Figure 8H). The reduction in body weight with the four PaPEs was largely due to a change in body fat mass, with little or no change in lean mass or water mass, as monitored by EchoMRI. E2 reduced fat mass more markedly, and did increase lean mass and water mass (Figure 8I). These differential effects may account for the fact that the body weight of E2-treated animals matched that of the PaPE-treated animals.
Figure 8. Assessment of the activities of PaPE-2, PaPE-3, and PaPE-4 in MCF-7 cells, in bovine aortic endothelial cells (BAECs), and in mice.
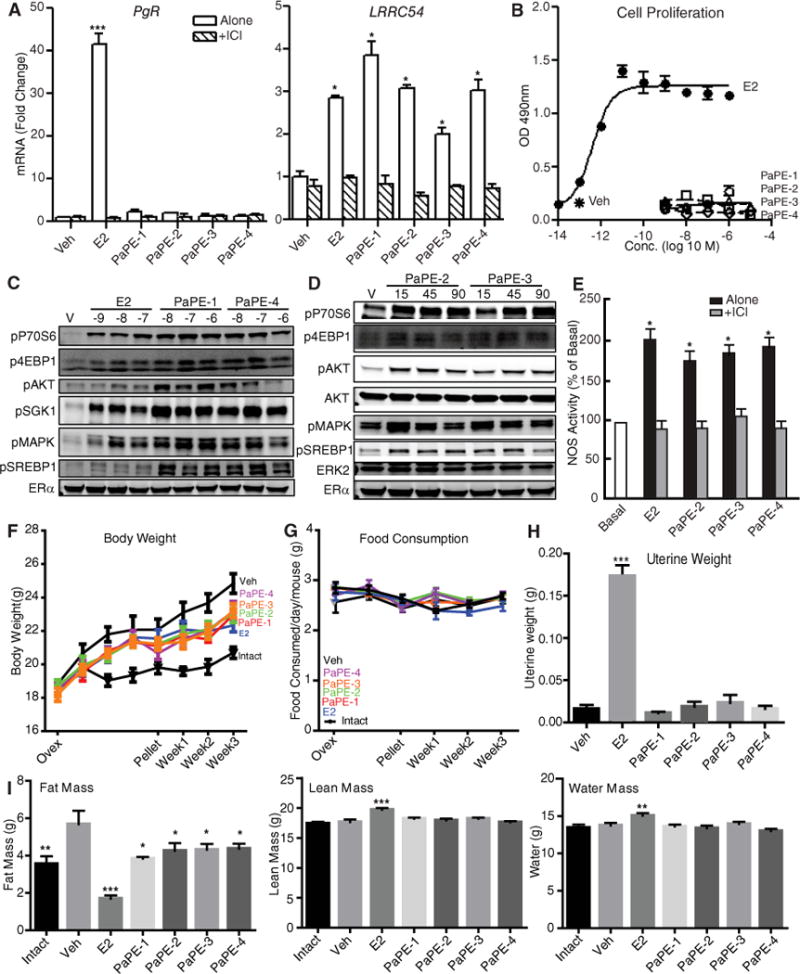
(A) Assessment of extranuclear-initiated LRRC52 gene expression compared to direct nuclear PgR gene expression in MCF-7 cells treated for 4 hours with control vehicle, E2, or the indicated PaPE. N=4 biological replicates. (B) Proliferation of MCF-7 cells after treatment with different concentrations of E2 or the PaPE for 6 days. N=4 biological replicates. (C) Stimulation of various cell signaling pathways by different concentrations of E2, PaPE-1, or PaPE-4 after 15 min in MCF-7 cells. ERα abundance is also shown. N=3 biological replicates. (D) Time course of cell signaling pathway activations by PaPE-2 or PaPE-3, monitored at the indicated times. ERα abundance is shown, and total ERK2 is used as a loading control. N=2 biological replicates. (E) Stimulation of NOS activity during 15 min treatment of BAECs with ligand either alone or with ICI 182,780. N= 4 biological replicates. (F) The PaPEs and E2 reduce weight gain after ovariectomy in C57BL/6 mice. Ovariectomized animals received pellets of E2, the PAPE, or vehicle, and body weight was monitored over the next 3 weeks. A group of intact non-ovariectomized mice were included for comparison. N= 4 mice/group. A two-way analysis of variance (ANOVA) model was fitted to assess the contribution of ligand treatment and time of ligand treatment on body weight or food consumption. When the effects were statistically significant at α=0.05, pairwise t-tests with a Bonferroni correction were employed to identify the treatments that were significantly different from each other. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001. (G) Food consumption of the mice in (F) was monitored over time. (H) Assessment of uterine weight gain in the ovariectomized C57BL/6 mice in (F) after 3 weeks of receiving E2 or PaPE-1, -2, -3 and -4. One-way ANOVA, Newman-Keuls post-test, * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001. (I) Fat mass, lean mass, and water mass were measured by EchoMRI at the end of the 3-week treatment period in the mice in (F). A one-way analysis of variance (ANOVA) model was fitted to assess the contribution of ligand treatment on gene expression, fat mass, lean mass and water mass. When the main effects were statistically significant at α=0.05, pairwise t-tests with a Newman-Keuls correction were employed to identify the treatments were significantly different from each other. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
DISCUSSION
The preferential extranuclear-initiated pathway activity of PaPEs results in a distinctive pharmacological outcome, favoring beneficial metabolic and vascular effects over reproductive tissue stimulation
In this study, we present a new approach to develop small molecule estrogens with a favorable profile of tissue-selective activity through preferential activation of the extranuclear-initiated signaling pathway over the nuclear-initiated pathway. This type of estrogen molecule was derived through a structural alteration process designed to preserve essential physical and functional features of estradiol (E2) or BPA, but purposefully lessen the high affinity binding required for nuclear ER signaling.
Our analyses of cellular pathways and gene expression changes in response to PaPE-1 and E2 treatments indicated that E2 and the PaPE-1 ligand regulated certain common as well as various different groups of genes and biological processes. In MCF-7 breast cancer cells, PaPE-1 regulated genes involved in nucleotide, amino acid and lipid metabolism, and induced RNA Pol II recruitment to chromatin, but PaPE-1 did not induce ERα or ERK2 recruitment to gene enhancers or stimulate expression of proliferation-associated genes, as seen with E2. However, like E2, PaPE-1 strongly activated MAPK and mTOR pathways, and PaPE-1 relied on these pathways for a considerable fraction of its gene regulation, based on the effect of MAPK and mTOR inhibitors. These results suggest a role of for kinase pathways for some of the transcriptional responses we observed, and that these kinases might be working through other transcription factors that are downstream of MAPK or mTOR signaling (such as CREB) to affect gene regulation.
The preferential regulation of cellular responses through the extranuclear signaling pathway by PaPE-1 and the three other PaPEs resulted in distinctive biological outcomes, favoring metabolic actions in liver and adipose tissues and actions on the vasculature, with negligible if any stimulatory activity in reproductive tissues. Thus, PaPE-1 lacked estrogen-like activity on uterus, thymus and mammary gland, but closely mimicked the actions of E2 in metabolic tissues and on the vasculature. Regarding the impact on adipose sites, it is notable that both PaPE-1 and E2 reduced central fat depots (perigonadal and perirenal) that are associated with the metabolic syndrome and obesity in postmenopausal women (31). All four PaPEs suppressed body weight gain after ovariectomy, although suppression by E2 was more marked and was accompanied by an increase in lean mass and water mass not seen with PaPEs. PaPE-1 greatly reduced expression of liver genes associated with lipid synthesis (such as FASN and ACACA), and decreased blood triglyceride concentrations. Like E2, PaPE-1 also decreased the expression of the mRNA encoding the transcription factor SREBP1c, which plays a key role in lipogenesis. These effects required ERα and were lost in ERαKO mice.
The design of PaPEs represents a new approach for developing estrogens with pathway and tissue-selective activity
Previous efforts to prepare agents having tissue-selective activity have utilized three different mechanisms: differential interaction with coregulators, differing activity on the two ER subtypes, and restricted tissue, cellular or sub-cellular distribution. SERMs (selective estrogen receptor modulators, such as tamoxifen, raloxifene, and bazedoxifene) have different amounts of partial agonist-antagonist activity in different target tissues, a result of differential engagement of coregulator proteins in a target cell- and gene-specific manner (32). ER subtype-selective ligands were developed to exploit the distinctly different tissue distributions of ERα and ERβ (33) so as to regulate the different sets of genes and physiological outcomes controlled by these two ERs (9, 34, 35), a major focus being on ERβ-selective compounds to avoid the proliferative drive of ERα in reproductive tissues (36–38).
To activate the extranuclear-initiated pathways of ER action in preference to nuclear-initiated actions, we originally developed a synthetic hormone-polymer conjugate, denoted estrogen dendrimer conjugate (EDC), that bound well to ER but had little genomic activity because it was excluded from the nucleus due to its charge and polymeric size (8, 10). While EDC provided robust ER-mediated protection against vascular injury and cortical bone loss without stimulation of the uterus and breast cancer (21, 22), its polymeric nature complicates its development as a pharmaceutical agent. Other approaches have been taken to obtain selective estrogen action by restricting the sites of hormone action (39). Recent studies using transgenic animals with ER engineered to have only nuclear effects (NOER mouse (40) or C451A-ERα mouse (41)) or extranuclear, membrane-initiated effects (MOER mouse (42)) have further highlighted that preferential activation of one or the other of these pathways can result in distinctive, often beneficial patterns of selectivity (40, 43–45); genetic engineering, however, is not a pharmaceutical approach. Hence, as described in this paper, we were prompted to undertake a different approach to discover small molecules that would have preferential activity on the extranuclear signaling pathway and might be used ultimately as novel pharmaceutical agents in women for hormone replacement therapy without detrimental effects on reproductive tissues.
Little is known in any system about the intimate molecular details of how a ligand binds to and dissociates from a receptor, and how the dynamics of these physical interactions are coupled to the dynamics of downstream signal transduction events. In general, as binding affinity drops, the rate of ligand dissociation increases whereas the rate of ligand association changes comparatively little. This has been characterized in some detail with a number of ER ligands having a spectrum of affinities (46). Despite wide differences in their binding affinities, ER agonist association rate constants all fell within the relatively narrow, 5-fold range (1.3–6.0 × 106 M−1 sec−1), whereas their dissociation rates covered a >5,000 fold range (from 5 × 10−5 to 2.7 × 10−1 sec−1), with rates being inversely related to ligand binding affinities. These dissociation rates corresponded to a half-life of the ER-ligand complex that ranged from several hours to under 1 minute for ligands with progressively decreasing affinities (46).
To investigate how ER ligand binding dynamics were coupled with signal transduction pathways, we altered the structure of the steroidal estrogen E2 to produce PaPE-1, -2, and-3, and modified the structure of the non-steroidal estrogen BPA to produce PaPE-4, with a particular aim in mind. We wanted to retain, as much as possible, the key physical, functional and structural features of active estrogens, so that they would still be recognized by the ER ligand binding domain, but to greatly lower the affinity of their interaction with ER.
We anticipated that the short lifetime of these ER-PaPE complexes might be sufficient to activate extranuclear signaling pathways but would be less effective in activating nuclear action, the former requiring the triggering of kinase cascades, but the latter requiring an ER-ligand complex of sufficient lifetime to alter chromatin architecture and promote transcription. In fact, by our measurements, the dissociation rates of PaPE-1 and E2 for ERα differed by nearly 2000 fold; the half-life of the ER-E2 complex was nearly 30 hours, whereas that of the PaPE-1 complex was less than 1 minute (Supplementary Figure S1B). These measurements were done with purified ER proteins in a cell-free system, and one should consider that, in a cellular context, the interaction of ER-ligand complexes with other proteins, such as coregulators, or signaling or scaffold proteins, such as seen with RAPTOR in these studies, might modulate the ligand binding properties of PaPEs.
There have been other attempts to produce estrogens having pathway selective activity (47, 48), but these compounds had either mixed endocrine effects or had limited selectivity and in vivo efficacy (49–51). Thus, the structural permutation and modification approaches we used to generate PaPEs appear to provide a robust way to produce small molecules having distinct pathway-preferential activity that is matched by a pattern of favorable biological actions. In fact, all four of the PaPEs described in this report provide favorable actions in metabolic and vascular tissues by selective activation of signaling pathways critical for ERα action in these tissues, yet they fail to activate these pathways and increase growth of the uterus or proliferation of mammary tissue. The tissue-selective actions of the PaPEs appear to result from the greater retention of their activity through the extranuclear-initiated pathway than through the nuclear pathway.
Other features of the PaPEs are of note: While the affinity of PaPE-1 and the other PaPEs for ER is approximately 50,000-fold less than that of E2, we were able to stimulate extranuclear ER effects using only a ca. 500-fold excess of PaPE-1 over E2, an observation suggesting that the extranuclear signaling pathway might depend to a lesser extent on the affinity of ligand for the receptor than the nuclear pathway. Also, whereas PaPE-1, -2, and -3, which were patterned after E2, all have physical characteristics (lipophilicity, polar surface area, volume, and so on) similar to that of E2, PaPE-4 is considerably larger and more polar than E2 and the other PaPEs, yet PaPE-4 has biological activities similar to those of the other three PaPEs, suggesting that the class of PaPE-like compounds can cover a rather broad range of physical and structural characteristics.
Our studies with PaPEs establish a new conceptual framework for the development of novel pathway preferential estrogens that might have promise as clinically useful pharmaceuticals for estrogen replacement therapy to improve the metabolic and vascular health of postmenopausal women without risk to the breast or uterus. Furthermore, our ligand structural permutation process, which defined a distinct class of tissue-selective, pathway-selective estrogens, might also have broad applicability to other nuclear hormone receptors such as the glucocorticoid and vitamin D receptors, and liver X receptor beta, for example, which are similar to ER in mediating both nuclear and extra-nuclear processes (52). Changes that reduce affinity but preserve essential bioactive structural features of drugs for other classes of drug receptors that operate through more than one signaling pathway might enable the development of drug analogs with an altered balance of cellular signaling pathway utilization and possibly favorable selective activity.
MATERIALS AND METHODS
Cell Culture and siRNA Treatments
MCF-7 cells were obtained from and grown as recommended by the American Type Culture Collection. Receptor abundance was measured by qPCR and Western blotting, and gene expression and proliferative response to E2 was monitored regularly. For experiments with E2 and PaPE treatment, cells were maintained in phenol red-free tissue culture medium for at least 5 days prior to use. ERα knockdown experiments utilized the SMART pool of 4 siRNAs from Dharmacon and siRNA from Santa Cruz Biotechnologies and were performed as described (10) with 30 nM siCtrl or siERα or siGPR30 for 72 h and resulted in knockdown of the corresponding mRNA and protein by greater than 80%. Primary bovine aortic endothelial cells were harvested, maintained and employed as previously described (22).
Animals and Ligand Treatments
Studies used wild-type and ERα knockout mice in C57BL/6 background. All experiments involving animals were conducted in accordance with National Institutes of Health standards for the use and care of animals, with protocols approved by the University of Illinois at Urbana-Champaign and the University of Texas Southwestern Medical Center. Wild-type C57BL/6 mice were purchased from Jackson Laboratories/National Cancer Institute. ERα knockout mice with complete excision of ERα and wild type littermates were obtained from Taconic and were used as described previously (53, 54).
In studies of metabolic parameters and gene expression in vivo, ligands were administered to ovariectomized recipient mice by subcutaneous implantation of pellets containing compound mixed with cholesterol to a total weight of 20 mg. Animals were single-housed during the study. For 3-week studies, estradiol (E2, Sigma-Aldrich) dosage (0.125 mg/pellet) was chosen based on our previous findings (38). In carotid artery reendothelialization experiments, compounds were delivered at a dose of 6 μg/day by a 4-week (Model 2006) Alzet minipump, as described (22). Total body weight and food intake were monitored every week after ovariectomy. Body fat and lean mass composition were monitored at the end of 3 weeks using EchoMRI-700 Body Composition Analyzer (Echo Medical Systems, Houston, Texas), which enables one to quantify longitudinal body composition in the live animal.
Western Blotting, and ChIP Assays
Western blot analysis used specific antibodies for ERα (HC-20, Santa Cruz); ERK2 (D-2, Santa Cruz); and pS6, pS6K, pmTOR, pRAPTOR, pRICTOR, pMAPK (Cell Signaling). Coimmunoprecipitation assays used antibodies for SRC3 (Santa Cruz, C-20) and ERα (Santa Cruz, F10). ChIP assays were carried out as described (9, 11). Antibodies used were for ERα (HC20), ERK2 (Santa Cruz, D2 and C14), and pSer5 RNA Pol II (Santa Cruz, sc-47701). ChIP DNA was isolated using QIAGEN PCR purification kit and used for ChIP-seq analysis and quantitative real-time PCR (qPCR). qPCR was used to calculate recruitment to the regions studied, as described (9).
ChIP-seq Analysis and Clustering
For genome-wide ChIP-seq, the ChIP DNA was prepared from 3 biological replicates and pooled. Libraries were prepared according to Illumina Solexa ChIP-seq sample processing methods, and single read sequencing was performed using the Illumina HiSeq 2000. Sequences generated were mapped uniquely onto the human genome (hg18) by Bowtie2 (9, 11). MACS (Model-based Analysis of ChIP-Seq) algorithm was used to identify enriched peak regions (default settings) with a p-value cutoff of 6.0e-7 and FDR of 0.01, as we have described (9). The seqMINER density array method with a 500-bp window in both directions was used for the generation of clusters, or groups of loci having similar compositional features (9). BED files for each cluster were used for further analysis with Galaxy Cistrome integrative analysis tools (Venn diagram, conservation, CEAS).
RNA-Seq Transcriptional Profiling
For gene expression analysis, total RNA was extracted from 3 biological replicates for each ligand treatment using Trizol reagent and further cleaned using the RNAeasy kit (QIAGEN). For time course studies, MCF-7 cells were treated with Veh (0.1% EtOH), 10 nM E2 or 1 μM PaPE for 4 h and 24 h. For inhibitor studies, MCF-7 cells were pretreated with Ctrl (0.1% DMSO), 1 μM PP242 or 1 μM AZD6244 for 30 min and then treated with Veh, 10 nM E2 or 1 μM PaPE-1 in the presence or absence of inhibitors for 4 h. Once the sample quality and replicate reproducibility were verified, 2 samples from each group were subjected to sequencing. RNA at a concentration of 100 ng/μL in nuclease-free water was used for library construction. cDNA libraries were prepared with the mRNA-TruSeq Kit (Illumina, Inc.). Briefly, the poly-A containing mRNA was purified from total RNA, RNA was fragmented, double-stranded cDNA was generated from fragmented RNA, and adapters were ligated to the ends.
The paired-end read data from the HiSeq 2000 were processed and analyzed through a series of steps. Base calling and de-multiplexing of samples within each lane were done with Casava 1.8.2. FASTQ files were trimmed using FASTQ Trimmer (version 1.0.0). TopHat2 (version 0.5) (55) was employed to map paired RNA-Seq reads to version hg19 of the Homo sapiens reference genome in the UCSC genome browser (56) in conjunction with the RefSeq genome reference annotation (57). Gene expression values (raw read counts) from BAM files were calculated using StrandNGS (version 2.1) Quantification tool. Partial reads were considered and option of detecting novel genes and exons was selected. Default parameters for finding novel exons and genes were specified. DESeq normalization algorithm using default values was selected. Differentially expressed genes were then determined by fold-change and p-value with Benjamini and Hochberg multiple test correction for each gene for each treatment relative to the vehicle control (9). We considered genes with fold-change >2 and Benjamini-Hochberg adjusted p-value less than or equal to 0.05 as statistically significant, differentially expressed. All RNA-Seq datasets have been deposited with the NCBI under GEO accession number GSE73663.
Motif and GO Category Analysis
Overrepresented GO biological processes were determined by the web-based DAVID Bioinformatics Resources database (58), ClueGO and web-based GREAT software (59). Motifs enrichment analysis was done using Seqpos (60).
Immunohistochemistry (IHC)
Hematoxylin and eosin (H&E) staining, and whole mount staining were performed on paraffin-embedded tissue sections (61). Images were quantified by monitoring average cell size from 3 randomly chosen fields in Fiji software (http://fiji.sc/wiki/index.php/Fiji).
Immunofluorescence Microscopy, Proximity Ligation Assays in Cells and Data Analysis
Cells treated with Vehicle (0.1% EtOH), 10 nM E2 or 1 μM PaPE-1 for 15 min were washed in PBS, fixed on glass coverslips and incubated with antibodies against ERα (F10, Santa Cruz), pSer5 RNA Pol II (Santa Cruz, 47701), or RAPTOR (Cell Signaling). The next day, a proximity ligation assay (PLA) was performed using the Duolink In Situ kit (Olink Bioscience) according to the manufacturer’s instructions, as described (62). Briefly, overnight incubation with primary antibodies was followed by hybridization with two PLA probes at 37 °C for 1 h, and then by ligation for 15 min and amplification for 90 min at 37 °C. A coverslip was mounted on each slide and image acquisition and analysis conducted. Samples were imaged using a 63×/1.4 Oil DIC M27 objective in a Zeiss LSM 700 or 710 laser scanning confocal microscope. Images were obtained in a sequential manner using a 488 Ar (10 mW) laser line for PLA signal. The individual channels for DAPI and PLA signal were obtained using a sequential scanning mode to prevent bleed-through of the excitation signal. Laser power, gain and offset were kept constant across the samples and scanned in a high resolution format of 512×512 or 1024×1024 pixels with 2/4 frames averaging. Further quantification of the images used Fiji software (http://fiji.sc/wiki/index.php/Fiji) (63). Briefly, images were converted to 8 bit for segmentation for each channel, and images were background subtracted using a rolling-ball method, with a pixel size of 100 and segmented using the DAPI channel.
Cell Proliferation Assays
Cells were seeded at 1000 cells/well in 96-well plates. On the second day, the cells were treated with 0.1% ethanol vehicle, 10 nM E2 or PaPE at the concentrations indicated and proliferation was assessed using WST-1 reagent (Roche) as described (9).
eNOS Activation
eNOS activation was assessed in intact primary endothelial cells by measuring 14C-L-arginine conversion to 14C-L-citrulline over 15 min using previously reported methods (22). Cells were treated with vehicle (yielding basal activity), E2, or PaPE alone or with the antiestrogen ICI 182,780 at the concentrations indicated.
Pharmacokinetic Analyses
For short-term studies, ovariectomized C57BL/6 mice were injected subcutaneously with 100 μg of PaPE-1 in 100 μl DMSO. Three mice were sacrificed at each time point, and 400 μl of blood was obtained from the abdominal aorta. Samples were centrifuged at 2000 g for 10 min, and serum was collected. A 50 μl portion of each sample was submitted to the Metabolomics Center at the University of Illinois for analysis. For longer-term studies, ovariectomized C57BL/6 mice were implanted subcutaneously with a pellet fabricated with 8 mg PaPE-1 and 12 mg cholesterol. Blood samples (30 μl) were collected by tail snipping every week until the third week of treatment. Samples were centrifuged at 2000 g for 10 min and serum was collected. Serum (10 μl) was mixed with 40 μl of PBS and submitted to the Metabolomics Center for analysis. For analysis, a mass standard of PaPE-1 (labeled with three deuterium atoms) was added to each sample before analysis by liquid chromatography-mass spectrometry using the 5500 QTrap with Agilent 1200 HPLC.
Carotid Artery Reendothelialization
Carotid artery reendothelialization was studied following perivascular electric injury in mice by assessing Evans blue dye uptake 72 h after injury. Endothelial denudation and recovery after injury in this model have been confirmed by immunohistochemistry for von Willebrand Factor (64). At the time of ovariectomy at 8–9 weeks of age, female mice received intraperitoneal osmotic minipumps prepared to deliver 6 μg/d E2 or PaPE-1. Carotid artery denudation was performed 21d later. In select studies additional treatments included subcutaneous injections of vehicle or ICI 182,780 (360 μg/mouse) administered 3d prior to carotid injury and on the day of injury. At the end of the study, uteri were also harvested and weighed.
Ligand Dissociation Assays
The fluorescence polarization or anisotropy characteristics of fluorescein attached to C530 in ERα, which is sensitive to the nature of the bound ligand, can be exploited to detect the dissociation of one ligand and the association of a second ligand, with the rate of ligand exchange being limited by the rate of dissociation of the initially bound ligand (65). PaPE-1 gives an anisotropy value about 20% lower than that of E2 when bound to ER, and OH-Tam gives an 80% lower anisotropy value than E2 when bound to the ER; so, these differences can be used to monitor the rate of dissociation of the initially bound ligand.
ERα-ligand binding domain (LBD), mutated to have one active cysteine at Cys530, was site-specifically labeled with 5-iodoacetamidofluorescein (65) and then diluted into t/g (50 mM Tris, 10% glycerol, pH 8) buffer with 0.01 M mercaptoethanol and 0.03 mg/ml ovalbumin added as a carrier protein to give 2 nM ER. To minimize homoFRET, a 5-fold excess (10 nM) of unlabeled ERα-LBD (10nM) was added, and the fluorescein-labeled and unlabeled ER dimers were allowed to exchange at room temperature in the dark, for 1 h, thereby producing dimers in which essentially only one monomer is fluorescein labeled. The ER was then bound with 100 nM E2, or 100 nM/RBA of PaPE-1; the RBA of PaPE-1 is 0.002%; therefore, 100 nM/RBA = 50 μM of PaPE-1 was used. These samples were allowed to complete ligand binding at room temperature in the dark for 2.5 h.
The anisotropy was measured on a Spex fluorolog II cuvette-based fluorimeter under constant wavelength conditions. The excitation was set at 488 nm and emission at 520 nm, under magic angle conditions, and three to five time points were taken for a zero time. To initiate the dissociation of PaPE-1, 300 nM of E2 was added to the PaPE-1 sample, and the time course of dissociation was subsequently followed by changes in anisotropy. Due to glycerol viscosity changes, there is a change in the protein anisotropy between room temperature and 5 °C; therefore, care was taken to prechill the protein as well as the cuvette chamber to 5 °C.
To measure the dissociation of E2, 300 nM E2 was added to the pre-exchanged sample of fluorescein-labeled and unlabeled apo-ER dimer and allowed to bind for 2.5 h, as above. The cuvette and chamber were chilled to 5 °C, and after taking the zero time points, E2 dissociation was initiated by adding 5 μM OH-Tam, and change in anisotropy was followed with time. The data for both dissociation experiments were fitted to an exponential decay function by linear regression using Prism 4.
Computational Modeling of the Complex of ERα with PaPE-1 or E2
Starting from the ERα+E2 crystal structure (PDB code: 1GWR), the structure preparation routine in MOE (MOE: Molecular Operating Environment, Chemical Computing Group) was used to fill missing loops, side chains, add explicit hydrogen atoms. A custom volume visualization code was used to create the binding volume for the ERα+E2 structure, shown in slate blue in Fig. 1B; the red dot is a structural water. The model of the binding of PaPE-1 was built by progressive generation of the PaPE-1 ligand structure from that of E2, coupled with progressive minimization of the ligand-binding domain: Atoms were first deleted from E2 to open the B-ring and convert the C ring into an aromatic ring, but the two ortho-methyl groups on the A-ring were not yet added. At this stage, the positions of the ligand oxygen atoms and all protein atoms were fixed, and energy minimization was performed using the MMFF94x force field with a termination gradient cutoff of 0.1 kcal/(mol·Å) to obtain a low energy conformation of the PaPE-1 ligand core. All atoms were then unfixed, and energy minimization was further performed while constraining protein backbones to optimize interactions with hydrogen bonding side chains. After the two A-ring ortho-methyl groups were added, another energy minimization was performed with constrained backbone atoms, and then a final unconstrained energy minimization was performed, all to the same gradient cutoff. The resulting positions of the ligand and hydrogen bonding residues are shown in yellow in Fig. 1B.
Statistical Analyses
Data from in vivo animal metabolism studies were analyzed using either a one-way ANOVA model to compare different ligand effects or a two-way-ANOVA model to compare time dependent changes. For every main effect that was statistically significant at α = 0.05, pairwise t-tests were conducted to determine which treatment levels were significantly different from each other. For these t-tests, the Bonferroni correction was employed to control the experimentwise type I error rate at α=0.05 followed by Bonferroni post hoc test using GraphPad Prism 6 for Windows (GraphPad Software, La Jolla, CA,). Differences were considered statistically significant if * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
Supplementary Material
Figure S1. Estrogen receptor and coactivator binding and interaction assays with ligands and ligand dissociation rates.
Figure S2. Verification of siRNA results in Fig. 2C with another siRNA.
Figure S3. PLAs with E2 and PaPE-1.
Figure S4. Pharmacokinetic studies for analysis of blood concentrations of PaPE-1 after injection or pellet implantation.
Figure S5. Effects of PaPE-1 require ERα.
Table S1. List of genes differentially expressed by cell treatment with E2 and PaPE-1.
Table S2. BED files for ERα, ERK2, and pSer5 RNA-Pol II ChIP-Seq data from experiments with MCF-7 cells.
Acknowledgments
We thank Dr. Alvaro Hernandez of the University of Illinois Keck Biotechnology Center for assistance with RNA-Seq analyses, and Rosa Ventrella, Luke Petry, Francis Lee and Liwen Xu for technical assistance.
FUNDING: This work was supported by a grant from The Breast Cancer Research Foundation (BSK), and by NIH grants R37 DK015556 (JAK), P50AT006268 (BSK) from the National Center for Complementary and Integrative Health (NCCIH), the Office of Dietary Supplements (ODS) and the National Cancer Institute (NCI), and the National Institute of Food and Agriculture, U.S. Department of Agriculture, award ILLU-698-909 (to ZME), and NIH HL087564 (PWS), the Associates First Capital Corporation Distinguished Chair in Pediatrics at UT Southwestern (to PWS), by a fellowship to HZ from Sun Yat Sen University, China, and by Division of Intramural Research, NIEHS 1ZIAES070065 (to KSK). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health, NCCIH, ODS, NCI, or the U.S. Department of Agriculture.
Footnotes
AUTHOR CONTRIBUTIONS: Z.M.E., S.H.K., P.W.S., J.A.K., and B.S.K. conceived and designed the project; Z.M.E., P.G., Y.C.Z., H.Z., K.L.C., K.E.C., and C.G.M. performed the experiments; K.S.K. provided key reagents and advice; all authors discussed and analyzed data. Z.M.E., P.W.S., J.A.K., and B.S.K. wrote the manuscript; all authors added parts and comments. All authors read and approved the manuscript.
COMPETING INTERESTS: The authors declare that they have no competing interests.
Data and Materials Availability: All RNA-Seq datasets have been deposited with the NCBI and are available under GEO accession number GSE73663. ChIP-Seq data for ERα, ERK2 and pSer5 RNA-Pol II from experiments with MCF-7 cells treated with Vehicle, E2 or PaPE-1 can be found as BED files in Supplementary Table S2. A United States Provisional Patent Application (serial number 62/275,416) entitled as “Estrogen Derived Compositions and Methods of Using the Same” was filed January 6, 2016 by the University of Illinois and the University of Texas Southwestern Medical Center.
REFERENCES AND NOTES
- 1.Bai ZL, Gust R. Breast Cancer, Estrogen Receptor and Ligands. Archiv Der Pharmazie. 2009;342:133–149. doi: 10.1002/ardp.200800174. [DOI] [PubMed] [Google Scholar]
- 2.Deroo BJ, Korach KS. Estrogen receptors and human disease. Journal of Clinical Investigation. 2006;116:561–570. doi: 10.1172/JCI27987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fox EM, Davis RJ, Shupnik MA. ER beta in breast cancer - Onlooker, passive player, or active protector? Steroids. 2008;73:1039–1051. doi: 10.1016/j.steroids.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Osipo C, Liu H, Meeke K, Jordan VC. The consequences of exhaustive antiestrogen therapy in breast cancer: Estrogen-induced tumor cell death. Experimental Biology and Medicine. 2004;229:722–731. doi: 10.1177/153537020422900804. [DOI] [PubMed] [Google Scholar]
- 5.Banerjee S, Chambliss KL, Mineo C, Shaul PW. Recent insights into non-nuclear actions of estrogen receptor alpha. Steroids. 2014;81:64–69. doi: 10.1016/j.steroids.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Hammes SR, Levin ER. Minireview: Recent advances in extranuclear steroid receptor actions. Endocrinology. 2011;152:4489–4495. doi: 10.1210/en.2011-1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clark S, Rainville J, Zhao X, Katzenellenbogen BS, Pfaff D, Vasudevan N. Estrogen receptor-mediated transcription involves the activation of multiple kinase pathways in neuroblastoma cells. J Steroid Biochem Mol Biol. 2014;139:45–53. doi: 10.1016/j.jsbmb.2013.09.010. [DOI] [PubMed] [Google Scholar]
- 8.Harrington WR, Kim SH, Funk CC, Madak-Erdogan Z, Schiff R, Katzenellenbogen JA, Katzenellenbogen BS. Estrogen dendrimer conjugates that preferentially activate extranuclear, nongenomic versus genomic pathways of estrogen action. Molecular endocrinology. 2006;20:491–502. doi: 10.1210/me.2005-0186. [DOI] [PubMed] [Google Scholar]
- 9.Madak-Erdogan Z, Charn TH, Jiang Y, Liu ET, Katzenellenbogen JA, Katzenellenbogen BS. Integrative genomics of gene and metabolic regulation by estrogen receptors alpha and beta, and their coregulators. Mol Syst Biol. 2013;9:676. doi: 10.1038/msb.2013.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Madak-Erdogan Z, Kieser KJ, Kim SH, Komm B, Katzenellenbogen JA, Katzenellenbogen BS. Nuclear and extranuclear pathway inputs in the regulation of global gene expression by estrogen receptors. Molecular endocrinology. 2008;22:2116–2127. doi: 10.1210/me.2008-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Madak-Erdogan Z, Lupien M, Stossi F, Brown M, Katzenellenbogen BS. Genomic collaboration of estrogen receptor alpha and extracellular signal-regulated kinase 2 in regulating gene and proliferation programs. Mol Cell Biol. 2011;31:226–236. doi: 10.1128/MCB.00821-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vasudevan N, Kow LM, Pfaff DW. Early membrane estrogenic effects required for full expression of slower genomic actions in a nerve cell line. Proc Natl Acad Sci U S A. 2001;98:12267–12271. doi: 10.1073/pnas.221449798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gee AC, Carlson KE, Martini PG, Katzenellenbogen BS, Katzenellenbogen JA. Coactivator peptides have a differential stabilizing effect on the binding of estrogens and antiestrogens with the estrogen receptor. Molecular endocrinology. 1999;13:1912–1923. doi: 10.1210/mend.13.11.0373. [DOI] [PubMed] [Google Scholar]
- 14.Wright JS, Shadnia H, Anderson JM, Durst T, Asim M, El-Salfiti M, Choueiri C, Pratt MA, Ruddy SC, Lau R, Carlson KE, Katzenellenbogen JA, O’Brien PJ, Wan L. A-CD estrogens. I. Substituent effects, hormone potency, and receptor subtype selectivity in a new family of flexible estrogenic compounds. J Med Chem. 2011;54:433–448. doi: 10.1021/jm100513m. [DOI] [PubMed] [Google Scholar]
- 15.Anstead GM, Carlson KE, Katzenellenbogen JA. The estradiol pharmacophore: ligand structure-estrogen receptor binding affinity relationships and a model for the receptor binding site. Steroids. 1997;62:268–303. doi: 10.1016/s0039-128x(96)00242-5. [DOI] [PubMed] [Google Scholar]
- 16.Lesuisse D, Albert E, Bouchoux F, Cerede E, Lefrancois JM, Levif MO, Tessier S, Tric B, Teutsch G. Biphenyls as surrogates of the steroidal backbone. Part 1: synthesis and estrogen receptor affinity of an original series of polysubstituted biphenyls. Bioorganic & medicinal chemistry letters. 2001;11:1709–1712. doi: 10.1016/s0960-894x(01)00267-0. [DOI] [PubMed] [Google Scholar]
- 17.Paris F, Balaguer P, Terouanne B, Servant N, Lacoste C, Cravedi JP, Nicolas JC, Sultan C. Phenylphenols, biphenols, bisphenol-A and 4-tert-octylphenol exhibit alpha and beta estrogen activities and antiandrogen activity in reporter cell lines. Molecular and cellular endocrinology. 2002;193:43–49. doi: 10.1016/s0303-7207(02)00094-1. [DOI] [PubMed] [Google Scholar]
- 18.Carlson KE, Choi I, Gee A, Katzenellenbogen BS, Katzenellenbogen JA. Altered ligand binding properties and enhanced stability of a constitutively active estrogen receptor: evidence that an open pocket conformation is required for ligand interaction. Biochemistry. 1997;36:14897–14905. doi: 10.1021/bi971746l. [DOI] [PubMed] [Google Scholar]
- 19.Jeyakumar M, Carlson KE, Gunther JR, Katzenellenbogen JA. Exploration of dimensions of estrogen potency: parsing ligand binding and coactivator binding affinities. The Journal of biological chemistry. 2011;286:12971–12982. doi: 10.1074/jbc.M110.205112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeyakumar M, Katzenellenbogen JA. A dual-acceptor time-resolved Foster resonance energy transfer assay for simultaneous determination of thyroid hormone regulation of corepressor and coactivator binding to the thyroid hormone receptor: Mimicking the cellular context of thyroid hormone action. Analytical biochemistry. 2009;386:73–78. doi: 10.1016/j.ab.2008.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bartell SM, Han L, Kim HN, Kim SH, Katzenellenbogen JA, Katzenellenbogen BS, Chambliss KL, Shaul PW, Roberson PK, Weinstein RS, Jilka RL, Almeida M, Manolagas SC. Non-Nuclear-Initiated Actions of the Estrogen Receptor Protect Cortical Bone Mass. Molecular Endocrinology. 2013 doi: 10.1210/me.2012-1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chambliss KL, Wu Q, Oltmann S, Konaniah ES, Umetani M, Korach KS, Thomas GD, Mineo C, Yuhanna IS, Kim SH, Madak-Erdogan Z, Maggi A, Dineen SP, Roland CL, Hui DY, Brekken RA, Katzenellenbogen JA, Katzenellenbogen BS, Shaul PW. Non-nuclear estrogen receptor alpha signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. The Journal of clinical investigation. 2010;120:2319–2330. doi: 10.1172/JCI38291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, Metzger D, Chambon P. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270:1491–1494. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- 24.Albert V, Hall MN. mTOR signaling in cellular and organismal energetics. Curr Opin Cell Biol. 2015;33:55–66. doi: 10.1016/j.ceb.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 25.Jia G, Aroor AR, Martinez-Lemus LA, Sowers JR. Overnutrition, mTOR signaling, and cardiovascular diseases. Am J Physiol Regul Integr Comp Physiol. 2014;307:R1198–1206. doi: 10.1152/ajpregu.00262.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zoller AL, Kersh GJ. Estrogen induces thymic atrophy by eliminating early thymic progenitors and inhibiting proliferation of beta-selected thymocytes. J Immunol. 2006;176:7371–7378. doi: 10.4049/jimmunol.176.12.7371. [DOI] [PubMed] [Google Scholar]
- 27.Yecies JL, Zhang HH, Menon S, Liu S, Yecies D, Lipovsky AI, Gorgun C, Kwiatkowski DJ, Hotamisligil GS, Lee CH, Manning BD. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. 2011;14:21–32. doi: 10.1016/j.cmet.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yecies JL, Manning BD. Transcriptional control of cellular metabolism by mTOR signaling. Cancer Res. 2011;71:2815–2820. doi: 10.1158/0008-5472.CAN-10-4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Higuchi N, Kato M, Shundo Y, Tajiri H, Tanaka M, Yamashita N, Kohjima M, Kotoh K, Nakamuta M, Takayanagi R, Enjoji M. Liver X receptor in cooperation with SREBP-1c is a major lipid synthesis regulator in nonalcoholic fatty liver disease. Hepatol Res. 2008;38:1122–1129. doi: 10.1111/j.1872-034X.2008.00382.x. [DOI] [PubMed] [Google Scholar]
- 30.Beaven SW, Matveyenko A, Wroblewski K, Chao L, Wilpitz D, Hsu TW, Lentz J, Drew B, Hevener AL, Tontonoz P. Reciprocal regulation of hepatic and adipose lipogenesis by liver X receptors in obesity and insulin resistance. Cell Metab. 2013;18:106–117. doi: 10.1016/j.cmet.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tchkonia T, Thomou T, Zhu Y, Karagiannides I, Pothoulakis C, Jensen MD, Kirkland JL. Mechanisms and metabolic implications of regional differences among fat depots. Cell Metab. 2013;17:644–656. doi: 10.1016/j.cmet.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McDonnell DP, Wardell SE. The molecular mechanisms underlying the pharmacological actions of ER modulators: implications for new drug discovery in breast cancer. Current opinion in pharmacology. 2010;10:620–628. doi: 10.1016/j.coph.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Swedenborg E, Power KA, Cai W, Pongratz I, Ruegg J. Regulation of estrogen receptor beta activity and implications in health and disease. Cellular and Molecular Life Sciences. 2009;66:3873–3894. doi: 10.1007/s00018-009-0118-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang EC, Charn TH, Park SH, Helferich WG, Komm B, Katzenellenbogen JA, Katzenellenbogen BS. Estrogen Receptors alpha and beta as determinants of gene expression: influence of ligand, dose, and chromatin binding. Molecular Endocrinology. 2008;22:1032–1043. doi: 10.1210/me.2007-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang EC, Frasor J, Komm B, Katzenellenbogen BS. Impact of estrogen receptor beta on gene networks regulated by estrogen receptor alpha in breast cancer cells. Endocrinology. 2006;147:4831–4842. doi: 10.1210/en.2006-0563. [DOI] [PubMed] [Google Scholar]
- 36.Moore SM, Khalaj AJ, Kumar S, Winchester Z, Yoon J, Yoo T, Martinez-Torres L, Yasui N, Katzenellenbogen JA, Tiwari-Woodruff SK. Multiple functional therapeutic effects of the estrogen receptor beta agonist indazole-Cl in a mouse model of multiple sclerosis. Proc Natl Acad Sci U S A. 2014;111:18061–18066. doi: 10.1073/pnas.1411294111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paterni I, Granchi C, Katzenellenbogen JA, Minutolo F. Estrogen receptors alpha (ERalpha) and beta (ERbeta): subtype-selective ligands and clinical potential. Steroids. 2014;90:13–29. doi: 10.1016/j.steroids.2014.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao Y, Gong P, Chen Y, Nwachukwu JC, Srinivasan S, Ko C, Bagchi MK, Taylor RN, Korach KS, Nettles KW, Katzenellenbogen JA, Katzenellenbogen BS. Dual suppression of estrogenic and inflammatory activities for targeting of endometriosis. Sci Transl Med. 2015;7:271ra279. doi: 10.1126/scitranslmed.3010626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Finan B, Yang B, Ottaway N, Stemmer K, Muller TD, Yi CX, Habegger K, Schriever SC, Garcia-Caceres C, Kabra DG, Hembree J, Holland J, Raver C, Seeley RJ, Hans W, Irmler M, Beckers J, de Angelis MH, Tiano JP, Mauvais-Jarvis F, Perez-Tilve D, Pfluger P, Zhang L, Gelfanov V, DiMarchi RD, Tschop MH. Targeted estrogen delivery reverses the metabolic syndrome. Nat Med. 2012;18:1847–1856. doi: 10.1038/nm.3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pedram A, Razandi M, Lewis M, Hammes S, Levin ER. Membrane-localized estrogen receptor alpha is required for normal organ development and function. Dev Cell. 2014;29:482–490. doi: 10.1016/j.devcel.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adlanmerini M, Solinhac R, Abot A, Fabre A, Raymond-Letron I, Guihot AL, Boudou F, Sautier L, Vessieres E, Kim SH, Liere P, Fontaine C, Krust A, Chambon P, Katzenellenbogen JA, Gourdy P, Shaul PW, Henrion D, Arnal JF, Lenfant F. Mutation of the palmitoylation site of estrogen receptor alpha in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc Natl Acad Sci U S A. 2014;111:E283–290. doi: 10.1073/pnas.1322057111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pedram A, Razandi M, Kim JK, O’Mahony F, Lee EY, Luderer U, Levin ER. Developmental phenotype of a membrane only estrogen receptor alpha (MOER) mouse. The Journal of biological chemistry. 2009;284:3488–3495. doi: 10.1074/jbc.M806249200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abot A, Fontaine C, Buscato M, Solinhac R, Flouriot G, Fabre A, Drougard A, Rajan S, Laine M, Milon A, Muller I, Henrion D, Adlanmerini M, Valera MC, Gompel A, Gerard C, Pequeux C, Mestdagt M, Raymond-Letron I, Knauf C, Ferriere F, Valet P, Gourdy P, Katzenellenbogen BS, Katzenellenbogen JA, Lenfant F, Greene GL, Foidart JM, Arnal JF. The uterine and vascular actions of estetrol delineate a distinctive profile of estrogen receptor alpha modulation, uncoupling nuclear and membrane activation. EMBO Mol Med. 2014;6:1328–1346. doi: 10.15252/emmm.201404112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pedram A, Razandi M, O’Mahony F, Harvey H, Harvey BJ, Levin ER. Estrogen reduces lipid content in the liver exclusively from membrane receptor signaling. Sci Signal. 2013;6:ra36. doi: 10.1126/scisignal.2004013. [DOI] [PubMed] [Google Scholar]
- 45.Rai D, Frolova A, Frasor J, Carpenter AE, Katzenellenbogen BS. Distinctive actions of membrane-targeted versus nuclear localized estrogen receptors in breast cancer cells. Molecular endocrinology. 2005;19:1606–1617. doi: 10.1210/me.2004-0468. [DOI] [PubMed] [Google Scholar]
- 46.Rich RL, Hoth LR, Geoghegan KF, Brown TA, LeMotte PK, Simons SP, Hensley P, Myszka DG. Kinetic analysis of estrogen receptor/ligand interactions. Proc Natl Acad Sci U S A. 2002;99:8562–8567. doi: 10.1073/pnas.142288199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kousteni S, Chen JR, Bellido T, Han L, Ali AA, O’Brien CA, Plotkin L, Fu Q, Mancino AT, Wen Y, Vertino AM, Powers CC, Stewart SA, Ebert R, Parfitt AM, Weinstein RS, Jilka RL, Manolagas SC. Reversal of bone loss in mice by nongenotropic signaling of sex steroids. Science. 2002;298:843–846. doi: 10.1126/science.1074935. [DOI] [PubMed] [Google Scholar]
- 48.Otto C, Fuchs I, Altmann H, Klewer M, Schwarz G, Bohlmann R, Nguyen D, Zorn L, Vonk R, Prelle K, Osterman T, Malmstrom C, Fritzemeier KH. In vivo characterization of estrogen receptor modulators with reduced genomic versus nongenomic activity in vitro. J Steroid Biochem Mol Biol. 2008;111:95–100. doi: 10.1016/j.jsbmb.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 49.Centrella M, McCarthy TL, Chang WZ, Labaree DC, Hochberg RB. Estren (4-estren-3alpha,17beta-diol) is a prohormone that regulates both androgenic and estrogenic transcriptional effects through the androgen receptor. Molecular endocrinology. 2004;18:1120–1130. doi: 10.1210/me.2003-0491. [DOI] [PubMed] [Google Scholar]
- 50.Krishnan V, Bullock HA, Yaden BC, Liu M, Barr RJ, Montrose-Rafizadeh C, Chen K, Dodge JA, Bryant HU. The nongenotropic synthetic ligand 4-estren-3alpha17beta-diol is a high-affinity genotropic androgen receptor agonist. Molecular pharmacology. 2005;67:744–748. doi: 10.1124/mol.104.005272. [DOI] [PubMed] [Google Scholar]
- 51.Windahl SH, Galien R, Chiusaroli R, Clement-Lacroix P, Morvan F, Lepescheux L, Nique F, Horne WC, Resche-Rigon M, Baron R. Bone protection by estrens occurs through non-tissue-selective activation of the androgen receptor. The Journal of clinical investigation. 2006;116:2500–2509. doi: 10.1172/JCI28809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ishikawa T, Yuhanna IS, Umetani J, Lee WR, Korach KS, Shaul PW, Umetani M. LXRbeta/estrogen receptor-alpha signaling in lipid rafts preserves endothelial integrity. J Clin Invest. 2013;123:3488–3497. doi: 10.1172/JCI66533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dupont S, Krust A, Gansmuller A, Dierich A, Chambon P, Mark M. Effect of single and compound knockouts of estrogen receptors alpha (ERalpha) and beta (ERbeta) on mouse reproductive phenotypes. Development. 2000;127:4277–4291. doi: 10.1242/dev.127.19.4277. [DOI] [PubMed] [Google Scholar]
- 54.Hewitt SC, Kissling GE, Fieselman KE, Jayes FL, Gerrish KE, Korach KS. Biological and biochemical consequences of global deletion of exon 3 from the ER alpha gene. FASEB J. 2010;24:4660–4667. doi: 10.1096/fj.10-163428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A, Lu YT, Roskin KM, Schwartz M, Sugnet CW, Thomas DJ, Weber RJ, Haussler D, Kent WJ. The UCSC Genome Browser Database. Nucleic Acids Res. 2003;31:51–54. doi: 10.1093/nar/gkg129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 59.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu T, Ortiz JA, Taing L, Meyer CA, Lee B, Zhang Y, Shin H, Wong SS, Ma J, Lei Y, Pape UJ, Poidinger M, Chen Y, Yeung K, Brown M, Turpaz Y, Liu XS. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 2011;12:R83. doi: 10.1186/gb-2011-12-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao Y, Park S, Bagchi MK, Taylor RN, Katzenellenbogen BS. The coregulator, repressor of estrogen receptor activity (REA), is a crucial regulator of the timing and magnitude of uterine decidualization. Endocrinology. 2013;154:1349–1360. doi: 10.1210/en.2012-2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Madak-Erdogan Z, Ventrella R, Petry L, Katzenellenbogen BS. Novel roles for ERK5 and cofilin as critical mediators linking ERalpha-driven transcription, actin reorganization, and invasiveness in breast cancer. Mol Cancer Res. 2014;12:714–727. doi: 10.1158/1541-7786.MCR-13-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Appachi S, Sun J, Chambliss KL, Katzenellenbogen JA, Katzenellenbogen BS, Shaul PW, Murphy E. Non-nuclear estrogen receptor activation is protective in cardiac ischemia-reperfusion injury in mice. Journal of Molecular and Cellular Cardiology. 2012;53:S25. doi: 10.1016/j.yjmcc.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Seetharam D, Mineo C, Gormley AK, Gibson LL, Vongpatanasin W, Chambliss KL, Hahner LD, Cummings ML, Kitchens RL, Marcel YL, Rader DJ, Shaul PW. High-density lipoprotein promotes endothelial cell migration and reendothelialization via scavenger receptor-B type I. Circ Res. 2006;98:63–72. doi: 10.1161/01.RES.0000199272.59432.5b. [DOI] [PubMed] [Google Scholar]
- 65.Tamrazi A, Carlson KE, Katzenellenbogen JA. Molecular sensors of estrogen receptor conformations and dynamics. Molecular endocrinology (Baltimore, Md) 2003;17:2593–2602. doi: 10.1210/me.2003-0239. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Estrogen receptor and coactivator binding and interaction assays with ligands and ligand dissociation rates.
Figure S2. Verification of siRNA results in Fig. 2C with another siRNA.
Figure S3. PLAs with E2 and PaPE-1.
Figure S4. Pharmacokinetic studies for analysis of blood concentrations of PaPE-1 after injection or pellet implantation.
Figure S5. Effects of PaPE-1 require ERα.
Table S1. List of genes differentially expressed by cell treatment with E2 and PaPE-1.
Table S2. BED files for ERα, ERK2, and pSer5 RNA-Pol II ChIP-Seq data from experiments with MCF-7 cells.