

Significance
Plasmodium vivax is a causative agent of malaria that results in high morbidity and mortality. P. vivax Duffy Binding Protein (PvDBP) is a leading vaccine candidate for P. vivax; however, PvDBP is highly variable, which prevents a strain transcending immune response, complicating vaccine design. Here we report the first, to our knowledge, broadly neutralizing antibody epitopes within PvDBP, and expand the known repertoire of neutralizing epitopes for this protein. The identification of broadly conserved inhibitory epitopes provides critical new motifs that should be retained in the next generation of P. vivax malaria vaccines and serve as a basis for rational structure-based vaccine design.
Keywords: malaria, Plasmodium vivax, Duffy Binding Protein, broadly neutralizing, epitopes
Abstract
Plasmodium vivax Duffy Binding Protein (PvDBP) is the most promising vaccine candidate for P. vivax malaria. The polymorphic nature of PvDBP induces strain-specific immune responses, however, and the epitopes of broadly neutralizing antibodies are unknown. These features hamper the rational design of potent DBP-based vaccines and necessitate the identification of globally conserved epitopes. Using X-ray crystallography, small-angle X-ray scattering, hydrogen-deuterium exchange mass spectrometry, and mutational mapping, we have defined epitopes for three inhibitory mAbs (mAbs 2D10, 2H2, and 2C6) and one noninhibitory mAb (3D10) that engage DBP. These studies expand the currently known inhibitory epitope repertoire by establishing protective motifs in subdomain three outside the receptor-binding and dimerization residues of DBP, and introduce globally conserved protective targets. All of the epitopes are highly conserved among DBP alleles. The identification of broadly conserved epitopes of inhibitory antibodies provides critical motifs that should be retained in the next generation of potent vaccines for P. vivax malaria.
The social and economic burden of human malaria caused by Plasmodium vivax is severely underestimated (1). Recent studies show comparable rates of severe malaria and of mortality between P. vivax and Plasmodium falciparum in Southeast Asia (2, 3), and frequent clinical manifestations of debilitating symptoms result in high morbidity (4, 5). This places a tremendous burden on the healthcare infrastructure (4, 5) and imparts hidden costs in the form of decreased economic productivity and standard of living (5). Thus, effective control methods for P. vivax malaria are desperately needed.
Both cost-effective and efficient, vaccines are among the leading avenues of intervention, and individuals living in regions with P. vivax develop naturally acquired humoral immunity that correlates with results from in vitro functional assays (6–9). A leading vaccine candidate for P. vivax is the Duffy Binding Protein (DBP) (10–19), a parasite cell surface protein in the Erythrocyte Binding-Like (EBL) invasion protein family (14, 20–28). DBP binds to the Duffy Antigen Receptor for Chemokines (DARC) on host reticulocytes through a conserved cysteine-rich Duffy Binding-Like (DBL) domain known as region II (DBP-II) (10–19). This interaction plays a major role in establishing P. vivax invasion and infection of reticulocytes.
DBP-II engages DARC in a stepwise fashion (18, 19) that invokes dimerization reminiscent of PfEBA-175, another EBL family member (23). During invasion, DBP-II binds to a single DARC molecule and dimerizes to form a heterotrimer, which then matures into a heterotetramer of a 2:2 complex of DBP-II and DARC (18, 19). DBP-II is a three-subdomain (SD) protein, with SD2 contributing key residues for dimerization and receptor binding (19). Duffy-independent invasion has been reported for certain isolates of P. vivax (29); however, these isolates contain a gene duplication of DBP, suggesting that increased expression of DBP may facilitate Duffy-negative invasion (30).
Antibodies obtained from naturally immune individuals block DARC receptor binding and potently neutralize P. vivax invasion (7, 9). In addition, antibodies that engage the dimer interface and/or receptor binding residues of DBL domains are potently neutralizing (9, 18, 19, 31). Polymorphisms in DBP and the presence of multiple strains in endemic regions present unique challenges (32–34). Analysis of 676 bp within DBP-II revealed 127 polymorphic sites with nucleotide diversity varying between 0.006 and 0.0109, resulting in 193 haplotypes (35). These factors induce strain-specific protection rather than strain-transcending immunity, which leaves individuals susceptible to continued infection and disease (6, 9, 17, 32, 33, 36).
There are currently two strategies for vaccine development to counteract this variation: (i) creation of a multiallele vaccine encompassing all variants encountered in endemic regions, and (ii) engineering of a single synthetic antigen lacking highly immunogenic polymorphic sites (8, 37, 38). Both approaches necessitate the identification of globally conserved epitopes that elicit broadly neutralizing antibodies (bnAbs). A bnAb response is required for protective immunity against highly variable and polymorphic targets, including HIV (39) and influenza (40), and will be indispensable in DBP-II vaccine design. Characterizing bnAb epitopes will ensure that these motifs are retained through vaccine design and will allow antigen engineering to enhance epitope immunogenicity and antibody affinity (41).
Three monoclonal antibodies (mAbs), 2D10, 2H2, and 2C6, block DBP from binding to erythrocytes (32). Here we present a crystal structure of DBP-II bound to a single-chain variable fragment (scFv) derived from a potent inhibitory mAb 2D10 that identifies a specific inhibitory epitope within SD3. We established epitopes for the inhibitory mAbs 2D10, 2H2, and 2C6 and the noninhibitory mAb 3D10 by small angle X-ray scattering (SAXS), hydrogen-deuterium exchange mass spectrometry (HDX-MS), an extensive surface mutant panel, and direct protein–protein interaction studies. All three inhibitory mAbs target residues in SD3 distinct from F261-K367 in SD2, which form the dimer interface of DBP and engage DARC 19–30 (19). These mAb epitopes are conserved among P. vivax strains, signifying their importance as broadly neutralizing epitopes and targets of strain-transcending global protection. Taken together, our findings expand the currently known inhibitory epitope repertoire and introduce globally conserved protective targets for P. vivax vaccine design.
Results
Structure of the DBP-II/2D10-scFv Complex.
We solved the crystal structure of an scFv derived from mAb 2D10 in complex with DBP-II to a resolution of 4.0 Å (Fig. 1, Table S1, and Fig. S1). At this resolution, the mAb epitope can be clearly identified, because the backbone density is clear and several side chains located in the epitope are ordered (Fig. S1). mAb 2D10 binds to a conformational epitope composed of amino acids 413–417 and 425–441 at the end of a three-helix bundle within SD3 (Fig. 1 and Table S2). All complementary determining regions (CDRs) make contacts; the heavy and light chains contribute buried surface areas of 649 Å2 and 855 Å2, respectively, for a total of 1,504 A2. The complex has a slightly greater-than-average shape complementarity at 0.70 (average range, 0.64–0.68; perfect complementarity, 1.00) (42). Binding of 2D10 does not change the structure of DBP-II, with an rmsd of 0.692 Å between bound and unbound DBP-II. Thus, the inhibitory effects of 2D10 are not due to disruption of the DBL fold, but rather to the epitope location and disruption of protein function.
Fig. 1.

Crystal structure of the DBP-II/2D10scFv complex. (A) Overall structure of the DBP-II/2D10scFv complex shown in ribbon representation. The DBP-II domain is colored in green. The scFv heavy chain (VH) is in blue, and the light chain (VL) is in orange. (B) Ribbon representation of DBP-II mapping the 2D10 epitopes. Residues contacted by the scFv are shown in stick form. Residues contacted by the heavy chain are colored blue, residues contacted by the light chain are in orange, and residues contacted by both are in beige. Residues not contacted by mAb are in green. Regions of disorder are shown as a dotted line. (C) Surface representation of the DBP-II. Color scheme is as in B.
Table S1.
Data collection and refinement statistics for the DBP-II 2D10 scFv complex
| Statistic | DBP-II/2D10scFv |
| Data collection | |
| Space group | P21 |
| Cell dimensions | |
| a, b, c, Å | 84.94, 63.94, 108.88 |
| α, β, γ, ° | 90, 100.092, 90 |
| Resolution, Å* | 20–4.0 (4.1–4.0) |
| Rsym* | 0.116 (0.275) |
| I/σI* | 6.74 (3.29) |
| Completeness, %* | 92.9 (91.0) |
| Redundancy* | 2.73 (2.70) |
| Refinement | |
| Resolution, Å | 20–4.0 |
| No. of reflections | 9,188 |
| Rwork/Rfree | 26.69/29.97 |
| No. of atoms | |
| Protein | 7,721 |
| Ligand/ion | 0 |
| Water | 0 |
| B-factors | |
| Protein | 87.28 |
| Ligand/ion | 0 |
| Water | 0 |
| Ramachandran, % | |
| Outliers | 0 |
| Allowed | 3.02 |
| Favored | 96.98 |
| rmsd | |
| Bond lengths, Å | 0.004 |
| Bond angles, ° | 0.678 |
Data were collected from a single crystal.
Highest-resolution shell is shown in parentheses.
Fig. S1.

2Fo-Fc electron density contoured at 1σ around the epitope clearly identifies contact sites in DBP. The mAb is removed for clarity, and only DBP and associated electron density are shown. In this orientation, clear electron density for residues 433–441 that compose part of the epitope for 2D10 is observed.
Table S2.
Contact residues between DBP and mAb 2D10
| Residues in DBP | Interface with 2D10 chains | Residues in 2D10 scFv light chain | Residues in 2D10 scFv heavy chain |
| E413 | Light | Y70 | S200 |
| K414 | Light | Q71 | W202 |
| D416 | Light | D72 | R219 |
| G417 | Light | K74 | Y221 |
| K425 | Light | Y76 | D224 |
| K428 | Light | L135 | D226 |
| V429 | Light | V136 | H228 |
| P430 | Light, heavy | E137 | E268 |
| P431 | Light | Y138 | E269 |
| Q433 | Light, heavy | L140 | T270 |
| N434 | Light, heavy | A271 | |
| K437 | Heavy | G274 | |
| S438 | Heavy | ||
| D440 | Heavy | ||
| Q441 | Heavy |
SAXS of the DBP-II/2D10Fab Complex.
We performed SAXS analysis on the DBP-II/2D10Fab complex (Fig. 2A). SAXS is an independent method for determining the structure and molecular weight (MW) of a complex. Because the SAXS data were collected on a Fab complex, we modeled the DBP-II/2D10Fab structure by aligning a 2D10Fab model on the scFv. The predicted scatter from the DBP-II/2D10Fab structure fit the SAXS profile, with a χ2 value of 2.30. A χ2 value of <3 is indicative of a correct fit (43). The MW for the DBP-II/2D10Fab structure was 94.9 kDa by SAXS MOW (44). This value is consistent with the mass of a complex (87 kDa) consisting of monomers of DBP-II (37 kDa) and a Fab (50 kDa). Thus, our SAXS analysis strongly validated the 2D10 epitope.
Fig. 2.

mAbs 2D10 and 2H2 share an epitope that is distinct from that of 2C6. (A) Plot of scattering intensity (I) against scattering momentum (Q) and statistical fit of theoretical scatter from the DBP-II/2D10scFv crystal structure (red line) with the experimental SAXS profile of the DBP-II/2D10Fab complex (red line) with a χ2 value of 2.3. (B) SAXS pairwise comparison of 2D10, 2H2, and 2C6. (C) Plot of scattering intensity (I) against scattering momentum (Q) and statistical fit of theoretical scatter from the DBP-II/2D10scFv crystal structure (red line) with the experimental SAXS profile of the DBP-II/2H2Fab complex (red line), with a χ2 value of 1.9. (D) Plot of scattering intensity (I) against scattering momentum (Q) and statistical fit of theoretical scatter from the DBP-II/2D10scFv crystal structure (red line) with the experimental SAXS profile of the DBP-II/2C6Fab complex (red line), with a χ2 value of 5.1.
Pairwise SAXS Comparison of 2D10, 2H2, and 2C6.
We obtained SAXS data for DBP-II/2H2Fab and DBP-II/2C6Fab and performed a pairwise comparison with the DBP-II/2D10Fab data to identify structurally similar complexes using the volatility of ratio (VR) metric (45). Structurally similar complexes will have a low VR, whereas structurally distinct complexes will have a high VR. The low VR of 1.9 between DBP-II/2D10Fab and DBP-II/2H2Fab complexes (Fig. 2B) demonstrate that these two complexes are similar. The high VR between DBP-II/2C6Fab and either DBP-II/2D10Fab (3.8) or DBP-II/2H2Fab (3.1) indicate that DBP-II/2C6Fab has an architecture distinct from that of the other two complexes.
mAbs 2H2 and 2D10 Have Similar Paratopes.
Sequence analysis of mAb 2D10 and 2H2 revealed high sequence identity throughout the entire variable regions of the light and heavy chains (Fig. S2). The sequence identity of the entire variable heavy chain was 89%, and that of the variable light chain was 96%. CDR1, CDR2, and CDR3 of the heavy chain shared sequence identity of 86%, 100%, and 11%, respectively. The three CDR regions in the light chain were highly similar: 100% for both CDR1 and CDR2, and 67% for CDR3. These alignments suggest that mAbs 2D10 and 2H2 share antigenic determinants on DBP-II. Furthermore, mAbs 2D10 and 2H2 likely share a common B-cell ancestor during lineage development.
Fig. S2.

Heavy and light chain alignments for mAbs 2D10, 2H2, and 2C6. Gray indicates similar residues; black indicates identical residues.
In contrast, mAb 2C6 shared little sequence identity to 2D10 or 2H2 in CDR1 (29%) and CDR3 (0%) of the heavy chain (Fig. S2). CDR2 of the heavy chain had a slightly higher sequence identity of 75%. The light chain CDR2 and CDR3 were completely distinct between 2C6 and 2D10 or 2H2, whereas light chain CDR1 showed 71% sequence identity to 2D10 and 2H2. These divergent CDRs further support the idea that 2D10 and 2H2 share epitopes, whereas 2C6 engages a unique epitope on DBP.
SAXS of the DBP-II/2H2Fab Complex Supports a Shared Binding Site with 2D10.
We compared the SAXS profiles of 2H2 and 2C6 Fab complexes with the DBP-II/2D10Fab model (Fig. 2C). The DBP-II/2H2Fab SAXS profile fit the DBP-II/2D10Fab model, with a χ2 value of 1.9. In contrast, the DBP-II/2C6Fab SAXS profile did not fit the DBP-II/2D10Fab model, returning a χ2 value of 5.1 (Fig. 2D), much higher than the significance cutoff value of 3 (43). The 2H2 and 2C6 complexes had an estimated MW of 82.6 kDa and 77 kDa, respectively as determined by SAXS MOW (44). This mass is consistent with the mass of a 1:1 complex (87 kDa) and indicates that the samples were not aggregated.
HDX-MS Determination of 2D10, 2H2, and 2C6 Epitopes.
Anticipating that HDX-MS would show increased protection at binding sites, we analyzed the epitopes for mAbs 2D10, 2H2, and 2C6. DBP-II contains multiple disulfide bonds, and conditions were optimized to obtain complete coverage of DBP-II (Fig. S3). The data successfully captured effects on HDX due to secondary structure elements (Fig. S3). The majority of peptides exhibited no significant differences in deuterium uptake, as represented by peptide 443–450 (Fig. 3 and Table S3); however, two identical peptides, 408–413 and 408–442, showed a clear decrease in exchange on binding of either 2D10 or 2H2 (Fig. 3 and Table S3). The latter peptides cover the complete epitope seen in the crystal structure of 2D10 in complex with DBP-II (Fig. 1). The results are consistent with the SAXS data and indicate that the two mAbs engage a similar region of DBP (Fig. 2). The complex of 2C6 with DBP resulted in a different array of peptides with reduction in deuterium uptake (Fig. 3 and Table S3); these peptides include 471–478 and 479–484 (Fig. 3), which are distinct and distant from the 2D10 epitope.
Fig. S3.

Sequence coverage of peptic digestion with an optimized quenching/reducing condition and HDX heat map for apo PvDBP. Each bar represents a peptide analyzed for deuterium content by mass spectrometry. Average deuterium uptake level and SD for the duplicate analysis across all exchange time points are listed on each bar. Bars are also color-coded, with warmer colors indicating higher deuterium uptake levels. Stable secondary structural features identified by crystallography (PDB ID code 3RRC) are represented by boxes underneath the corresponding segments.
Fig. 3.

Comparison of the kinetics of HDX for five regions of DBP in the presence of various mAbs (holo state, depicted in red) and in the absence of the mAb (apo state, depicted in blue). Each region (column) is represented by a peptic peptide and its charge state, as measured by mass spectrometry. Each row represents a state bound with a mAb; the antibody is listed on the right. Those regions showing reduced rates or extents of exchange for the holo state (red) are considered to contain the epitopes. Those regions showing no difference are examples of region that do not contain the epitopes, and can be viewed as controls.
Table S3.
Differential HDX analysis of a peptide set that provides comprehensive sequence coverage for PvDBP
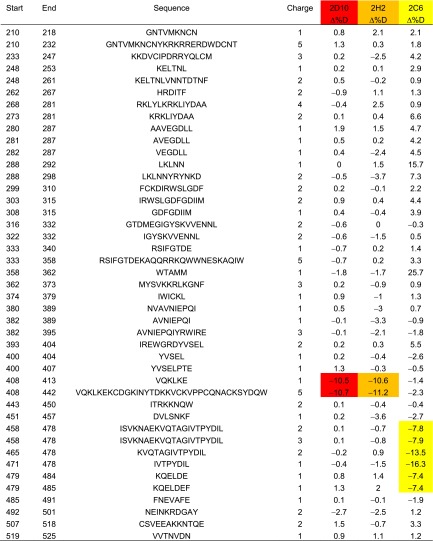 |
The ΔD% values represent the average difference of deuterium incorporation between apo (DBP alone) and the corresponding holo (DBP-antibody complex) states across all experimental time points. Negative values of ΔD% indicate less deuterium incorporation in the holo state. Peptides with significantly less D% induced by binding to 2D10, 2H2, or 2C6 are highlighted with a red, orange, or yellow background, respectively.
Mutation of the 2D10 Epitope Affects Binding to both 2D10 and 2H2.
The foregoing analyses revealed that mAbs 2D10 and 2H2 recognize a distinct epitope on DBP-II, which we assessed through mutational studies. Residues at the epitope center were substituted to either alanine or arginine (mutants 6 and 9, respectively). Sal-1 DBP-II and both mutants were expressed and purified, after which these proteins were adsorbed onto ELISA plates along with a BSA negative control. Both 2D10 and 2H2 recognized Sal-1 DBP-II (Fig. 4A) but did not bind either of the mutants (Fig. 4 B and C), consistent with the epitope seen in the crystal structure (Fig. 1). In addition, we probed the proteins with conformationally dependent mAbs 2C6 and 3D10, which engage distinct regions of DBP-II, and found that 2C6 and 3D10 bound Sal-1 DBP-II and both alanine and arginine mutants equally well, demonstrating that all proteins used were folded correctly (Fig. 4 A–C).
Fig. 4.

Determination of mAb epitopes by surface mutant libraries and ELISA. mAbs 2D10, 2H2, 3D10, and 2C6 were tested for binding against a panel of Sal-1 DBP surface mutants as described in Experimental Procedures. (A) Sal-1 DBP wild type. (B) Mutant 6 (K414A, P430A, P431A, N434A, K437A, Q441A). (C) Mutant 9 (N434R, K437R). (D) Mutant 1 (N218S, R221G, K222S, R223G). (E) Mutant 12 (K479A, Q480A). (F) Mutant 8 (K428A, P430A). All error bars are ±1 SD, calculated from three replicate experiments.
Mutational Screens Identify Critical Binding Residues for mAbs 2C6 and 3D10.
Difficulties in crystallizing DBP/mAb complexes of 2C6 and 3D10 led us to pursue alternative methods to map epitopes. We created an extensive panel of DBP-II variants in which predicted epitopes (46) and surface residues were altered. Each variant was expressed, purified, adsorbed on ELISA plates, and probed with mAbs 2C6 and 3D10, using mAbs 2D10 and 2H2 as controls (Fig. 4 D–F and Figs. S4 and S5).
Fig. S4.

Alignment of mutant sequences. An alignment of the sequences of all mutants tested in this study and wild type Sal-1 DBP, with mutated residues highlighted in black and the sub domains of DBP (SD1, red; SD2, blue; and SD3, green) outlined above the alignment.
Fig. S5.

Binding of monoclonal antibodies to a panel of Sal-1 DBP mutants. mAbs 2D10, 2H2, 3D10, and 2C6 were tested for binding against a panel of Sal-1 DBP mutants and BSA as described in Experimental Procedures. (A) BSA. (B) Mutant 2. (C) Mutant 3. (D) Mutant 4. (E) Mutant 5. (F) Mutant 7. (G) Mutant 10. (H) Mutant 11. (I) Mutant 13. (J) Mutant 14. (K) Mutant 15. (L) Mutant 16.
Mutant 1 contains changes at residues 218 and 221–223 in the N terminus of SD1 of DBP-II. This mutant showed ablated binding to 3D10, but not to 2D10, 2H2, or 2C6 (Fig. 4D). 3D10 is noninhibitory, with an IC50 several orders of magnitude weaker than that of 2D10 and 2H2 (32). The decreased potency can be attributed to the fact that this motif is not implicated in primary DARC receptor binding or DBP-II dimerization (19).
Mutant 12 contains changes at residues 479 and 480 in the C terminus of SD3 of DBP-II and shows ablated binding to 2C6, but not to 2D10, 2H2, or 3D10 (Fig. 4E). The loss of binding in this mutant is consistent with the HDX-MS results for 2C6. The mutated site is on a different face of the SD3 helical bundle compared with 2D10 and 2H2, and is located on the end closer to SD2.
Curiously, one of the mutants, mutant 8, showed preferential ablation of 2H2 binding over 2D10 binding (Fig. 4F). Mutant 8 contains altered residues N-terminal to amino acids changed in mutants 6 and 9 (Fig. S4). This demonstrates that 2D10 and 2H2 do not have identical epitopes, but instead share overlapping binding regions on DBP-II.
Antibody Competition ELISA Validates Mapped Epitopes.
We performed competition ELISAs with mAbs 2D10, 2H2, 3D10, and 2C6 to assess their ability to compete with immobilized 2D10 for binding to DBP-II (Fig. 5). 2D10 served as a positive control, and was able to compete with itself in a dose-dependent manner. 2H2 also was able to compete with 2D10 in a dose-dependent manner for binding to DBP-II. Neither 3D10 nor 2C6 competed with 2D10, because their epitopes are distinct (Figs. 4 D and E and 6A). Taken together, our results demonstrate that 2D10 and 2H2 bind to similar epitope surfaces on DBP-II and likely have similar inhibitory mechanisms.
Fig. 5.

Competition ELISA demonstrates that 2D10 and 2H2 share the same epitope. mAbs 2D10, 2H2, 3D10, and 2C6 were tested at different molar ratios to 2D10 (0.1×, 1×, and 10×) for the ability to compete with 2D10 for binding to DBP. All error bars are ±1 SD, calculated from three replicate experiments.
Fig. 6.

Epitopes of 2D10, 2H2, 2C6, and 3D10 mapped on PvDBP reveal that the epitopes are broadly conserved. (A) Epitopes are mapped on the surface of PvDBP with 2D10 in orange, 2H2 specific residues in purple, 3D10 in blue, and 2C6 in green. (B) Sequence of the Sal-1 DBP-II region with identified polymorphic sites indicated by dots above the individual residues. Structurally, mutationally, and HDX-MS (boxes)-identified epitopes are highlighted for 2D10 in orange, 2H2 in purple, for 2C6 in green, and for 3D10 in blue.
Discussion
The central role of DBP in P. vivax infection cements its importance as a vaccine target for P. vivax malaria (11). Anti–DBP-II Abs can inhibit DBP-DARC receptor binding and parasite invasion (7, 9, 18, 19, 32); however, high dN/dS ratios in DBP-II, a pattern seen when selective pressure drives allelic diversity as a mechanism for immune invasion (47–49), result in diverse polymorphic populations within endemic regions that manifest as mixed infections with multiple strains. These polymorphisms result in strain-specific immune responses that render individuals susceptible to future infections by alternate strains and require vaccine efforts to preemptively account for this vulnerability (50). Two approaches to overcoming this variability involve multiallele vaccines and artificially engineered synthetic immunogens (8, 37, 38, 47). Both approaches require the identification of broadly neutralizing epitopes as described here.
We have identified the first, to our knowledge, structure of DBP-II bound to an inhibitory mAb 2D10. The epitope lies within residues 413–441 at the tail end of the helical bundle that composes SD3 (Fig. 1). Through extensive analysis involving SAXS, sequence analyses, HDX-MS, and ELISA, we have demonstrated that a second inhibitory mAb, 2H2, has a similar paratope and epitope as 2D10. We also mapped the epitope of a third inhibitory mAb, 2C6, to a distinct helical face on SD3 (Fig. 4). In addition to the three highly inhibitory mAbs, we mapped the epitope of a noninhibitory mAb, 3D10, to the N-terminal end of SD1 (Fig. 4). HDX-MS and epitope mapping via in silico designed mutant panels are valuable new approaches to identifying mAb epitopes that are difficult to study via crystallography. In addition, the epitopes for inhibitory mAbs 2D10, 2H2, and 2C6 in SD3 suggest that a secondary receptor-binding event may occur between DARC and DBP-II outside of previously identified primary DARC-binding sites in SD2 (18, 19). The tail end of SD3 may contact extracellular loops linking the seven transmembrane regions of DARC that may play a role in DBP binding (51).
The polymorphic nature of DBP-II is reminiscent of highly variable viral surface proteins. bnAbs that engage the conserved receptor-binding site of HIV have been identified, some of which have the ability to neutralize 90% of known HIV strains (39). Similarly, bnAbs that recognize the conserved stem region of hemagglutinin are broadly protective against influenza (40). Comparison of the DBP mAb epitopes with naturally occurring polymorphisms revealed a lack of high-frequency polymorphisms in epitopes (Fig. 6B). This is consistent with the ability of these mAbs to bind divergent DBP alleles (32). DBP sequences that represent global diversity (35) were analyzed for retention of the 2D10, 2H2, and 2C6 epitopes. The 2D10 and 2H2 epitopes (residues 413–441) were invariant in 92.5% of the DBP sequences examined, whereas the 2C6 epitope (residues 465–485) was invariant in 94.4% of the DBP sequences examined. Together, 2D10, 2H2, and 2C6 recognize broadly conserved epitopes and likely function as bnAbs that will be effective against multiple pathogenic strains.
The identification of these protective motifs will aid tremendously in the design of DBP-II based vaccines that will impart global strain-transcending protection (8, 38, 47). Our findings characterize motifs that should be included in all future DBP-II vaccine designs and shed light on the mechanistic and structural process of how DBP engages DARC. Future studies will seek to map additional conserved protective motifs on DBP-II. The identification of additional DBP–DARC interaction sites will also be explored to fully characterize the DBP–DARC interaction. The results from both endeavors will significantly bolster the ability to create broadly protective vaccines against P. vivax malaria.
Experimental Procedures
Protein Expression and Purification.
Sal-1 DBP-II, BirA-tagged Sal-1 DBP-II, and DBP-II mutants were prepared as described previously (18, 19). Inclusion bodies were solubilized in 6 M guanidinium hydrochloride and refolded via rapid dilution into 400 mM l-arginine, 50 mM Tris (pH 8.0), 10 mM EDTA, 0.1 mM PMSF, 2 mM reduced glutathione, and 0.2 mM oxidized glutathione. Refolded proteins were captured on SP Sepharose Fast Flow resin (GE Healthcare) and purified by size-exclusion chromatography (GF200; GE Healthcare). For crystallization, Sal-1 DBP-II was further purified by ion exchange chromatography (HiTrapS - GE Healthcare) and size-exclusion chromatography (GF75; GE Healthcare) into Buffer A [10 mM Hepes (pH 7.4) and 100 mM NaCl].
Fab Generation and Purification.
Fab fragments for 2D10, 2H2, and 2C6 were generated from IgG using immobilized papain and protein A resin (Thermo Fisher Scientific) as described previously (31). The eluted Fab fragments were purified by size-exclusion chromatography in Buffer A.
Antibody Sequencing.
RNA was extracted from hybridoma cells using QiaShredder (Qiagen) and the RNeasy Mini Kit (Qiagen). The 5′ RACE Kit (Invitrogen) was used to obtain cDNA for the heavy chain and light chain variable regions. RACE primers for the heavy chain were GSP1 for the IgG1 isotype, TGCATTTGAACTCCTTGCC, and GSP2 for the IgG1 isotype, CTTTGGGGGGAAGATGAAG. RACE primers for the light chain were GSP1 for Cκ, CACTCATTCCTGTTGAAGC, and GSP2 for Cκ, CTTGTGAGTGGCCTCACAGG. cDNA was TOPO cloned (Invitrogen) and sequenced.
Synthesis and Expression of mAb 2D10 scFv.
The light chain variable region was linked to the heavy chain variable region using a (GGGGS)3 linker and cloned into the pHLSec expression vector using restriction sites AgeI/KpnI (52). scFv was obtained by transient transfection in HEK293F cells, and purified by Ni-NTA and size-exclusion chromatography into Buffer A.
Protein Crystallization and Data Collection.
DBP-II and 2D10scFv were mixed in a 0.9:1 molar ratio and incubated at 4 °C for 30 min. The complex was purified by size-exclusion chromatography (GF75; GE Healthcare) in Buffer A. Crystals were grown by hanging-drop vapor diffusion, mixing 1 µL of complex at 18 mg/mL with either 3 μL or 4 µL of reservoir [1% (wt/vol) tryptone, 50 mM Hepes sodium salt (pH 7.0), and 12% (wt/vol) PEG 3350]. Crystals were flash-frozen with 30% PEG 400 as a cryoprotectant in liquid nitrogen. Data were collected at beamline 4.2.2 of the Advanced Light Source (ALS) and processed with XDS (53).
Structure Solution and Analysis.
The DBP-II/2D10scFv structure was solved by molecular replacement in PHASER (54) using PDB 4NUV and a modeled scFv domain from the PIGS server (55) as starting models. Rigid body refinement in PHENIX (56) resulted in Rwork/Rfree of 41.24%/42.48%. DEN refinement in CNS (57), followed by refinement in PHENIX and rebuilding in COOT (58) led to a final model with Rfactor/Rfree of 26.69%/29.97% with good geometry, as reported by MOLPROBITY (59) (Table S1). Interaction interfaces were determined using PDBePISA (60). Software used in the project was installed and configured by SBGrid (61). Atomic coordinates and structural factors have been deposited into the Protein Data Bank (ID code 5F3J).
SAXS.
Data for 2D10, 2H2, and 2C6 DBP-II/Fab were collected at the SIBYLS beamline 12.3.1 at ALS using standard procedures (62), and analyzed using the ATSAS package, version 2.5.2–1 (63). Data quality was assessed using PRIMUS. Experimental profiles were compared with the structure using CRYSOL. Ab initio model generation was performed in DAMMIF, and the filtered average envelope of 10 models was obtained by DAMAVER. SUPCOMB20 was used to align structures and SAXS reconstructions. The MW estimate was obtained using SAXS-MOW (44). Pairwise SAXS comparisons were performed using the volatility of ratio (VR) metric (45).
Hydrogen-Deuterium Exchange Mass Spectrometry.
Each holo-state sample was prepared by incubating 10 µM PvDBP with 12 µM of mAb in PBS for 30 min at 25 °C. The apo-state sample was 10 µM PvDBP in PBS. Continuous HDX on apo- and holo-state samples were performed at 10, 30, 60, 120, 900, 3,600, and 14,400 s, as described previously (64). To better reduce and denature the highly disulfide-bonded antigen for comprehensive coverage, an intensive quenching and reducing protocol was developed that included a high concentration of the reducing agent, increased temperature, and prolonged incubation. Porcine pepsin, guanidine hydrochloride, water, acetonitrile, formic acid, trifluoroacetic acid, sodium hydroxide, and PBS were purchased from Sigma-Aldrich. Tris-(2-carboxyethyl) phosphine hydrochloride was purchased from Pierce Biotechnology. D2O was purchased from Cambridge Isotope Laboratories.
ELISA Binding Assays with Anti-DBP mAbs.
The ELISA assays were performed as described previously (25). In brief, BSA, Sal1 DBP-II, and DBP-II mutants were coated on plates overnight at 4° C, washed with PBS/Tween-20, and blocked with 2% BSA in PBS/Tween-20 for 1 h at room temperature. The plates were washed with PBS/Tween-20 and then incubated with each mAb (2D10, 2H2, 3D10, and 2C6) for 1 h at room temperature. Then the plates were washed again with PBS/Tween-20 and incubated with an anti-mouse antibody conjugated to Alexa Fluor 488 for 30 min at room temperature. After a final washing step, the fluorescence was measured using a POLARstar Omega plate reader (BMG Labtech).
Biotinylation of BirA-Tagged Sal-1 DBP-II.
BirA-tagged Sal-1 DBP-II was buffer-exchanged into biotinylation buffer [100 mM Tris (pH 7.5), 200 mM NaCl, and 5 mm MgCl2]. Then 50 μL of BiomixA (Avidity), 50 μL of BiomixB (Avidity), and 100 µL of d-biotin (Avidity) were added to the protein along with BirA ligase, followed by overnight incubation at 4 °C. The reaction mix was buffer-exchanged into PBS before use.
Competition ELISA with Anti-DBP mAbs.
Here 10 μg of mAb 2D10 per well was coated on plates overnight at 4 °C, then washed with PBS/Tween-20, and blocked with 2% BSA in PBS/Tween-20 for 1 h at room temperature. The plates were washed again with PBS/Tween-20, incubated with 100 µL mixtures of 2.5 μg of biotinylated Sal-1 DBP-II and either 1 μg, 10 µg, or 100 μg of competitor for 1 h at room temperature. The plates were washed with again PBS/Tween-20 and then incubated with 100 µL of a 1:5,000 dilution of streptavidin-HRP (Thermo Fisher Scientific) for 1 h at room temperature. Then 100 μL of 3,3′,5,5′-Tetramethylbenzidine (Sigma-Aldrich) was added, and the reaction was quenched with 100 µL of 2 M H2SO4. Absorbance at 450 nm was measured using a POLARstar Omega (BMG Labtech) plate reader.
DBP Epitope Conservation Analysis.
A total of 599 sequences representing global variation in DBP-II (35) were obtained from GenBank (accession codes XM_001608337.1, DQ156519, AF289480–AF289483, AF289635–AF289653, AF291096, AY970837–AY970925, AF469515–AF469602, U50575–U50590, DQ156513, DQ156515, DQ156522–DQ156523, AF215737–AF215738, AF220657, AF220659–AF220667, FJ491142–FJ491241, EF219451, EF368159–EF368180, EF379127–EF379132, EF379134, GU143914–GU144013, DQ156520, EU812839–EU812960, and EU860428–EU860438), aligned using ClustalW, and inspected in JalView. All 599 sequences contained complete coverage for the 2D10 and 2H2 epitopes, and 195 sequences contained complete coverage of the 2C6 epitope.
Acknowledgments
We thank J. Nix and Advanced Light Source Beamline 4.2.2 (supported by US Department of Energy Contract DE-AC02-05CH11231), and the Rheumatic Diseases Core Center [under National Institutes of Health (NIH) Grant P30 AR048335]. This work was supported by NIH Grant R56 AI080792 (to N.H.T.), NIH Contract HHSN272201400018C (to N.H.T., J.H.A., and M.L.G.), NIH Grant R01 AI064478 (to J.H.A. and N.H.T.), and NIH Grant T32 AI007172 (to E.C.); and the Burroughs Wellcome Fund Grant 1013514 (to N.H.T.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 5F3J). All epitopes determined here have been deposited with the Immune Epitope Database, www.iedb.org (submission ID 1000705).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1600488113/-/DCSupplemental.
References
- 1.Carlton JM, Sina BJ, Adams JH. Why is Plasmodium vivax a neglected tropical disease? PLoS Negl Trop Dis. 2011;5(6):e1160. doi: 10.1371/journal.pntd.0001160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tjitra E, et al. Multidrug-resistant Plasmodium vivax associated with severe and fatal malaria: A prospective study in Papua, Indonesia. PLoS Med. 2008;5(6):e128. doi: 10.1371/journal.pmed.0050128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Genton B, et al. Plasmodium vivax and mixed infections are associated with severe malaria in children: A prospective cohort study from Papua New Guinea. PLoS Med. 2008;5(6):e127. doi: 10.1371/journal.pmed.0050127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guerra CA, et al. The international limits and population at risk of Plasmodium vivax transmission in 2009. PLoS Negl Trop Dis. 2010;4(8):e774. doi: 10.1371/journal.pntd.0000774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Price RN, et al. Vivax malaria: Neglected and not benign. Am J Trop Med Hyg. 2007;77(6) Suppl:79–87. [PMC free article] [PubMed] [Google Scholar]
- 6.King CL, et al. Naturally acquired Duffy-binding protein-specific binding inhibitory antibodies confer protection from blood-stage Plasmodium vivax infection. Proc Natl Acad Sci USA. 2008;105(24):8363–8368. doi: 10.1073/pnas.0800371105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grimberg BT, et al. Plasmodium vivax invasion of human erythrocytes inhibited by antibodies directed against the Duffy binding protein. PLoS Med. 2007;4(12):e337. doi: 10.1371/journal.pmed.0040337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ntumngia FB, et al. Immunogenicity of a synthetic vaccine based on Plasmodium vivax Duffy binding protein region II. Clin Vaccine Immunol. 2014;21(9):1215–1223. doi: 10.1128/CVI.00205-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chootong P, et al. Mapping epitopes of the Plasmodium vivax Duffy binding protein with naturally acquired inhibitory antibodies. Infect Immun. 2010;78(3):1089–1095. doi: 10.1128/IAI.01036-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller LH, Mason SJ, Dvorak JA, McGinniss MH, Rothman IK. Erythrocyte receptors for (Plasmodium knowlesi) malaria: Duffy blood group determinants. Science. 1975;189(4202):561–563. doi: 10.1126/science.1145213. [DOI] [PubMed] [Google Scholar]
- 11.Miller LH, Mason SJ, Clyde DF, McGinniss MH. The resistance factor to Plasmodium vivax in blacks: The Duffy-blood-group genotype, FyFy. N Engl J Med. 1976;295(6):302–304. doi: 10.1056/NEJM197608052950602. [DOI] [PubMed] [Google Scholar]
- 12.Wertheimer SP, Barnwell JW. Plasmodium vivax interaction with the human Duffy blood group glycoprotein: Identification of a parasite receptor-like protein. Exp Parasitol. 1989;69(4):340–350. doi: 10.1016/0014-4894(89)90083-0. [DOI] [PubMed] [Google Scholar]
- 13.Adams JH, et al. The Duffy receptor family of Plasmodium knowlesi is located within the micronemes of invasive malaria merozoites. Cell. 1990;63(1):141–153. doi: 10.1016/0092-8674(90)90295-p. [DOI] [PubMed] [Google Scholar]
- 14.Adams JH, et al. A family of erythrocyte binding proteins of malaria parasites. Proc Natl Acad Sci USA. 1992;89(15):7085–7089. doi: 10.1073/pnas.89.15.7085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chitnis CE, Chaudhuri A, Horuk R, Pogo AO, Miller LH. The domain on the Duffy blood group antigen for binding Plasmodium vivax and P. knowlesi malarial parasites to erythrocytes. J Exp Med. 1996;184(4):1531–1536. doi: 10.1084/jem.184.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ranjan A, Chitnis CE. Mapping regions containing binding residues within functional domains of Plasmodium vivax and Plasmodium knowlesi erythrocyte-binding proteins. Proc Natl Acad Sci USA. 1999;96(24):14067–14072. doi: 10.1073/pnas.96.24.14067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.VanBuskirk KM, Sevova E, Adams JH. Conserved residues in the Plasmodium vivax Duffy-binding protein ligand domain are critical for erythrocyte receptor recognition. Proc Natl Acad Sci USA. 2004;101(44):15754–15759. doi: 10.1073/pnas.0405421101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Batchelor JD, Zahm JA, Tolia NH. Dimerization of Plasmodium vivax DBP is induced upon receptor binding and drives recognition of DARC. Nat Struct Mol Biol. 2011;18(8):908–914. doi: 10.1038/nsmb.2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Batchelor JD, et al. Red blood cell invasion by Plasmodium vivax: Structural basis for DBP engagement of DARC. PLoS Pathog. 2014;10(1):e1003869. doi: 10.1371/journal.ppat.1003869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salinas ND, Tolia NH. Red cell receptors as access points for malaria infection. Curr Opin Hematol. 2016;23(3):215–223. doi: 10.1097/MOH.0000000000000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malpede BM, Tolia NH. Malaria adhesins: Structure and function. Cell Microbiol. 2014;16(5):621–631. doi: 10.1111/cmi.12276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paing MM, Tolia NH. Multimeric assembly of host-pathogen adhesion complexes involved in apicomplexan invasion. PLoS Pathog. 2014;10(6):e1004120. doi: 10.1371/journal.ppat.1004120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tolia NH, Enemark EJ, Sim BK, Joshua-Tor L. Structural basis for the EBA-175 erythrocyte invasion pathway of the malaria parasite Plasmodium falciparum. Cell. 2005;122(2):183–193. doi: 10.1016/j.cell.2005.05.033. [DOI] [PubMed] [Google Scholar]
- 24.Salinas ND, Tolia NH. A quantitative assay for binding and inhibition of Plasmodium falciparum Erythrocyte Binding Antigen 175 reveals high affinity binding depends on both DBL domains. Protein Expr Purif. 2014;95:188–194. doi: 10.1016/j.pep.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salinas ND, Paing MM, Tolia NH. Critical glycosylated residues in exon three of erythrocyte glycophorin A engage Plasmodium falciparum EBA-175 and define receptor specificity. MBio. 2014;5(5):e01606–e01614. doi: 10.1128/mBio.01606-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sim BK, Chitnis CE, Wasniowska K, Hadley TJ, Miller LH. Receptor and ligand domains for invasion of erythrocytes by Plasmodium falciparum. Science. 1994;264(5167):1941–1944. doi: 10.1126/science.8009226. [DOI] [PubMed] [Google Scholar]
- 27.Malpede BM, Lin DH, Tolia NH. Molecular basis for sialic acid-dependent receptor recognition by the Plasmodium falciparum invasion protein erythrocyte-binding antigen-140/BAEBL. J Biol Chem. 2013;288(17):12406–12415. doi: 10.1074/jbc.M113.450643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin DH, Malpede BM, Batchelor JD, Tolia NH. Crystal and solution structures of Plasmodium falciparum erythrocyte-binding antigen 140 reveal determinants of receptor specificity during erythrocyte invasion. J Biol Chem. 2012;287(44):36830–36836. doi: 10.1074/jbc.M112.409276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ménard D, et al. Plasmodium vivax clinical malaria is commonly observed in Duffy-negative Malagasy people. Proc Natl Acad Sci USA. 2010;107(13):5967–5971. doi: 10.1073/pnas.0912496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Menard D, et al. Whole genome sequencing of field isolates reveals a common duplication of the Duffy binding protein gene in Malagasy Plasmodium vivax strains. PLoS Negl Trop Dis. 2013;7(11):e2489. doi: 10.1371/journal.pntd.0002489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen E, Paing MM, Salinas N, Sim BK, Tolia NH. Structural and functional basis for inhibition of erythrocyte invasion by antibodies that target Plasmodium falciparum EBA-175. PLoS Pathog. 2013;9(5):e1003390. doi: 10.1371/journal.ppat.1003390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ntumngia FB, et al. Conserved and variant epitopes of Plasmodium vivax Duffy binding protein as targets of inhibitory monoclonal antibodies. Infect Immun. 2012;80(3):1203–1208. doi: 10.1128/IAI.05924-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cole-Tobian JL, et al. Strain-specific Duffy binding protein antibodies correlate with protection against infection with homologous compared to heterologous Plasmodium vivax strains in Papua New Guinean children. Infect Immun. 2009;77(9):4009–4017. doi: 10.1128/IAI.00158-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.VanBuskirk KM, et al. Antigenic drift in the ligand domain of Plasmodium vivax Duffy binding protein confers resistance to inhibitory antibodies. J Infect Dis. 2004;190(9):1556–1562. doi: 10.1086/424852. [DOI] [PubMed] [Google Scholar]
- 35.Nóbrega de Sousa T, Carvalho LH, Alves de Brito CF. Worldwide genetic variability of the Duffy binding protein: Insights into Plasmodium vivax vaccine development. PLoS One. 2011;6(8):e22944. doi: 10.1371/journal.pone.0022944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ceravolo IP, et al. Naturally acquired inhibitory antibodies to Plasmodium vivax Duffy binding protein are short-lived and allele-specific following a single malaria infection. Clin Exp Immunol. 2009;156(3):502–510. doi: 10.1111/j.1365-2249.2009.03931.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ntumngia FB, et al. Immunogenicity of single versus mixed allele vaccines of Plasmodium vivax Duffy binding protein region II. Vaccine. 2013;31(40):4382–4388. doi: 10.1016/j.vaccine.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen E, Salinas ND, Ntumngia FB, Adams JH, Tolia NH. Structural analysis of the synthetic Duffy Binding Protein (DBP) antigen DEKnull relevant for Plasmodium vivax malaria vaccine design. PLoS Negl Trop Dis. 2015;9(3):e0003644. doi: 10.1371/journal.pntd.0003644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou T, et al. NISC Comparative Sequencing Program Structural repertoire of HIV-1–neutralizing antibodies targeting the CD4 supersite in 14 donors. Cell. 2015;161(6):1280–1292. doi: 10.1016/j.cell.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ekiert DC, Wilson IA. Broadly neutralizing antibodies against influenza virus and prospects for universal therapies. Curr Opin Virol. 2012;2(2):134–141. doi: 10.1016/j.coviro.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dormitzer PR, Grandi G, Rappuoli R. Structural vaccinology starts to deliver. Nat Rev Microbiol. 2012;10(12):807–813. doi: 10.1038/nrmicro2893. [DOI] [PubMed] [Google Scholar]
- 42.Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234(4):946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 43.Petoukhov MV, Svergun DI. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys J. 2005;89(2):1237–1250. doi: 10.1529/biophysj.105.064154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fischer H, de Oliveira Neto M, Napolitano HB, Polikarpov I, Craievich AF. Determination of the molecular weight of proteins in solution from a single small-angle X-ray scattering measurement on a relative scale. J Appl Cryst. 2010;43(1):101–109. [Google Scholar]
- 45.Hura GL, et al. Comprehensive macromolecular conformations mapped by quantitative SAXS analyses. Nat Methods. 2013;10(6):453–454. doi: 10.1038/nmeth.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haste Andersen P, Nielsen M, Lund O. Prediction of residues in discontinuous B-cell epitopes using protein 3D structures. Protein Sci. 2006;15(11):2558–2567. doi: 10.1110/ps.062405906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ntumngia FB, Adams JH. Design and immunogenicity of a novel synthetic antigen based on the ligand domain of the Plasmodium vivax Duffy binding protein. Clin Vaccine Immunol. 2012;19(1):30–36. doi: 10.1128/CVI.05466-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cole-Tobian J, King CL. Diversity and natural selection in Plasmodium vivax Duffy binding protein gene. Mol Biochem Parasitol. 2003;127(2):121–132. doi: 10.1016/s0166-6851(02)00327-4. [DOI] [PubMed] [Google Scholar]
- 49.Baum J, Thomas AW, Conway DJ. Evidence for diversifying selection on erythrocyte-binding antigens of Plasmodium falciparum and P. vivax. Genetics. 2003;163(4):1327–1336. doi: 10.1093/genetics/163.4.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Welsh RM, Fujinami RS. Pathogenic epitopes, heterologous immunity and vaccine design. Nat Rev Microbiol. 2007;5(7):555–563. doi: 10.1038/nrmicro1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tournamille C, et al. Close association of the first and fourth extracellular domains of the Duffy antigen/receptor for chemokines by a disulfide bond is required for ligand binding. J Biol Chem. 1997;272(26):16274–16280. doi: 10.1074/jbc.272.26.16274. [DOI] [PubMed] [Google Scholar]
- 52.Aricescu AR, Lu W, Jones EY. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 10):1243–1250. doi: 10.1107/S0907444906029799. [DOI] [PubMed] [Google Scholar]
- 53.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marcatili P, Rosi A, Tramontano A. PIGS: Automatic prediction of antibody structures. Bioinformatics. 2008;24(17):1953–1954. doi: 10.1093/bioinformatics/btn341. [DOI] [PubMed] [Google Scholar]
- 56.Adams PD, et al. PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58(Pt 11):1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 57.Schröder GF, Levitt M, Brunger AT. Super-resolution biomolecular crystallography with low-resolution data. Nature. 2010;464(7292):1218–1222. doi: 10.1038/nature08892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 59.Davis IW, et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35(Web Server Issue):W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372(3):774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 61.Morin A, et al. Collaboration gets the most out of software. eLife. 2013;2:e01456. doi: 10.7554/eLife.01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hura GL, et al. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat Methods. 2009;6(8):606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Petoukhov MV, et al. New developments in the ATSAS program package for small-angle scattering data analysis. J Appl Cryst. 2012;45(Pt 2):342–350. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yan Y, Grant GA, Gross ML. Hydrogen-deuterium exchange mass spectrometry reveals unique conformational and chemical transformations occurring upon [4Fe-4S] cluster binding in the type 2 L-serine dehydratase from Legionella pneumophila. Biochemistry. 2015;54(34):5322–5328. doi: 10.1021/acs.biochem.5b00761. [DOI] [PMC free article] [PubMed] [Google Scholar]