

Threats to genomic integrity arising from DNA damage are mitigated by DNA glycosylases, which initiate the base excision repair (BER) pathway by locating and excising aberrant nucleobases1,2. How these enzymes find small modifications within the genome is a current area of intensive research. A hallmark of these and other DNA repair enzymes is their use of base flipping to sequester modified nucleotides from the DNA helix and into an active site pocket2-5. Consequently, base flipping is generally regarded as an essential aspect of lesion recognition and a necessary precursor to base excision. Here, we present the first DNA glycosylase mechanism that does not require base flipping for either binding or catalysis. Using the DNA glycosylase AlkD, we crystallographically monitored excision of an alkylpurine substrate as a function of time, and reconstructed the steps along the reaction coordinate through structures representing substrate, intermediate, and product complexes. Instead of directly interacting with the damaged nucleobase, AlkD recognizes aberrant base pairs through interactions with the phosphoribose backbone while the lesion remains stacked in the DNA duplex. Quantum mechanical calculations revealed that these contacts include catalytic charge-dipole and CH-π interactions that preferentially stabilize the transition state. We show in vitro and in vivo how this unique means of recognition and catalysis enables AlkD to repair large adducts formed by yatakemycin, a member of the duocarmycin family of antimicrobial natural products exploited in bacterial warfare and chemotherapeutic trials6,7. Bulky adducts of this or any type are not excised by DNA glycosylases that utilize a traditional base-flipping mechanism5. Hence, these findings represent a new paradigm for DNA repair and provide novel insights into catalysis of base excision.
Alkylation of DNA by endogenous metabolites, environmental toxins, and chemotherapeutic agents is a major source of genotoxic damage8. By virtue of their positive charge, N3- and N7-alkylpurines are prone to spontaneous depurination at physiological pH, and both N3-methyladenine (3mA) and apurinic (AP) sites interfere with DNA replication and transcription9,10. As the initial step in the BER pathway, DNA glycosylases remove 3mA and other cationic and neutral nucleobases from the genome. Both enzymatic and non-enzymatic depurination of these lesions has been shown to proceed through a stepwise pathway initiated by cleavage of the glycosidic bond, followed by addition of the nucleophilic water to the oxocarbenium (dR+) intermediate (Fig. 1a)11. The resulting AP site is then converted to an undamaged nucleotide by a common set of lesion-independent BER enzymes1,2.
Figure 1. Crystallographic reconstruction of the reaction trajectory for 3mA excision.
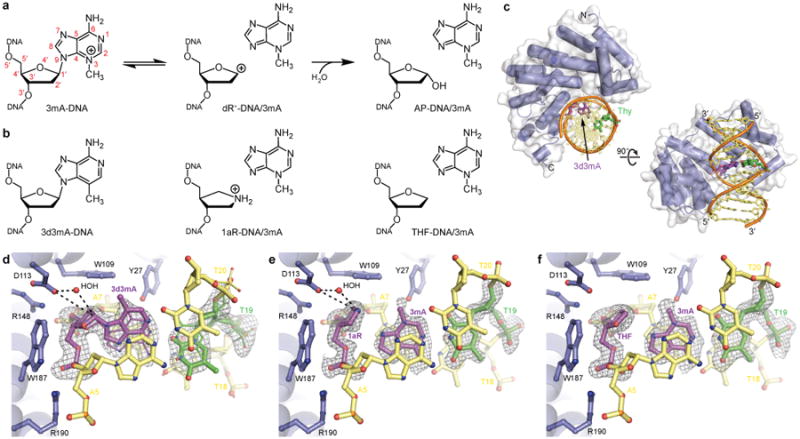
a , Proposed reaction scheme illustrating stepwise depurination of 3mA-DNA. b, Corresponding structural analogs used for crystallography. c, AlkD/3d3mA-DNA complex. d, Substrate-like complex containing 3d3mA-DNA. Watson-Crick (56%) and sheared (44%) conformations of the 3d3mA•T base pair are represented with thin and thick bonds, respectively. e, Intermediate-like complex containing 1aR-DNA and 3mA nucleobase. f, Product-like complex containing THF-DNA and 3mA nucleobase. AlkD is colored blue, the lesion is purple, the opposing thymine is green, and the flanking nucleotides are yellow. Charge-dipole and hydrogen-bonding interactions are shown as dashed lines. Annealed omit electron density in panels d–f is contoured to 2.5σ.
Despite their structural diversity, DNA glycosylases are generally thought to accomplish base flipping through the use of two conserved protein elements—a nucleobase binding pocket and a DNA intercalating residue5. The nucleobase binding pocket provides a means of damage recognition through shape and charge complementary with the modified base, while also creating a scaffold for the residues that catalyze hydrolysis of the N-glycosidic bond. With few exceptions, an aspartate or a glutamate residue plays a dual role in stabilizing developing positive charge on the sugar as the glycosidic bond is broken and in deprotonating the nucleophilic water that forms the AP product12. Additional residues in the pocket enhance the leaving-group potential of the base. For neutral alkylpurines, this enhancement comes from a general acid that protonates the nucleobase to provide the same instability inherent to cationic purines, which are in essence pre-activated for depurination11. Outside the binding pocket, DNA intercalating residues stabilize the catalytically active conformation of the DNA by filling the void in the duplex created when the lesion is flipped13.
We previously determined that the alkylpurine DNA glycosylase AlkD from Bacillus cereus14 uniquely recognizes damaged DNA without a nucleobase binding pocket or an intercalating residue15,16. However, the modified nucleotides in these structures were not in contact with the protein, leaving us to only speculate on the catalytic mechanism. To elucidate how AlkD excises positively charged substrates without a base binding pocket or an intercalating residue, we determined a new AlkD crystal structure with DNA containing 3-deaza-3-methyladenine (3d3mA), a comparatively stable isostere of 3mA (Fig. 1b and Extended Data Table 1). As in the previous AlkD/DNA complexes, the DNA duplex is bound by the concave surface of the protein. The helix is bent by 30° away from the enzyme while the minor groove surrounding the lesion is widened by 4 Å (Fig. 1c). This distortion induces an equilibrium in which the 3d3mA•T base pair is in either a Watson-Crick or highly sheared conformation. Shearing displaces the 3d3mA nucleobase by 4 Å into the minor groove and toward AlkD, but leaves the base partially stacked in the duplex (Extended Data Fig. 1). In this conformation, the deoxyribose of 3d3mA is in contact with three residues—Trp109, Asp113, and Trp187 (Fig. 1d)—all of which are critical for lesion excision and are invariant in the AlkD family15-18. The carboxylate and two indole side chains cradle the backbone of the lesion and the two flanking nucleotides. Asp113 is in line with the N-glycosidic bond of 3d3mA and thus ideally positioned to stabilize developing positive charge on the deoxyribose as the nucleobase is excised (Fig. 1d). This arrangement also allows Asp113 to position and deprotonate the nucleophilic water for subsequent addition to the oxocarbenium intermediate. Similar interactions between catalytic carboxylate residues and the deoxyribose are achieved by other DNA glycosylases, but only after the lesion has been flipped into the nucleobase binding pocket. In contrast, hypothetical rotation of the 3d3mA nucleotide in the AlkD complex would disrupt these catalytic contacts. The lack of base flipping, however, precludes a general acid from gaining access to protonate the nucleobase substrate, which limits AlkD to excision of cationic lesions.
Unlike 3mA, 3d3mA is uncharged and therefore refractory to depurination at physiological pH10. To our surprise, the electron density in the new AlkD/DNA complexes revealed a mixture of intact 3d3mA nucleotide together with AP site and free 3d3mA nucleobase, indicating that cleavage of the N-glycosidic bond had occurred (Fig. 2a and Extended Data Fig. 2). By flash freezing crystals at various times and determining their structures, we were able to visualize the glycosylase reaction at 1.4–2.0 Å resolution, starting with intact 3d3mA-DNA substrate and ending with AP-DNA product (Fig. 2a and Extended Data Tables 1–2). Quantifying the fractional occupancies of substrate and product over the course of the reaction gave a rate constant for in crystallo base excision of 4.6×10−6 s−1 (Fig. 2b). For comparison, cationic 3mA lesions are excised by AlkD at least 800-fold more rapidly19. The unexpected excision of 3d3mA can be explained by pH-dependent protonation at N7, which would confer positive charge on 3d3mA, activating it for excision by AlkD (Fig. 2c). Given the moderately acidic (pH 5.7) crystallization buffer and the calculated pKa (3.8) of 3d3mA, ∼1% of the lesion should be protonated, consistent with the slow rate of in crystallo cleavage. In contrast, we did not observe excision of 3d3mA in AlkD/3d3mA-DNA complexes crystallized at pH 7.0 (Extended Data Fig. 3 and Extended Data Table 3). We also did not observe cleavage in crystals grown at pH 5.7 in which AlkD bound 3d3mA-DNA in a non-catalytic orientation that placed the lesion on the opposite face of the duplex and away from the protein (Extended Data Fig. 4)16. Thus, the AlkD/3d3mA-DNA structure presented here represents a bona fide enzyme•substrate complex that enabled visualization of the endpoints of the glycosylase reaction.
Figure 2. Crystallographic snapshots of 3d3mA excision by AlkD.

a , Enzymatic conversion of 3d3mA-DNA substrate (purple) to AP-DNA product and 3d3mA nucleobase (pink). The excised 3d3mA nucleobase is rotated by 180° relative to its position in the non-hydrolyzed substrate. Annealed omit electron density is contoured to 2.5σ. b, Time course of 3d3mA excision determined from fractional occupancies of substrate and product in the crystal structures. c, Proposed reaction scheme showing protonation and excision of 3d3mA.
We probed the intervening step of the reaction trajectory by determining a structure representing the oxocarbenium intermediate, using DNA containing 1′-aza-2′,4′-dideoxyribose (1aR) and 3mA nucleobase (Fig. 1b and Extended Data Table 3). Relative to the position of the 3d3mA nucleotide, the cationic 1aR moiety is shifted slightly toward the surface of AlkD, which enhances electrostatic interactions with Asp113 and the nucleophilic water (Fig. 1e). These same interactions would stabilize the high-energy oxocarbenium intermediate formed upon cleavage of the glycosidic bond. A nearly identical arrangement is present in the product-like complex containing tetrahydrofuran (THF)-DNA and 3mA nucleobase (Fig. 1b,f and Extended Data Table 3). The only notable exception is a small rotation of the neutral THF ring away from Asp113. In both ternary complexes, the 3mA nucleobase is retained in the DNA duplex and paired with the complementary thymine, maintaining stacking interactions with the flanking bases (Fig. 1e,f). While base stacking is altered upon shearing of the 3d3mA•T base pair, it is never fully disrupted and is completely restored following cleavage of the N-glycosidic bond. In stark contrast to the traditional base-flipping mechanism, there is no evidence from these structures that a void in the duplex is created at any point along the reaction trajectory that would require stabilization by a DNA intercalating residue. Furthermore, these structures indicate that after shearing of the 3d3mA•T base pair, minimal movement of the protein and the DNA is necessary for either bond cleavage or nucleophile addition to occur.
The manner in which Trp109 and Trp187 contribute to binding and catalysis17,18 has heretofore been unknown. The new AlkD/DNA structures reveal that both residues form CH-π interactions20 with C2′, C4′, and C5′ of the 3d3mA nucleotide (Fig. 3a). To gain insight into how these weakly polar contacts contribute to recognition and excision of 3mA, we employed a reductive computational approach using only the three catalytic residues, the nucleophilic water, and the lesion. Electrostatic potential calculations showed that substantial cationic character is present on both the 3mA nucleobase and the deoxyribose (Fig. 3b and Extended Data Fig. 5). Importantly, increased positive charge on the deoxyribose correlated with stronger calculated binding energies, suggesting a means by which AlkD might recognize cationic alkylpurine lesions through backbone contacts (Extended Data Fig. 5). This would enhance detection of altered base pairing or base stacking21 without the need to directly interact with the modified nucleobase. The electrostatic potential calculations also revealed that additional positive charge is transferred to the deoxyribose upon elongation of the N-glycosidic bond (Extended Data Fig. 5). Correspondingly, preferential binding and stabilization of this transition state (TS)-like structure resulted in a theoretical 103–104-fold rate enhancement of glycosidic bond cleavage. Roughly half of this enhancement was attributed to the increasingly polar CH-π interactions provided by Trp109 and Trp187 (Extended Data Fig. 5). While interactions of this type are widespread among proteins and are prevalent in protein/DNA complexes22,23, to our knowledge, this is the first indication that CH-π interactions might function in a catalytic capacity in DNA repair. These interactions are reminiscent of the π-π and cation-π interactions used by base-flipping enzymes to recognize extrahelical nucleobases24,25, but are distinctly different in their involvement of the deoxyribose, and may be fundamental to lesion excision in the absence of base flipping.
Figure 3. 3mA recognition and excision through charge-dipole and CH-π interactions.

a , Simulated AlkD/3mA-DNA complex (stereo image). Charge-dipole and hydrogen-bonding interactions are depicted as black dashed lines. CH-π interactions are shown as purple arrows. b, Electrostatic potential surfaces of catalytic residues and 3mA nucleoside prior to protein-DNA binding (E+S), in the enzyme•substrate (E•S) complex, and in an approximate enzyme•transition state (E•TS) complex. All structures were determined computationally. Potentials were scaled to −0.22–0.55 atomic units on an isodensity surface of 0.05 electrons/bohr3.
The unique mechanism reported here explains our previous finding that AlkD, but not human AAG, removes bulky, cationic pyridyloxobutyl (POB) adducts16. While extrahelical POB adducts are accommodated by the large nucleobase binding pockets of alkyltransferase-like proteins26, the relatively small active site pockets of AAG and other base-flipping DNA glycosylases impose tighter steric limitations. AlkD, however, does not rely on a base-flipping mechanism and therefore is not restricted in this fashion, suggesting it may have a cellular role in repair of bulky lesions. Specificity for bulky alkyl adducts would explain the coexistence of multiple alkylpurine glycosylases in numerous bacterial species14. Consistent with this notion, an AlkD ortholog from Streptomyces sp. TP-A0356 has been found to excise 815-Da N3-yatakemycyladenine (YTMA) adducts from DNA27 (Fig. 4a). Yatakemycin (YTM) is a minor-groove DNA-alkylating agent belonging to the duocarmycin family of antibiotic and antitumor drugs6. The gene cluster responsible for biosynthesis of YTM encodes the DNA glycosylase YtkR2, which confers resistance to YTM by excising YTMA lesions27.
Figure 4. Excision of N3-yatakemycyladenine by AlkD.

a, YTMA. b, Modeled AlkD/YTMA-DNA complex. c, Solvent-filled cavity between AlkD and 3d3mA-DNA into which YTM was modeled. d, In vitro YTMA excision monitored by separation of full-length YTMA-DNA substrate from cleaved AP-DNA product by denaturing gel electrophoresis. e, In vitro excision of 3mA (black) and YTMA (purple) by diverse alkylpurine DNA glycosylases. f, Growth of wild-type (black) and alkD-knockout (red) strains of B. anthracis in the presence of no drug (control), 2 mM MMS, and 10 nM YTM. Error bars in all panels represent the standard deviation of the mean from three replicate experiments.
Based on the sequence similarity between YtkR2 and AlkD, we hypothesized that AlkD would also have activity for YTMA. There is an extended solvent-filled volume between the protein and the minor groove in the AlkD/3d3mA-DNA complex that can accommodate YTM modeled onto the C3 position of 3d3mA without introducing steric clashes or disrupting catalytic interactions (Fig. 4b,c). Using a standard in vitro DNA glycosylase assay, we found that AlkD excised YTMA from DNA with the same efficiency as YtkR2 (Fig. 4d,e). In contrast, the alkylpurine DNA glycosylases AAG, MAG, and AlkA failed to excise YTMA but readily removed 3mA (Fig. 4d,e). To determine the specificity of AlkD for YTMA in cells, we constructed a Bacillus anthracis strain lacking alkD and tested its sensitivity against YTM and methyl methanesulfonate (MMS). MMS primarily produces 3mA and N7-methylguanine adducts that are removed by canonical alkylpurine DNA glycosylases5. In the absence of alkylating agent, deletion of alkD had no effect on the growth of B. anthracis (Fig. 4f and Extended Data Fig. 6). Similarly, alkD cells were no more sensitive to MMS than wild-type cells (Fig. 4f and Extended Data Fig. 6), most likely as a result of activity from the other alkylpurine glycosylases (AAG, AlkA, and AlkC) still present in the deletion strain. Conversely, deletion of alkD caused a significant increase in sensitivity to YTM, consistent with AlkD catalyzed excision of YTMA in vivo (Fig. 4f and Extended Data Fig. 6). This suggests that the primary role of AlkD in the cell is to repair larger alkyl adducts such as YTMA that are not normally corrected by the base excision repair pathway.
This work establishes that, contrary to dogma, substrate recognition and catalysis by DNA glycosylases can occur in the absence of base flipping. There is evidence that other enzymes are able to detect DNA damage and even discriminate between different chemical modifications prior to flipping28-30. That AlkD is limited to removing inherently labile cationic lesions strongly suggests that the main function of the nucleobase binding pocket in canonical DNA glycosylases is to increase the leaving-group potential of the nucleobase substrate, as opposed to merely discriminating against non-substrate nucleobases. With this capability, however, comes a limit on the size of the adduct that can be excised. Ultimately, the greatest benefit of a non-base-flipping mechanism may be the ability to repair bulky lesions.
Methods
Oligodeoxynucleotide synthesis
3d3mA phosphoramidite was purchased from Berry and Associates. 1aR phosphoramidite was synthesized with minor modification of a previously described method31. Both were incorporated into oligodeoxynucleotides using standard solid-phase techniques. The resulting products were purified by reverse-phase HPLC and verified by mass spectrometry. All other oligodeoxynucleotides were purchased from Integrated DNA Technologies and used without further purification.
Protein purification
Human (Δ79)AAG32, Saccharomyces cerevisiae MAG30, Escherichia coli AlkA33, and B. cereus AlkD15 were purified as previously described. The gene encoding YtkR2 (Genbank IADZ13541) was synthesized by DNA 2.0 and ligated into a modified pET27 expression vector encoding a Rhinovirus 3C cleavable hexahistidine tag. YtkR2 overproduction in E. coli HMS174(DE3) cells was induced at 16 °C upon addition of 0.5 mM IPTG. Cells were harvested from LB medium by centrifugation, resuspended in buffer L [50 mM Tris•hydrochloride pH 7.5, 500 mM sodium chloride, and 20% (v/v) glycerol], and lysed on ice by gentle sonication. Cleared lysate was applied to a Ni-NTA column equilibrated in buffer L. The column was then washed with buffer L containing 2 mM histidine and eluted with buffer L containing 100 mM EDTA. Pooled fractions were supplemented with 2 mM DTT prior to overnight cleavage of the affinity tag. Cleaved YtkR2 was diluted 10-fold in buffer H [50 mM Tris•hydrochloride pH 7.5, 20% (v/v) glycerol, 2 mM DTT, and 0.1 mM EDTA] before being applied to a heparin Sepharose column equilibrated in buffer H. The column was then washed with buffer H containing 50 mM sodium chloride and eluted by linearly increasing to buffer H containing 1 M sodium chloride. Pooled fractions were passed through a Ni-NTA column equilibrated in buffer L to remove trace protein contaminants. The column was subsequently rinsed with buffer L containing 2 mM histidine to elute weakly bound YtkR2. The flow-through and the rinse were then combined, concentrated by ultracentrifugation, and applied to a Superdex 200 column equilibrated in buffer S [20 mM Bis-tris propane pH 6.5, 400 mM sodium chloride, 20% (v/v) glycerol, 2 mM DTT, and 0.1 mM EDTA]. YtkR2 was eluted with additional buffer S, concentrated to 3 mg/mL by ultracentrifugation, flash-frozen in liquid nitrogen, and stored at −80 °C.
Crystallization of AlkD/DNA
DNA annealing reactions contained 0.54 mM 9-mer A [d(AAGCAXACC)/d(TGGTTTGCT)], 9-mer B [d(AAGCCXCCC)/d(TGGGTGGCT)], or 12-mer [d(CCCGAXAGTCCG)/d(CGGACTTTCGGG)] oligodeoxynucleotides, 10 mM MES pH 6.5, and 40 mM sodium chloride. Reactions containing 1aR•T-DNA or THF•T-DNA were supplemented with 27 mM 3mA nucleobase. Strands were annealed by heating to 85 °C and slowly cooling to 20 °C over several hours. AlkD/DNA complexes were formed by mixing 0.45 mM protein and 0.54 mM DNA solutions in equal volumes and incubating at 4 °C for 30 min. Complexes containing 9-mer DNA were crystallized using the sitting drop vapor diffusion method. Drops were prepared from 2 μL of protein-DNA solution [0.22 mM AlkD and 0.27 mM DNA], 2 μL of reservoir solution [22–25% (w/v) PEG 8,000, 50 mM HEPES pH 7.0, and 50 mM calcium chloride], and 1 μL of additive solution [5% (w/v) benzamidine•hydrochloride] and equilibrated against an additional 500 μL of reservoir solution at 21 °C. Crystals were harvested after 24 h, briefly soaked in reservoir solution supplemented with 15% (v/v) glycerol, and flash-cooled in liquid nitrogen. Complexes containing 12-mer DNA were crystallized using the hanging drop vapor diffusion technique. Drops were assembled from 2 μL of protein-DNA solution [0.22 mM AlkD and 0.27 mM DNA], 2 μL of reservoir solution [15–19% (w/v) PEG 4,000, 42 mM sodium acetate pH 4.6, 85 mM ammonium acetate, and 5% (v/v) glycerol], and 1 μL of seed solution [submicroscopic crystals of AlkD/1mA•T-DNA] and equilibrated against an additional 500 μL of reservoir solution at 21 °C. Crystals were harvested after 4–360 h, briefly soaked in reservoir solution supplemented with 15% (v/v) glycerol, and flash-cooled in liquid nitrogen.
X-ray data collection and structure refinement
X-ray diffraction data were collected at beamlines 21-ID-F and 21-ID-G at the Advanced Photon Source and processed using HKL200034. Data collection statistics are provided in Extended Data Tables 1–3. A previously determined model of AlkD (PDB accession code 3BVS) was positioned with Phaser35, while DNA was manually built in Coot36, guided by inspection of 2mFo−DFc and mFo−DFc electron density maps. The entirety of the 12-mer oligodeoxynucleotide duplex was readily apparent, as were AlkD residues 1–229 and two non-native residues (−1–0) from the cleaved N-terminal affinity tag, but not the last eight residues (230–237) at the C-terminus. As with the 12-mer, the complete 9-mer duplexes were visible in the density maps. However, three residues (52–54) between helix C and helix D and twelve residues (226–237) at the C-terminus could not be reliably modeled. Atomic coordinates, temperature factors, and fractional occupancies were refined in PHENIX37. The final AlkD/DNA models were validated using MolProbity38 and contained no residues in the disallowed regions of the Ramachandran plot. Refinement and validation statistics are given in Extended Data Tables 1–3.
All structure images were created in PyMOL39. mFo−DFc omit electron density maps were calculated using PHENIX by removing the lesion and the opposing thymine and then performing simulated annealing on the remaining AlkD/DNA complex to minimize model bias. Maps were carved around the omitted atoms with a 1.5–2.0 Å radius and contoured to 2.5σ. YTM was manually docked in the AlkD/3d3mA-DNA complex after defining the cavity between the protein and the DNA with Hollow40. Simulated structures shown in Fig. 3a and Extended Data Fig. 1c were generated from AlkD/3d3mA-DNA crystal structures by manually removing or transmuting atoms and without performing subsequent computational optimization.
Preparation of YTMA-DNA
YTM was purified from Streptomyces sp. TP-A0356 culture as previously described6. Adduction reactions containing 150 μM YTM, 10 μM fluorescein (FAM)-labeled DNA [FAM-d(CGGGCGGCGGCAAAGGGCGCGGGCC)/d(GGCCCGCGCCCTTTGCCGCCGCCCG)], 10 mM MES pH 6.5, 40 mM sodium chloride, and 10% (v/v) DMSO were incubated at 25 °C for 24 h and periodically mixed by inversion. YTM-modified DNA was then separated from free YTM by passage through a G25 size exclusion column equilibrated in 10 mM MES pH 6.5 and 40 mM sodium chloride and stored at −80 °C.
Quantitation of base excision
Reaction mixtures containing 5 μM enzyme, 10 μg of methylated calf thymus DNA or 100 nM YTM-modified oligodeoxynucleotide duplex, 50 mM Bis-tris propane pH 6.5, 100 mM sodium chloride, 5% (v/v) glycerol, 2 mM DTT, 0.1 mM EDTA, and 0.1 mg/mL BSA were incubated at 25 °C for 6 h. Excision of 3mA was quantitated by measuring cleaved nucleobase using a previously described HPLC-MS/MS method19. Excision of YTMA was quantitated by separating the 25-mer substrate from the 12-mer product using standard electrophoretic techniques19. Strand breakage of 25-mer abasic oligodeoxynucleotide was induced by heating the reaction mixture at 70 °C for 30 min in the presence of 0.2 M sodium hydroxide.
Preparation of alkDΔ cells
The 1-kb flanking regions surrounding alkD were inserted into the knockout-plasmid pLM4 using standard molecular biology techniques41. The modified plasmid was propagated in E. coli K1077 before introduction by electroporation into Bacillus anthracis Sterne cells42,43. B. anthracis colonies harboring the plasmid were grown on LB plates supplemented with 20 μg/mL kanamycin at 42.5 °C for 1–2 days to generate merodiploids with plasmid DNA integrated into their chromosomal DNA. Merodiploids were then grown in LB medium for 1 day at 30 °C to facilitate elimination of redundant DNA from their genomes. Cultures were serially diluted and grown on LB plates without kanamycin for 1 day at 30 °C so that colonies lacking alkD could be identified by PCR screening.
Determination of YTM and MMS resistance
Overnight cultures of B. anthracis Sterne (wild-type and alkdΔ) grown at 30 °C were diluted 1:100 in 100 μL of LB medium in the presence or absence of 2–40 nM YTM or 1–10 mM MMS in a 96-well round-bottom plate. The plate was incubated at 30 °C with shaking for 20 h, and cell density was measured at 600 nm every hour using a Synergy 2 multi-detector microplate reader. Before each measurement, the plate was gently vortexed to ensure full resuspension of sedimented cells. Experiments were performed in triplicate.
Overnight cultures of B. anthracis Sterne (wild-type and alkdΔ) grown at 30 °C were diluted 1:100 in 5 mL of LB medium and incubated at 30 °C with shaking until early logarithmic phase. Culture aliquots (5 μL) were then 10-fold serially diluted (1:10–1–1:10–5) and spotted on LB plates prepared with or without 2–40 nM YTM or 1–10 mM MMS. Plates were incubated at 37 °C and imaged after 2 days. Experiments were performed in duplicate.
Calculation overview
All calculations were performed using Gaussian 0944. Atomic coordinates for computationally optimized structures are provided in Supplementary Data 1.
pKa calculation
The aqueous pKa microacidity constant of N7-protonated 3-deaza-3,9-dimethyladenine (3d3m9mA) was calculated using the isodesmic reaction method45 with N7-protonated 9-methyladenine (9mA) employed as the reference acid (aqueous pKa = 2.96)46. Neutral and N7-protonated 9mA and 3d3m9mA were optimized to minima using Truhlar's M06-2X density functional47 with the split-valence 6-311+G(2d,p) basis set and the SMD solvation model48. Optimized structures were confirmed minima by vibrational frequency analyses. Single point evaluations were carried out on the optimized structures in the presence of implicit solvent (water) at the M06-2X/aug-cc-pVTZ level. Thermal corrections to Gibbs free energies were taken from the frequency calculations and applied to electronic energies determined at the M06-2X/aug-cc-pVTZ level. Proton exchange energetics (ΔGexchg) between 9mA and 3d3m9mA were then determined using the corrected electronic energies.
Constrained optimization of AlkD/lesion structures
The crystal structure of AlkD bound to 3d3mA-DNA served as the starting point for all computational structural optimizations. Atomic coordinates were extracted for AlkD residues W109, D113, and W187 as well as HOH308 and 3d3mA. Hydrogen atoms were added manually to satisfy all valences, and D113 was assumed to be in carboxylate form. To reduce computational cost during structural optimizations and subsequent calculations, protein residues were truncated to their respective side chains (β-carbons as methyl groups), and 3d3mA was truncated to the corresponding 2′-deoxynucleoside. The starting point for the structure of 3mA was generated by replacing carbon with nitrogen at the 3-position of the purine ring in 3d3mA. The starting point for the TS approximation (TSA) was generated by elongating the glycosidic bond of optimized 3mA by 0.5 Å. This distance was chosen based on the computationally determined TS for excision of uracil by UDG49. Finally, the starting point for dR+ was generated by deleting the 3d3mA nucleobase from the corresponding 2′-deoxynucleoside, leaving an open valence on C1′.
Structural optimizations were performed in the gas phase using the M06-2X functional in conjunction with the 6-31+G(d) basis set. In all cases, the Cartesian coordinates for all non-hydrogen atoms in AlkD residues W109, D113, and W187 as well as HOH308 were frozen using the freeze code −1 in the molecular coordinates specification. Additionally, the 3′- and 5′-oxygen atoms of all 2′-deoxynucleosides were frozen to simulate the experimentally observed lack of motion in the phosphodiester backbone in the full AlkD/DNA complexes. All other atoms, with the exception of C1′ and N9 in the TSA, were allowed to move during optimizations. Stationary points derived from geometry optimizations on these reduced-dimension potential energy surfaces were verified as minima by vibrational frequency analyses. Frequency analyses were performed at the same level of theory and on the same reduced-dimension surfaces as the geometry optimizations. Using this approach, freezing Cartesian coordinates results in zeroing of elements in the Hessian associated with the frozen atoms. As such, the expected imaginary frequency resulting from elongation of the glycosidic bond in the TSA was not observed. All structures, including the TSA, afforded zero imaginary frequencies.
Calculation of binding energy and catalytic rate enhancement
Binding energies of the truncated AlkD residues and the catalytic water to 3d3mA, 3mA, TSA, and dR+ were determined using the M06-2X functional. Structural optimizations were carried out as described above using the 6-31+G(d) basis set, and single point evaluations were carried out using the 6-311++G(3df,2p) basis set. The basis set superposition error (BSSE) was accounted for using the counterpoise method50, wherein AlkD residues and the catalytic water were defined as the first fragment and the 2′-deoxynucleosides were defined as the second fragment. Because heavy atoms in AlkD residues were frozen at their experimentally determined atomic positions, binding energies were evaluated as differences in zero point-uncorrected electronic energies. This approach reproduced experimental gas-phase binding energies to within 1 kcal/mol for two prototypical CH-π systems, including neutral CH4/benzene (experimental 1.03–1.13 kcal/mol versus calculated 1.6 kcal/mol)51 and cationic (CH3)4N+/benzene (experimental 9.4 kcal/mol versus calculated 10.3 kcal/mol)52. To determine the individual contributions of W109, D113, W187, and HOH308 to ensemble binding energies, the counterpoise method was again employed as described above, but with the atoms in W109, D113, W187, or HOH308 replaced with the corresponding Gaussian ghost atoms. The contribution of each residue to the total binding energy (defined as BEindividual(x) = BEensemble – BEensemble,ghost(x)) was computed as the difference between the ensemble binding energy and that where W109, D113, W187, or HOH308 had been replaced with ghost atoms. Complexes containing ghost atoms were not reoptimized, which may affect individual binding energies. However, using this method, between 92–99% of the ensemble binding energy was accounted for in each complex.
The barrier to bond elongation (defined as ΔE = ETSA – E3mA) was calculated in the presence (ΔE = 18.1 kcal/mol) and absence (ΔE = 23.0 kcal/mol) of W109, D113, W187, and HOH308. The difference in these values (ΔΔE = 4.9 kcal/mol) was taken as the catalytic contribution of AlkD to 3mA excision. This process was repeated for complexes in which atoms in each residue or the catalytic water had been replaced with ghost atoms. For each series, ΔΔE values were computed as outlined above, and the difference between each of these ΔΔE values and that of the full ensemble was taken as the individual contribution of the omitted residue to catalysis. Estimated rate enhancements were calculated from these values by substituting ΔΔE for ΔΔG‡ in the Eyring equation at 25 °C. The barrier to bond elongation (ΔE = 23.0 kcal/mol) calculated with this approach is consistent with the activation enthalpy for depurination of 3mA (ΔH‡ = 23.5 kcal/mol) extrapolated from empirically determined half-lives53.
Quantitation of charge transfer and generation of electrostatic potential maps
Charge transfer from AlkD residues W109, D113, and W187 and HOH308 to the lesions was quantitated by performing Merz-Singh-Kollman (MK) population analyses54,55 on each complex and on each individual component (either AlkD and the catalytic water or the lesion). To correct for potential BSSE in population analyses, the appropriate Gaussian ghost atoms were utilized to maintain consistent basis between fragments and complexes. Partial atomic charges derived from MK population analyses were summed for all atoms in AlkD and for the lesion, affording the corresponding group charges. The arithmetic difference between group charges in the AlkD/lesion complex and AlkD or the lesion alone were taken as the amount of charge transferred upon complex formation and expressed as percentages. Electrostatic potential maps were generated from densities computed at the M06-2X/6-311++G(3df,2p) level with ghost atoms omitted for visual clarity. Potentials were scaled to −0.22–0.55 atomic units on an isodensity surface of 0.05 electrons/bohr3.
Extended Data
Extended Data Table 1. X-ray data collection and refinement statistics.
| 3d3mA•T-DNA 100% substrate 4 hours | 3d3mA•T-DNA AP•T-DNA/3d3mA 71% substrate/29% product 24 hours | 3d3mA•T-DNA AP•T-DNA/3d3mA 51% substrate/49% product 48 hours | 3d3mA•T-DNA AP•T-DNA/3d3mA 33% substrate/67% product 72 hours | |
|---|---|---|---|---|
| Data collection | ||||
|
| ||||
| Space group | P21 | P21 | P21 | P21 |
| Cell dimensions | ||||
| a, b, c (Å) | 38.19, 93.65, 48.08 | 38.25, 93.56, 47.98 | 38.34, 93.40, 48.12 | 38.39, 93.28, 48.08 |
| α, β, γ (°) | 90.00, 113.05, 90.00 | 90.00, 112.74, 90.00 | 90.00, 112.85, 90.00 | 90.00, 112.72, 90.00 |
| Resolution (Å) | 50.00–1.97 (2.04–1.97)* | 50.00–1.88 (1.95–1.88) | 50.00–1.57 (1.60–1.57) | 50.00–1.54 (1.57–1.54) |
| Rmerge | 0.068 (0.521) | 0.084 (0.590) | 0.063 (0.422) | 0.058 (0.184) |
| Avg. I/σI | 18.4 (2.9) | 18.4 (3.2) | 21.3 (3.1) | 34.0 (10.3) |
| Completeness (%) | 99.8 (99.5) | 99.9 (99.9) | 99.9 (98.9) | 99.4 (93.9) |
| Redundancy | 4.1 (4.0) | 5.3 (4.5) | 4.2 (3.7) | 7.5 (6.8) |
|
| ||||
| Refinement | ||||
|
| ||||
| Resolution (Å) | 40.01–1.97 (2.06–1.97) | 35.28–1.87 (1.94–1.87) | 35.34–1.57 (1.61–1.57) | 35.06–1.54 (1.58–1.54) |
| No. reflections | 21,918 (2,701) | 25,801 (2,791) | 43,431 (3,010) | 45,847 (3,225) |
| Rwork | 0.156 (0.217) | 0.153 (0.216) | 0.158 (0.208) | 0.153 (0.169) |
| Rfree† | 0.189 (0.305) | 0.196 (0.267) | 0.189 (0.238) | 0.183 (0.211) |
| No. atoms | ||||
| Protein | 1,951 | 1,951 | 1,951 | 1,951 |
| DNA | 487 | 487 | 487 | 488 |
| Water | 177 | 207 | 356 | 413 |
| Avg. B-factors‡ (Å2) | ||||
| Protein | 28.5 | 27.7 | 15.7 | 18.4 |
| DNA | 50.1 | 46.2 | 34.0 | 38.0 |
| Water | 39.0 | 38.6 | 31.8 | 35.2 |
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.007 | 0.007 | 0.006 | 0.006 |
| Bond angles (°) | 0.990 | 1.021 | 1.052 | 1.043 |
| Ramachandran distribution (%) | ||||
| Favored | 97.8 | 97.8 | 97.4 | 97.8 |
| Allowed | 2.2 | 2.2 | 2.6 | 2.2 |
| Disallowed | 0.0 | 0.0 | 0.0 | 0.0 |
| PDB accession code | 5CL3 | 5CL4 | 5CL5 | 5CL6 |
Statistics for the highest resolution shell are shown in parentheses.
Rfree was determined from the 5% of reflections excluded from refinement.
Equivalent isotropic B-factors were calculated in conjunction with TLS-derived anisotropic B-factors.
Extended Data Table 2.
X-ray data collection and refinement statistics.
| 3d3mA•T-DNA AP•T-DNA/3d3mA 18% substrate/82% product 96 hours | AP•T-DNA/3d3mA 100% product 144 hours | AP•T-DNA/3d3mA 100% product 240 hours | AP•T-DNA/3d3mA 100% product 360 hours | |
|---|---|---|---|---|
| Data collection | ||||
|
| ||||
| Space group | P21 | P21 | P21 | P21 |
| Cell dimensions | ||||
| a, b, c (Å) | 38.39, 93.15, 48.20 | 38.50, 93.45, 48.77 | 37.90, 92.87, 48.92 | 38.43, 93.08, 48.05 |
| α, β, γ (°) | 90.00, 112.81, 90.00 | 90.00, 114.19, 90.00 | 90.00, 113.77, 90.00 | 90.00, 112.61, 90.00 |
| Resolution (Å) | 50.00–1.44 (1.46–1.44)* | 50.00–1.38 (1.40–1.38) | 50.00–1.54 (1.57–1.54) | 50.00–1.54 (1.60–1.54) |
| Rmerge | 0.069 (0.509) | 0.047 (0.318) | 0.050 (0.523) | 0.049 (0.138) |
| Avg. I/σI | 23.3 (3.5) | 31.9 (5.1) | 32.0 (3.0) | 41.1 (16.6) |
| Completeness (%) | 96.4 (93.8) | 99.5 (96.1) | 99.5 (94.3) | 96.9 (92.3) |
| Redundancy | 6.4 (6.1) | 7.3 (5.2) | 6.2 (4.8) | 7.7 (7.4) |
|
| ||||
| Refinement | ||||
|
| ||||
| Resolution (Å) | 35.40–1.44 (1.47–1.44) | 35.13–1.38 (1.41–1.38) | 35.03–1.54 (1.58–1.54) | 35.05–1.54 (1.58–1.54) |
| No. reflections | 54,660 (3,702) | 64,110 (4,402) | 45,352 (3,149) | 44,651 (3,037) |
| Rwork | 0.128 (0.150) | 0.133 (0.160) | 0.160 (0.227) | 0.148 (0.147) |
| Rfree† | 0.166 (0.216) | 0.158 (0.231) | 0.186 (0.268) | 0.169 (0.194) |
| No. atoms | ||||
| Protein | 1,951 | 1,951 | 1,951 | 1,951 |
| DNA | 488 | 488 | 488 | 488 |
| Water | 363 | 405 | 248 | 400 |
| Avg. B-factors‡ (Å2) | ||||
| Protein | 17.0 | 18.2 | 26.6 | 16.2 |
| DNA | 38.1 | 39.7 | 53.6 | 35.1 |
| Water | 33.2 | 33.9 | 41.1 | 33.5 |
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.008 | 0.008 | 0.006 | 0.006 |
| Bond angles (°) | 1.219 | 1.157 | 0.972 | 1.041 |
| Ramachandran distribution (%) | ||||
| Favored | 98.3 | 98.3 | 97.8 | 97.9 |
| Allowed | 1.7 | 1.7 | 2.2 | 2.1 |
| Disallowed | 0.0 | 0.0 | 0.0 | 0.0 |
| PDB accession code | 5CL7 | 5CL8 | 5CL9 | 5CLA |
Statistics for the highest resolution shell are shown in parentheses.
Rfree was determined from the 5% of reflections excluded from refinement.
Equivalent isotropic B-factors were calculated in conjunction with TLS-derived and individual anisotropic B-factors.
Extended Data Table 3.
X-ray data collection and refinement statistics.
| 3d3mA•T-DNA 9-mer A | 3d3mA•T-DNA 9-mer B | 1aR•T-DNA/3mA | THF•T-DNA/3mA | |
|---|---|---|---|---|
| Data collection | ||||
|
| ||||
| Space group | C2 | C2 | P21 | P21 |
| Cell dimensions | ||||
| a, b, c (Å) | 127.44, 55.41, 47.97 | 127.40, 55.45, 47.56 | 38.20, 92.83, 48.31 | 38.49, 93.29, 48.16 |
| α, β, γ (°) | 90.00, 105.70, 90.00 | 90.00, 106.36, 90.00 | 90.00, 113.71, 90.00 | 90.00, 112.84, 90.00 |
| Resolution (Å) | 50.00–1.77 (1.83–1.77)* | 50.00–1.73 (1.79–1.73) | 50.00–1.54 (1.57–1.54) | 50.00–1.73 (1.79–1.73) |
| Rmerge | 0.088 (0.414) | 0.072 (0.362) | 0.057 (0.255) | 0.067 (0.443) |
| Avg. I/σI | 15.0 (2.7) | 21.3 (5.1) | 78.6 (7.0) | 23.2 (3.4) |
| Completeness (%) | 96.7 (80.0) | 100.0 (99.9) | 97.2 (92.2) | 99.9 (99.6) |
| Redundancy | 4.6 (4.2) | 5.3 (4.8) | 7.1 (6.4) | 4.9 (4.6) |
|
| ||||
| Refinement | ||||
|
| ||||
| Resolution (Å) | 46.20–1.77 (1.82–1.77) | 45.65–1.73 (1.78–1.73) | 35.00–1.54 (1.58–1.54) | 40.09–1.73 (1.78–1.73) |
| No. reflections | 30,638 (2,200) | 33,412 (2,770) | 44,194 (3,013) | 32,539 (2,620) |
| Rwork | 0.163 (0.228) | 0.154 (0.196) | 0.149 (0.153) | 0.155 (0.211) |
| Rfree† | 0.203 (0.277) | 0.186 (0.252) | 0.179 (0.179) | 0.192 (0.288) |
| No. atoms | ||||
| Protein | 1,898 | 1,898 | 1,951 | 1,951 |
| DNA | 364 | 364 | 487 | 487 |
| Water | 253 | 284 | 415 | 271 |
| Avg. B-factors‡ (Å2) | ||||
| Protein | 24.2 | 23.6 | 14.6 | 21.2 |
| DNA | 38.3 | 32.2 | 33.1 | 39.5 |
| Water | 34.0 | 34.3 | 31.9 | 35.5 |
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.007 | 0.007 | 0.006 | 0.007 |
| Bond angles (°) | 1.030 | 1.074 | 1.049 | 1.009 |
| Ramachandran distribution (%) | ||||
| Favored | 97.7 | 97.3 | 97.9 | 97.8 |
| Allowed | 2.3 | 2.7 | 2.1 | 2.2 |
| Disallowed | 0.0 | 0.0 | 0.0 | 0.0 |
| PDB accession code | 5CLB | 5CLC | 5CLD | 5CLE |
Statistics for the highest resolution shell are shown in parentheses.
Rfree was determined from the 5% of reflections excluded from refinement.
Equivalent isotropic B-factors were calculated in conjunction with TLS-derived anisotropic B-factors.
Extended Data Figure 1. Remodeled DNA structures.

DNA binding by AlkD bends the helical axis by 30° and widens the minor groove by 4 Å. Remodeling of this type reduces the energetic barrier to base pair opening but does not force base flipping56-58. In the AlkD/3d3mA-DNA complex, this distortion creates an equilibrium between Watson-Crick and sheared conformations of the 3d3mA•T base pair. In the sheared conformation, the 3d3mA nucleobase is displaced by 4 Å into the widened minor groove but remains partially stacked in the DNA duplex. a, Sheared 3d3mA•T base pair in the AlkD/DNA complex. b, Watson-Crick 3d3mA•T base pair in the AlkD/DNA complex. c, Simulated Watson-Crick A•T base pair in the AlkD/DNA complex. Adenine was generated by removing the methyl substituent from 3d3mA and changing carbon to nitrogen at the 3-position. Computational optimization was not performed. d, Watson-Crick A•T base pair in B-form DNA (PDB accession code 1BNA). The asterisks in panels a and b indicate the position of the methyl substituent at C3. All structures are depicted as stereo images.
Extended Data Figure 2. 3d3mA substrate and product structures.

The AlkD/3d3mA-DNA complex was crystallized under moderately acidic (pH 5.7) conditions, causing a small fraction (∼1%) of 3d3mA to become protonated and activated for enzymatic excision. Reaction progress was monitored by flash freezing crystals at various times after preparing the protein/DNA complex and refining the fractional occupancies of substrate and product in the structures. a, Structure after 4 h containing only 3d3mA-DNA substrate. The 3d3mA•T base pair is present as a mixture of Watson-Crick (thin bonds) and sheared (thick bonds) conformations. b, Structure after 48 h containing a mixture of 3d3mA-DNA substrate together with AP-DNA product and 3d3mA nucleobase. c, Structure after 360 h containing only AP-DNA product and 3d3mA nucleobase. All panels show annealed omit electron density contoured to 2.5σ.
Extended Data Figure 3. Additional substrate structures.

AlkD was crystallized with two alternate 9-mer 3d3mA-DNA constructs to verify that crystal packing contacts were not significantly influencing the conformation of the DNA. As in the catalytic 12-mer substrate complex, 3d3mA remained stacked in the duplex in both 9-mer structures. However, unlike in the 12-mer complex, a mixture of Watson-Crick and sheared conformations was not observed. Instead, the 3d3mA•T base pair in each construct is either entirely in the Watson-Crick conformation or entirely in the sheared conformation. Formation of the AP product was not observed in either 9-mer construct, likely as a consequence of the neutral (pH 7.0) crystallization conditions, under which the fraction (<0.1%) of 3d3mA activated for depurination would be drastically reduced. a, Watson-Crick conformation. b, Sheared conformation. Both panels show annealed omit electron density contoured to 2.5σ.
Extended Data Figure 4. Comparison of catalytic and non-catalytic AlkD/3d3mA-DNA complexes.
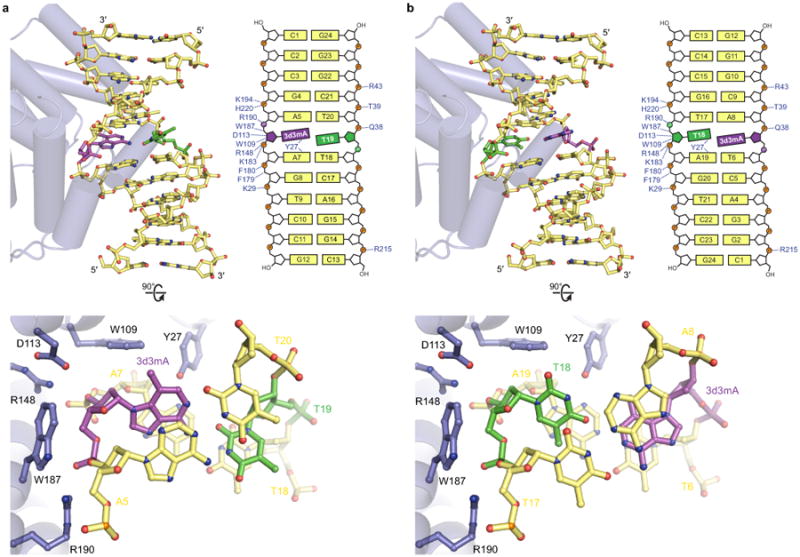
AlkD binds 3d3mA-DNA in two orientations that differ only in the position of the 3d3mA•T base pair about its dyad axis. Either 3d3mA (catalytic complex) or the opposing thymine (non-catalytic complex) resides against the protein surface. Both orientations use a common set of interactions that induce bending of the helical axis and widening of the minor groove, causing shearing of the 3d3mA•T base pair. In either complex, the nucleotide adjacent to the protein surface shows the same degree of displacement into the minor groove. The asymmetric position of the 3d3mA nucleotide in the duplex allows crystal packing interactions to select for a single orientation from a likely mixture of orientations in solution. Different DNA constructs were used to exploit these packing interactions and crystallize complexes in each binding orientation separately. Positive charge on the deoxyribose of 3mA would produce stronger electrostatic interactions with Trp109, Asp113, and Trp187 that would likely favor the catalytic binding orientation as well as a sheared conformation of a 3mA•T base pair. a, Catalytic orientation. The Watson-Crick conformer of the 3d3mA•T base pair was omitted for clarity. b, Non-catalytic orientation (PDB accession code 3JX7).
Extended Data Figure 5. Electrostatic potential surfaces and computed binding energies.

Charge transfer from the catalytic ensemble (Trp109, Asp113, Trp187, and Asp113-associated water) to the modified nucleosides stabilizes the AlkD/DNA complexes. a, 3d3mA substrate. b, 3mA substrate. c, TS approximation. d, dR+ intermediate. Positive charge located on the deoxyribose moiety of the unbound nucleosides relative to that on the unbound dR+ intermediate is indicated below the lesions. Charge transferred from the catalytic ensemble to the lesions upon complex formation is indicated above the arrows. Electrostatic potentials were scaled to −0.22–0.55 atomic units on an isodensity surface of 0.05 electrons/bohr3. e, Modified nucleosides shown in panels a–d. f, Computed binding energies (kcal/mol), differential bond elongation energies (ΔΔE‡, kcal/mol), and corresponding rate enhancements (r.e.).
Extended Data Figure 6. Growth curves and spot assays.
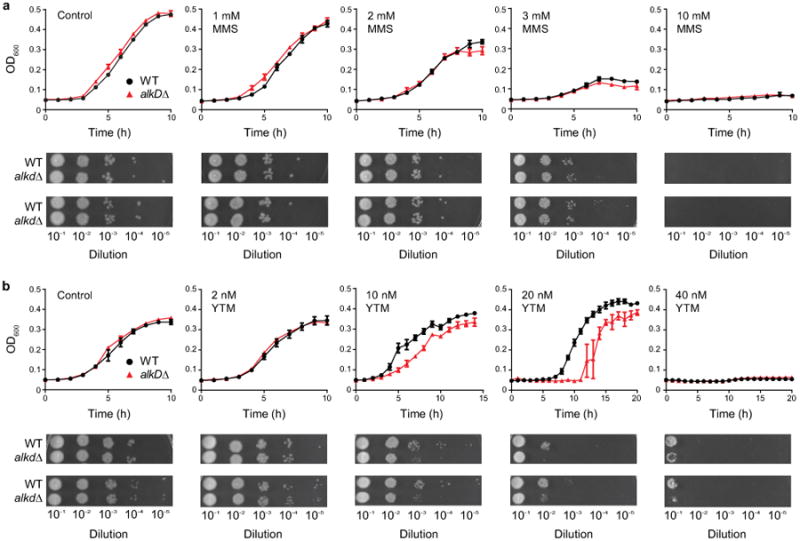
Wild-type (black) and alkD-knockout (red) strains of B. anthracis were treated with varying concentrations of MMS (a) and YTM (b). Control experiments contained no drug. Deletion of AlkD caused no observable phenotype with MMS but resulted in increased sensitivity to YTM. Error bars represent the standard deviation of the mean from three replicate growth curves. Spot assays were performed in duplicate.
Supplementary Material
Acknowledgments
We thank C. Rizzo and T. Johnson Salyard for assistance with oligodeoxynucleotide synthesis and E. Skaar and L. Mike for assistance with genetic manipulation of B. anthracis. This work was funded by the National Science Foundation (MCB-1122098 and MCB-1517695 to B.F.E.) and the National Institutes of Health (R01ES019625 to B.F.E. and R01CA067985 to S.S.D.). Support for the Vanderbilt Robotic Crystallization Facility was also provided by the National Institutes of Health (S10RR026915). Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Argonne National Laboratory, was supported by the U.S. Department of Energy (DE-AC02-06CH11357). Use of LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (085P1000817). E.A.M. and Z.D.P. were partially supported by the Vanderbilt Training Program in Environmental Toxicology (T32ES07028).
Footnotes
Supplementary Information is available in the online version of the paper.
Author Contributions: E.A.M. and B.F.E. conceived the project; E.A.M., R.S., Z.D.P., and B.F.E. designed experiments; E.A.M. performed biochemical and structural experiments; R.S. performed cellular experiments; Z.D.P. performed computational experiments; P.K.Y., S.S.D., and Y.I. supplied reagents; E.A.M., R.S., Z.D.P., and B.F.E. analyzed data and wrote the paper; all authors commented on the manuscript.
Author Information: Atomic coordinates and structure factors were deposited in the Protein Data Bank under accession codes 5CL3, 5CL4, 5CL5, 5CL6, 5CL7, 5CL8, 5CL9, 5CLA, 5CLB, 5CLC, 5CLD, and 5CLE.
The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper.
References
- 1.Fromme JC, Verdine GL. Base excision repair. Adv Protein Chem. 2004;69:1–41. doi: 10.1016/S0065-3233(04)69001-2. [DOI] [PubMed] [Google Scholar]
- 2.Hitomi K, Iwai S, Tainer JA. The intricate structural chemistry of base excision repair machinery: implications for DNA damage recognition, removal, and repair. DNA Repair. 2007;6:410–428. doi: 10.1016/j.dnarep.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 3.Slupphaug G, et al. A nucleotide-flipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature. 1996;384:87–92. doi: 10.1038/384087a0. [DOI] [PubMed] [Google Scholar]
- 4.Stivers JT. Site-specific DNA damage recognition by enzyme-induced base flipping. Prog Nucleic Acid Res Mol Biol. 2004;77:37–65. doi: 10.1016/S0079-6603(04)77002-6. [DOI] [PubMed] [Google Scholar]
- 5.Brooks SC, Adhikary S, Rubinson EH, Eichman BF. Recent advances in the structural mechanisms of DNA glycosylases. Biochim Biophys Acta. 2013;1834:247–271. doi: 10.1016/j.bbapap.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Igarashi Y, et al. Yatakemycin, a novel antifungal antibiotic produced by Streptomyces sp TP-A0356. J Antibiot. 2003;56:107–113. doi: 10.7164/antibiotics.56.107. [DOI] [PubMed] [Google Scholar]
- 7.Alberts SR, Suman VJ, Pitot HC, Camoriano JK, Rubin J. Use of KW-2189, a DNA minor groove-binding agent, in patients with hepatocellular carcinoma: a north central cancer treatment group (NCCTG) phase II clinical trial. J Gastrointest Cancer. 2007;38:10–14. doi: 10.1007/s12029-007-9007-6. [DOI] [PubMed] [Google Scholar]
- 8.Friedberg EC, et al. DNA repair and mutagenesis. 2nd. ASM Press; 2006. [Google Scholar]
- 9.Larson K, Sahm J, Shenkar R, Strauss B. Methylation-induced blocks to in vitro DNA replication. Mutat Res. 1985;150:77–84. doi: 10.1016/0027-5107(85)90103-4. [DOI] [PubMed] [Google Scholar]
- 10.Plosky BS, et al. Eukaryotic Y-family polymerases bypass a 3-methyl-2′-deoxyadenosine analog in vitro and methyl methanesulfonate-induced DNA damage in vivo. Nucleic Acids Res. 2008;36:2152–2162. doi: 10.1093/nar/gkn058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drohat AC, Maiti A. Mechanisms for enzymatic cleavage of the N-glycosidic bond in DNA. Org Biomol Chem. 2014;12:8367–8378. doi: 10.1039/c4ob01063a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stivers JT, Jiang YL. A mechanistic perspective on the chemistry of DNA repair glycosylases. Chem Rev. 2003;103:2729–2759. doi: 10.1021/cr010219b. [DOI] [PubMed] [Google Scholar]
- 13.Hendershot JM, O'Brien PJ. Critical role of DNA intercalation in enzyme-catalyzed nucleotide flipping. Nucleic Acids Res. 2014;42:12681–12690. doi: 10.1093/nar/gku919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alseth I, et al. A new protein superfamily includes two novel 3-methyladenine DNA glycosylases from Bacillus cereus, AlkC and AlkD. Mol Microbiol. 2006;59:1602–1609. doi: 10.1111/j.1365-2958.2006.05044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rubinson EH, Metz AH, O'Quin J, Eichman BF. A new protein architecture for processing alkylation damaged DNA: the crystal structure of DNA glycosylase AlkD. J Mol Biol. 2008;381:13–23. doi: 10.1016/j.jmb.2008.05.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rubinson EH, Gowda AS, Spratt TE, Gold B, Eichman BF. An unprecedented nucleic acid capture mechanism for excision of DNA damage. Nature. 2010;468:406–411. doi: 10.1038/nature09428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dalhus B, et al. Structural insight into repair of alkylated DNA by a new superfamily of DNA glycosylases comprising HEAT-like repeats. Nucleic Acids Res. 2007;35:2451–2459. doi: 10.1093/nar/gkm039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mullins EA, Rubinson EH, Eichman BF. The substrate binding interface of alkylpurine DNA glycosylase AlkD. DNA Repair. 2014;13:50–54. doi: 10.1016/j.dnarep.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mullins EA, et al. An HPLC-tandem mass spectrometry method for simultaneous detection of alkylated base excision repair products. Methods. 2013;64:59–66. doi: 10.1016/j.ymeth.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plevin MJ, Bryce DL, Boisbouvier J. Direct detection of CH/π interactions in proteins. Nat Chem. 2010;2:466–471. doi: 10.1038/nchem.650. [DOI] [PubMed] [Google Scholar]
- 21.Yang W. Poor base stacking at DNA lesions may initiate recognition by many repair proteins. DNA Repair. 2006;5:654–666. doi: 10.1016/j.dnarep.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 22.Brandl M, Weiss MS, Jabs A, Suhnel J, Hilgenfeld R. C-H ··· π-interactions in proteins. J Mol Biol. 2001;307:357–377. doi: 10.1006/jmbi.2000.4473. [DOI] [PubMed] [Google Scholar]
- 23.Wilson KA, Kellie JL, Wetmore SD. DNA-protein π-interactions in nature: abundance, structure, composition and strength of contacts between aromatic amino acids and DNA nucleobases or deoxyribose sugar. Nucleic Acids Res. 2014;42:6726–6741. doi: 10.1093/nar/gku269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Metz AH, Hollis T, Eichman BF. DNA damage recognition and repair by 3-methyladenine DNA glycosylase I (TAG) EMBO J. 2007;26:2411–2420. doi: 10.1038/sj.emboj.7601649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilkinson OJ, et al. Alkyltransferase-like protein (Atl1) distinguishes alkylated guanines for DNA repair using cation-π interactions. Proc Natl Acad Sci USA. 2012;109:18755–18760. doi: 10.1073/pnas.1209451109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tubbs JL, et al. Flipping of alkylated DNA damage bridges base and nucleotide excision repair. Nature. 2009;459:808–813. doi: 10.1038/nature08076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu H, et al. Self-resistance to an antitumor antibiotic: a DNA glycosylase triggers the base-excision repair system in yatakemycin biosynthesis. Angew Chem. 2012;51:10532–10536. doi: 10.1002/anie.201204109. [DOI] [PubMed] [Google Scholar]
- 28.Qi Y, et al. Encounter and extrusion of an intrahelical lesion by a DNA repair enzyme. Nature. 2009;462:762–766. doi: 10.1038/nature08561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Imamura K, Averill A, Wallace SS, Doublie S. Structural characterization of viral ortholog of human DNA glycosylase NEIL1 bound to thymine glycol or 5-hydroxyuracil-containing DNA. J Biol Chem. 2012;287:4288–4298. doi: 10.1074/jbc.M111.315309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adhikary S, Eichman BF. Analysis of substrate specificity of Schizosaccharomyces pombe Mag1 alkylpurine DNA glycosylase. EMBO Rep. 2011;12:1286–1292. doi: 10.1038/embor.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chu AM, Fettinger JC, David SS. Profiling base excision repair glycosylases with synthesized transition state analogs. Bioorg Med Chem Lett. 2011;21:4969–4972. doi: 10.1016/j.bmcl.2011.05.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O'Brien PJ, Ellenberger T. Human alkyladenine DNA glycosylase uses acid-base catalysis for selective excision of damaged purines. Biochemistry. 2003;42:12418–12429. doi: 10.1021/bi035177v. [DOI] [PubMed] [Google Scholar]
- 33.Bjelland S, Birkeland NK, Benneche T, Volden G, Seeberg E. DNA glycosylase activities for thymine residues oxidized in the methyl group are functions of the AlkA enzyme in Escherichia coli. J Biol Chem. 1994;269:30489–30495. [PubMed] [Google Scholar]
- 34.Otwinowski Z, Minor W. Processing of X-ray diffraction data. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 35.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emsley P, Lohkamp B, Scott W, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davis IW, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.The PyMOL molecular graphics system, Version 1.7.4. Schrödinger, LLC; [Google Scholar]
- 40.Ho BK, Gruswitz F. HOLLOW: generating accurate representations of channel and interior surfaces in molecular structures. BMC Struct Biol. 2008;8:49. doi: 10.1186/1472-6807-8-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mike LA, et al. Two-component system cross-regulation integrates Bacillus anthracis response to heme and cell envelope stress. PLoS Pathog. 2014;10:e1004044. doi: 10.1371/journal.ppat.1004044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sterne M. Avirulent anthrax vaccine. Onderstepoort J Vet Sci Animal Ind. 1937;21:41–43. [PubMed] [Google Scholar]
- 43.Stauff DL, Skaar EP. Bacillus anthracis HssRS signalling to HrtAB regulates haem resistance during infection. Mol Microbiol. 2009;72:763–778. doi: 10.1111/j.1365-2958.2009.06684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gaussian 09, Revision A.02. Gaussian, Inc; [Google Scholar]
- 45.Ho J, Coote ML. A universal approach for continuum solvent pKa calculations: are we there yet? Theor Chem Acc. 2009;125:3–21. [Google Scholar]
- 46.Kapinos LE, Operschall BP, Larsen E, Sigel H. Understanding the acid–base properties of adenosine: the intrinsic basicities of N1, N3 and N7. Chem Eur J. 2011;17:8156–8164. doi: 10.1002/chem.201003544. [DOI] [PubMed] [Google Scholar]
- 47.Zhao Y, Truhlar DG. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Acc. 2007;120:215–241. [Google Scholar]
- 48.Marenich AV, Cramer CJ, Truhlar DG. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B. 2009;113:6378–6396. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- 49.Dinner AR, Blackburn GM, Karplus M. Uracil-DNA glycosylase acts by substrate autocatalysis. Nature. 2001;413:752–755. doi: 10.1038/35099587. [DOI] [PubMed] [Google Scholar]
- 50.Boys SF, Bernardi F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors Mol Phys. 1970;19:553–566. [Google Scholar]
- 51.Shibasaki K, Fujii A, Mikami N, Tsuzuki S. Magnitude of the CH/π interaction in the gas phase: experimental and theoretical determination of the accurate interaction energy in benzene-methane. J Phys Chem A. 2006;110:4397–4404. doi: 10.1021/jp0605909. [DOI] [PubMed] [Google Scholar]
- 52.Meot-Ner M, Deakyne CA. Unconventional ionic hydrogen-bonds. 1. CHδ+···X. Complexes of quaternary ions with n- and π-donors. J Am Chem Soc. 1985;107:469–474. [Google Scholar]
- 53.Osborne MR, Phillips DH. Preparation of a methylated DNA standard, and its stability on storage. Chem Res Toxicol. 2000;13:257–261. doi: 10.1021/tx990182e. [DOI] [PubMed] [Google Scholar]
- 54.Singh UC, Kollman PA. An approach to computing electrostatic charges for molecules. J Comput Chem. 1984;5:129–145. [Google Scholar]
- 55.Besler BH, Merz KM, Kollman PA. Atomic charges derived from semiempirical methods. J Comput Chem. 1990;11:431–439. [Google Scholar]
- 56.Ramstein J, Lavery R. Energetic coupling between DNA bending and base pair opening. Proc Natl Acad Sci USA. 1988;85:7231–7235. doi: 10.1073/pnas.85.19.7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Adhikary S, Cato MC, McGary KL, Rokas A, Eichman BF. Non-productive DNA damage binding by DNA glycosylase-like protein Mag2 from Schizosaccharomyces pombe. DNA Repair. 2013;12:196–204. doi: 10.1016/j.dnarep.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dalhus B, et al. Sculpting of DNA at abasic sites by DNA glycosylase homolog Mag2. Structure. 2013;21:154–166. doi: 10.1016/j.str.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.