

Abstract
Advances in technology and data analysis have made it possible to take a new look at human immunology. These advances run the gamut from systems biology approaches, which are likely in the vanguard of how we can start “to put the pieces together” of immune function, to a deeper understanding of specific diseases and vaccines and the immune repertoire. In our own experience, we have also found that asking simple questions about human immunity has often given us very surprising answers, causing a rethink of established dogma. Thus, we have developed a new perspective on the nature of the αβ TCR repertoire and also the likely role of T-cell repertoire (TCR) cross-reactivity in generating T memory independent of specific antigen interactions. These findings show that human immunology is not just a necessary step for “translating” basic immunology to treat diseases or develop better vaccines, but is also an important complement to the inbred mouse model.
Although we have learned a great deal about basic immunology from the laboratory mouse model, it has significant limitations in terms of genetics, environmental exposure, and evolutionary distance from human beings. These differences, together with the necessarily artificial constraints of disease models (Casanova and Abel 2005), are likely the reason that it has been so difficult to apply immunotherapies developed in mouse models to many human diseases (Davis 2008). Although there have been notable successes (Feldmann and Maini 2001; Pentcheva-Hoang et al. 2009; Grosso and Jure-Kunkel 2013), these are rare exceptions rather than rules (Seok et al. 2013). This suggests that it should be a priority to make direct studies of human immunology to both better “translate” immunology and to learn whatever this may teach us about the basic biology of another system. In the latter respect, the human immune system has some significant advantages, including a much broader scope in terms of naturally acquired infectious diseases, spontaneous autoimmunity, and environmental and genetic variations than are available to us with laboratory mice. Technical, regulatory, timescale, and ethical challenges have of course discouraged many from taking human immunology studies, but what we hope to show in this chapter is that although these limitations are quite real, there are ways to approach important and basic immunological questions using human material that can significantly add to our knowledge of immunology in general. Specifically, we have used both peptide–major histocompatibility complex (MHC) tetramers and systems biology approaches to study the human immune system. With respect to the αβ T-cell repertoire, we find many T cells in adults, but not newborns, that have a memory phenotype for pathogen epitopes for which the individual has not been exposed (Su et al. 2013). In the case of an inducible autoimmune disease, celiac sprue, we have found that an intriguing cascade of different T-cell types (CD4+, CD8+, and γδ+) are induced by gluten ingestion (Han et al. 2013). In general, we are finding that studying the human immune system directly in selected human cohorts is very complementary to mouse studies and in some cases provides a much broader view. We are also applying systems biology approaches to human immunology, because this promises a means to understand the role of different immune system components in vaccine responses and disease. Here, we have found a role for apoptosis in predicting successful responders to an influenza vaccine (Furman et al. 2013a) and also that higher testosterone levels in men correlates with poor vaccine response (Furman et al. 2013b). Ongoing work also involves the analysis of immune biomarkers in twins to assess the role of genetics versus the environment and in comparisons of cohorts in the developing world versus those in the United States and in different age groups to determine the effects of the environment and aging, respectively (Brodin et al., in prep.).
SYSTEMS APPROACHES TO IMMUNOLOGY
The advent of broad, high-throughput assay methods to expansively survey gene expression and other aspects of the immune system has raised the possibility that one could learn important things about the immune system in a human blood sample (Davis 2008). This is important because blood samples contain a microcosm of the immune system in the circulating white blood cells they contain and they are among the easiest clinical samples to obtain. This has made blood a major target of our efforts, because in the appropriate clinical setting, one can obtain blood samples from all ages, sick or healthy, and in large quantities from blood donors. Although it is much more difficult to obtain other types of human samples, a critical discovery of Wilson and colleagues (unpubl.) is that 6 or 7 d after vaccination for influenza, specific plasmablasts enter the circulatory system and thus can be captured at that time—a principle that has been extended to other vaccines and infectious diseases and autoimmunity (as discussed below). This is also true for some T-cell types, and it was shown by Sollid and colleagues (Sollid et al. 2012) that 6 d after gluten challenge, celiac patients who had been on a gluten-free diet had specific CD4+ T cells entering the bloodstream (Raki et al. 2007). Thus, there appears to be a general mobilization of specific lymphocytes into the circulatory system 6 or 7 d after a significant immunological event. This creates a unique opportunity to study these cells, which are otherwise difficult to access, as we illustrate here. Early systems' immunology studies were pioneered by Sékaly and Pulendran and their respective colleagues (Querec et al. 2006; Gaucher et al. 2008), who focused on analyzing changes in gene expression with vaccination, initially with the yellow fever vaccine (YF-17D), one of the most effective vaccines known. Vaccination is an important way to understand human immunology because it is widely used and poses fewer safety risks. In addition, there are many varieties of vaccines, and there is a clear need to understand how they work because we now find ourselves trying to develop next-generation vaccines for the diseases such as HIV, TB, and others where classical approaches have proven to be largely inadequate. It is also useful as a way to stimulate the immune system broadly to see how the different components of the system “move” especially with respect to one another. Here, Querec and colleagues found that vaccination triggered changes in dendritic cells (DCs) that correlated with effective antibody responses (Querec et al. 2006). This is consistent with the known role of DCs in presenting antigens robustly to T cells and B cells, as Gaucher and colleagues found (Gaucher et al. 2008). In a later study of influenza vaccination, a much less successful vaccine with respect to the correlates of protection based on antibody titers, Nakaya et al. (2011) found that a robust innate immune response preceded and may be causal to a robust antibody response to flu. In our studies, we have focused on using both gene expression using a module approach together with a variety of other data, including dozens of serum cytokines and phenotypic analysis of white blood cells (WBCs) in old and young subjects before vaccination (Furman et al. 2013a). In addition, in collaboration with PJ Utz (Price et al. 2013), we also surveyed an array of flu peptides that gave interesting insights into the preexisting antibody repertoire of the subjects in this study (Furman et al. 2013a; Price et al. 2013). Although limited to linear epitopes, we saw robust serological responses, some of which correlated positively with the neutralizing antibody response to the flu strains in the vaccine but others correlated negatively. This is an interesting reminder that some antibody responses can be a diversion from effective ones. In Figure 1, we show the results of analysis of different biomarkers that have a statistically significant correlation with either a positive or negative effect on the probability that a subject will give a good antibody response to a seasonal influenza vaccine. This is a study of 89 individuals, 30 of whom are young adults (20–30 yr of age) and 59 the older elderly (60− 89+ yr). The top positive predictor of vaccine responsiveness is a gene module that is heavily weighted with genes involved in apoptosis, and a minor negative predictor is soluble Fas ligand, also a factor in apoptosis. This indicates that apoptosis is an important factor in flu vaccine responsiveness and perhaps others as well. This is interesting because one indicator of immune senescence in elderly people is the accumulation of large numbers of memory phenotype T cells and B cells. This could potentially be explained by a drop in the efficiency with which these cells are “pruned” to allow new responses to occur. Another important link between apoptosis and immune responses is the activation of the inflammasome. These protein complexes induce proinflammatory caspases causing cell death via pyroptosis and apoptosis and the production of inflammatory cytokines IL-1β and IL-18 that regulate both T-cell and B-cell responses (Lamkanfi and Dixit 2012). Other data have shown that apoptotic mechanisms are important for the direct killing of bacteria-infected cells in murine lungs, suggesting another way in which this mechanism may be important for immunological health (Weber et al. 2012).
Figure 1.
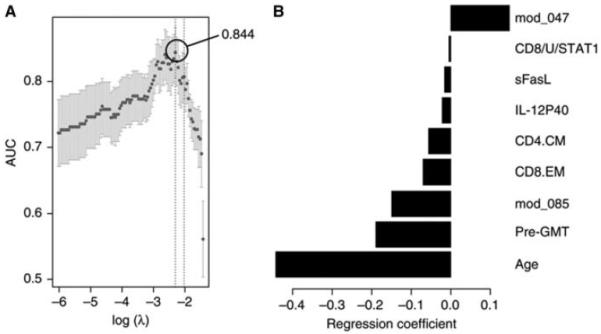
Baseline variables that correlate positively or negatively with a robust serological response in a seasonal influenza vaccine. A total of 241 variables in 89 subjects were assessed for their contribution to a robust serological response to a seasonal flu vaccine (TIV) before immunization. This includes 109 gene modules, serum cytokines, cell subsets, age, preimmune titer (pre-GMT), and in vitro cytokine stimulation of peripheral blood mononuclear cells (PBMCs) as measured by the signal transducer and activator of transcription (STAT) phosphorylation. The cross-validated region under each curve (cvAUROC) (A) and regression coefficients for each of the nine features that emerged as significant (B) are shown. Only a gene module enriched in apoptotic genes was a positive predictor, and the rest were negative. (Adapted from Furman et al. 2013a.)
Together, these variables gave us an estimated 83% probability of success in predicting which subjects, old or young, would respond well to a standard influenza vaccine response in the years surveyed. Still, age and preexisting titer are the major variables, as shown in Fig. 1. But there are many other questions to resolve in studies of this kind, for example, whether these markers carry over to other vaccines. or different formulations of the same vaccines (such as adjuvants, which currently approved flu vaccines in the United States do not have), live versus “dead” vaccines. And a great unknown is how to assess T-cell contributions to immunity—we know that they must contribute, with a simple example being that the live attenuated form of flu vaccine (LAIV) is quite effective, perhaps equally effective as that of the killed vaccine, but it is much poorer at eliciting “protective antibodies.” So it seems likely that their effectiveness must involve some enhanced T cell or other components, but the component has not been identified yet. This points to the inadequacy of the usual gold standard—neutralizing antibodies. Yes, they can be important, but are they the whole story? Very unlikely. The real gold standard is live virus “challenge” studies, which are much more difficult to perform, and here, a recent report using this kind of approach has identified CD4+ T-cell responses as the best indicator of a successful immune response to the virus, as indicated by reduced disease severity (Xian et al. 2012).
So at this time, it is clear that we have just fragments of the story regarding what influenza and other vaccines do and why different people respond to them differently. It would also be helpful to have better technologies to assess the diverse types of T-cell response, candidates for which we describe below. And it also would be extremely interesting to know how individuals with disregulated immune systems (such as patients with autoimmunity) respond to a standard vaccination such as flu, or how children with broad exposure to the many pathogens in third-world countries respond compared with those raised in the relatively hygienic confines of developed countries.
Another set of questions that is amenable to a system's approach is the differences between men and women with respect to their immune responses and susceptibility to disease. In particular, it has long been known that women and female mice tend to have stronger immune responses to infectious diseases and vaccination (Klein and Poland 2013) than males and that they are also much more susceptible to autoimmune diseases (Amur et al. 2012), but the molecular mechanism(s) of this effect is not known. To address this, we surveyed a cohort of 87 nominally healthy adults from ages 20 to 89+ with respect to gene expression, serum cytokines, and their serological response to influenza vaccination (Furman et al. 2013a). We found that females showed significantly higher serum levels of particular inflammatory cytokines (LEPTIN, IL-1RA, CRP, GM-CSF, and IL-5) regardless of age, but that the elevation of these cytokines did not correlate significantly with the antibody response to the influenza vaccine. However, what did correlate was a gene module containing genes that regulate lipid metabolism and others that down-regulate particular immune cell activation pathways. Because some of those genes were known to be regulated by testosterone, we then measured testosterone levels in males in the cohort and found that those in the highest-level group had very depressed antibody responses to the vaccine, compared with those with lower levels or with the female group (Fig. 2). This fits somewhat with data in animals showing that testosterone has anti-inflammatory properties and can suppress the lethal effects of lipopolysaccharide (LPS) injection, for example (Laubach et al. 1998). It is also consistent with the data showing that women are twice as likely to die from sepsis than men. Thus, there are likely beneficial effects of testosterone as well as unhelpful ones, and this should inform vaccine development going forward as well as provide some basic data to further investigate the mechanisms of these effects.
Figure 2.
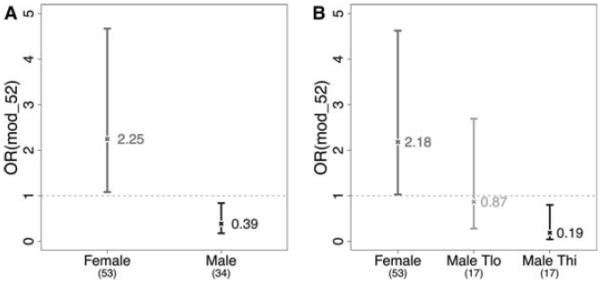
Male and female vaccine responses correlate with gene module 52 and testosterone levels. The graphs provide odds ratios (OR) for gene module 52 and high and low testosterone levels in males as correlating with a robust serological response in a seasonal influenza vaccine. (Reprinted from Furman et al. 2013b.)
CELIAC DISEASE
Celiac disease (CD) is one of the most common human autoimmune diseases, with an estimated prevalence of 1% among people of European ancestry. It is characterized by small intestinal mucosal injury and nutrient malabsorption in genetically susceptible individuals, due to dietary gluten ingestion (Green and Cellier 2007; Meresse et al. 2012). Similar to many human autoimmune diseases, CD has a clear association with human leukocyte antigen (HLA) Class II alleles. Inflammation depends on CD4+ T-cell responses to dietary gluten in HLA-DQ2/8-positive individuals. Aside from its clinical importance, some unique features of CD make it an especially attractive system in the study of human immunology. The triggering antigen in CD is known, and exposure to the antigen can be readily controlled.
Although the importance of dietary gluten in the initiation of CD4+ T-cell responses has been well established, the link between CD4+ T-cell activation and disease has been unclear. No gluten-induced enteropathy is seen in mouse models expressing a gluten-specific TCR (de Kauwe et al. 2009; Du Pre et al. 2011), suggesting that specific CD4+ T cells alone are unable to induce tissue damage (Fallang et al. 2009; Sollid et al. 2012). The cells thought to be responsible for gut inflammation in CD are intestinal intraepithelial lymphocytes (IELs), composed of both CD8+ αβ T cells and γδ T cells. The link between dietary gluten and the CD4+ response is well established, but the link between dietary gluten and the recruitment and activation of IELs in celiac disease is unknown. Prevailing models had proposed that celiac IEL were activated as bystanders due to inflammation initiated by gluten-specific CD4+ T cells.
We recently described the presence of large numbers of activated CD8+ αβ T cells and γδ T cells bearing intestinal epithelial homing markers in the peripheral blood, peaking at ~6 d following gluten exposure in patients with CD. The appearance of these cells parallels the peripheral blood appearance of gluten-specific CD4+ T cells (Raki et al. 2007). In addition, using single-cell TCR sequencing, we found that these gut-homing, activated CD8+ αβ T cells and γδ T cells were highly clonal and showed strong TCR convergence, consistent with an antigen-driven response (Fig. 3). This indicates that the IELs in CD are not the result of nonspecific bystander activation but rather are activated and recruited to the intestine in a deliberate and antigen-driven fashion. These findings suggest that celiac disease is much more complex than previously thought and involves many different components of the adaptive immune system.
Figure 3.
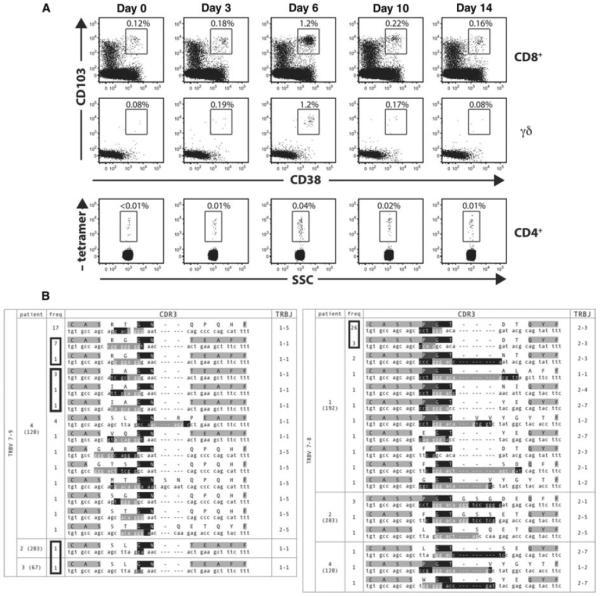
Kinetics of gut homing CD8+ and γδ T cells reveals clonal expansion upon gluten challenge in celiac disease. Five celiac sprue patients were analyzed before and after gluten challenge after having been on a gluten-free diet. A. We noted the appearance of activated CD8+ and γδ T cells bearing a gut homing receptor (αEβ7) entering the circulation in parallel with previously described gliaden-specific CD4+ T cells. These αEβ7+CD38+CD8+ TCRs were sequenced in five separate patients after gluten challenge, two of whom underwent rechallenge. And αEβ7+CD38+γδ TCRs were sequenced in three patients, one of whom underwent rechallenge. Single-cell sequencing revealed oligoclonal expansions and homologies across patients. (Adapted from Han et al. 2013.)
A particularly important question asks for the antigen that is triggering celiac IELs. Because celiac IEL populations are induced by gluten, it was originally thought that their TCRs recognize gluten. But despite extensive study, only a few reports of gluten-derived peptide epitopes recognized by CD8+ T cells in CD are extant, and there is no significant genetic association of CD with particular HLA Class I alleles. Therefore, most investigators believe that IELs do not mediate tissue damage through gluten recognition.
If these IELs are not responding to gluten, to what are they responding? Besides gluten, other possibilities for IEL ligands include self-antigens and infectious pathogens. The possibility of self-antigen recognition is supported by the very selective destruction of intestinal epithelial cells and the presence of autoantibodies, including antibodies to tissue transglutaminase (Jabri and Sollid 2009; Meresse et al. 2012; Sollid and Jabri 2013). Furthermore, the role of an infectious cofactor in CD has been proposed based on epidemiologic data showing that neonatal infection seems to predispose individuals to the development of CD (Sandberg-Bennich et al. 2002).
Another important question is how a CD4+ T-cell response to gluten enables the concurrent activation of IEL. that may be responding to a different antigen. Here, a number of aspects of CD8+ T-cell responses to viruses have been shown to depend on CD4+ T-cell help, including primary effector responses, the generation of memory, and recruitment to sites of infection (Janssen et al. 2003; Shedlock and Shen 2003; Sun and Bevan 2003; Nakanishi et al. 2009). This process has been termed licensing, referring to the ability of CD4+ T cells to “license” cognate effector CD8+ T-cell responses. From these precedents, we have speculated that CD4+ T cells may be “licensing” self-antigen-specific CD8+ T cells to become activated and recruited to the intestine, subsequently leading to tissue damage (Han et al. 2013).
These findings may be relevant to other autoimmune diseases. Similar to CD, most autoimmune diseases with HLA associations are associated with MHC Class II alleles, including Type 1 diabetes, multiple sclerosis, rheumatoid arthritis, and ulcerative colitis (Trowsdale 2011). Despite the association of these diseases with Class II alleles rather than Class I alleles, CD8+ effector T cells have an important role in the pathogenesis of these diseases and are appreciated to be the primary effectors driving tissue damage (Wang et al. 1996; Wong et al. 1996; Coppieters et al. 2012). Thus, the scenario we outline for CD may be generalizable to other forms of autoimmunity, in that an initial misdirected CD4+ T-cell response may “license” effector CD8+ and γδ T cells to cause tissue destruction at a particular site.
MEMORY PHENOTYPE T CELLS IN UNEXPOSED ADULTS
It has long been assumed that memory T cells are derived from the naive T-cell repertoire and that each pathogen or vaccine largely selects just those cells specific for its components from this pool. And, indeed one can see evidence for this in mouse models of disease. But for many years, there has been data that does not quite fit with this paradigm and suggests that cross-reactivity and/or heightened general responsiveness could also have a role. In particular, it has long been noted that mice challenged with one vaccine or pathogen can be protected from another infectious organism that is unrelated to the initial immunogen (Yang et al. 1989; Selin et al. 1994). In particular, Welsh and colleagues have documented this phenomenon extensively, referred to as “heterologous immunity,” and at least in one case isolated T-cell clones that are induced by lymphocytic choriomeningitus (LCMV) and are cross-reactive to pichinde virus (Brehm et al. 2002). In humans, similar results have been obtained by Aaby and colleagues who found that children in Africa who were immunized with a measles vaccine had a 35% reduction in mortality to diseases other than measles, and an entirely different study tracking individuals in Denmark who had received a vaccinia vaccine had a significantly reduced overall hospitalization rate for infection compared with those who did not (Aaby et al. 2010; Sorup et al. 2011). But these results, particularly the human studies, have been controversial, and explanations other than cross-reactivity have been proposed.
In our own recent work, we were surveying the frequency and characteristics of antigen-specific CD4+ T cells using DR 0401 tetramers, and we noted that there was a surprisingly high frequency of memory T cells for HIV in healthy blood bank donors who were HIV seronegative. For this particular T-cell epitope, the frequency of memory phenotype T cells (MPTs) ranged from 12% to 93% in a survey of 26 adults, with an average of more than 50%. That the serological assay for HIV is an accurate reflection of who is or isn't infected is shown by the fact that, because blood banks instituted this test, the risk of HIV transmission from blood products has become exceptionally low and is estimated to occur in 1 in 2.6 million blood donations nationwide (Lackritz et al. 1995). Extending this to other viral epitopes for which there are reliable serological assays, cytomegalovirus (CMV) and herpes simplex virus (HSV), we found very similar results in individuals who had never had these diseases (which, in the case of HIV particularly, is well established, because the consequences of infected donors in the blood supply is very serious). Specifically, as shown in Figure 4, an average of 57% of CMV-epitope-specific T cells and 63% of HSV-specific T cells also show a memory phenotype in naive individuals, with every one of the seranegative individuals having a detectable percentage of MPTs. An important question at this point becomes whether or not these MPTs are functional memory cells. Here, we have performed extensive analyses and found that these cells have all of the expected characteristics of memory T cells that we can measure in this system in that they can (1) make cytokines rapidly, (2) show evidence of clonal expansion, and (3) have gene expression patterns that mimic authentic memory T cells. These characteristics are shown in Figure 4 (from Su et al. 2013). Thus, all of the current evidence points to these being authentic memory T cells, although a direct test of their protective capacity is beyond what can be done ethically in human beings.
Figure 4.
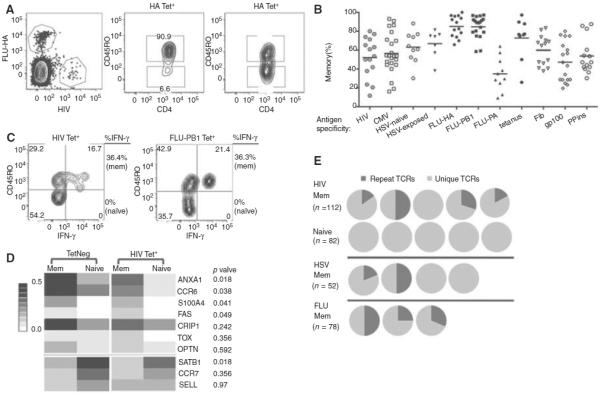
Characterization of the memory phenotype in the CD4+ T-cell repertoire show typical memory features. (A) Representative HA- and HIV-1-tetramer labeling (left) and CD45RO-antibody staining (right). Plots are representative of 15 individuals. (B) Percentage of CD45RO+ memory cells within each tetramer-tagged population. Each symbol represents an antigen-specific population from one individual, and the bar indicates the mean of experiments performed independently with blood obtained at different times. (C) Contour plots showing IFN-γ response of HIV-1- and PB1-specific T cells to stimulation by PMA and ionomycin. Experiments were repeated twice with blood from two different individuals. (D) Gene expression of tetramer-negative and HIV-1-tetramer-labeled cells. Forty-eight tetramer-negative memory cells (TetNeg mem), 46 tetramer-negative naive cells (TetNeg naive), 53 HIV-1-specific memory cells (HIV Tet+ mem), and 52 HIV-1-specific naive cells (HIV Tet+ naive) were analyzed. The heatmap summarizes the fraction of cells expressing a particular gene out of the total number of cells assayed. The genes have been grouped by whether they associate with memory (top) or naive (bottom) T cells and then ordered by ascending p values, which compare the differences between HIV memory and HIV naive T cells. (E) TCRβ sequencing of tetramer-labeled CD45RO+ and CD45RO− cells. Each pie chart represents TCR sequences from one individual. Dark-gray color represents the fraction of cells expressing a TCR identical to that of another cell. Light-gray color represents cells expressing unique TCRs. HIV-1- and HSV-specific T cells were obtained from individuals negative for these infections. (Adapted from Su et al. 2013.)
Also very intriguing is our finding that cord blood cells obtained from newborns had frequencies of HIV-specific T cells that were very similar to the unexposed adults, suggesting that a complete T-cell repertoire is present at birth, but these cells were all or almost all of the naive phenotype. This indicates that this acquisition of MPTs in adults represents some type of normal developmental progression, and so the question becomes how this might come about. One possible mechanism is homeostatic proliferation, which in mouse CD8+ T cells has been shown to produce a similar phenotype even in germ-free conditions, although not nearly as extensive as we see in most individuals in our studies (Haluszczak et al. 2009; Rudd et al. 2011; Akue et al. 2012; Decman et al. 2012). Furthermore, this propensity may be most pronounced in CD8+ T cells, because mouse CD4+ T cells have very few memory phenotype cells for antigens for which they have not been exposed (Moon et al. 2007; Chu et al. 2010) (at least in younger mice). An alternative, although not a mutually exclusive model, is that these MPTs arise through TCR cross-reactivity, a well-known phenomenon with T cells, significantly aided by the characteristic flexibility in the binding site that allows not only a given TCR to bind similar peptides presented by the same MHC but also some very different ones as well (Reinherz et al. 1999; Reiser et al. 2003; Newell et al. 2011). Coupled with a large number of antigens in infectious diseases and vaccines that most adults are exposed to, it seems reasonable to believe that there are ample opportunities for cross-reactivity among unrelated pathogen epitopes. To test this idea, we first made T-cell clones from HIV-tetramer-positive T cells and stimulated them with a collection of 24 peptides that showed significant sequence homology with the HIV peptide and came from microbial genomes obtained by searching the NCBI-blast nonredundant protein sequence database. Even with this crude approach, we saw abundant cross-reactivity, with five clones out of 24 able to be stimulated by eight of these peptides with distinct specificity. We next performed a more elaborate experiment in which we vaccinated two individuals with an influenza vaccine containing A/California/7/2009 H1N1 and identified a dominant epitope derived from the HA region of the H1N1 virus (HA 391–410). Again, T-cell clones were made from responding T cells and these were tested with a battery of 46 peptides chosen for their close homology with the HA epitope and derived from microbial genomes.
A number of these peptides cross-reacted with several T-cell clones, including one for Finegoldia magna, a common human skin bacterium, which costained with the flu peptide tetramer and a tetramer made from this closely related peptide. Going back to the blood samples from the vaccination time course showed that in one of the two patients, there was a parallel increase and decrease in both specificities. This is a formal demonstration that individuals' exposure to one microbe's antigens can confer some degree of immunity to another, and likely many others, considering how many T-cell epitopes there are in even a small viral genome such as influenza.
TECHNOLOGY DEVELOPMENT
Another factor that has inhibited human immunology is the many technical challenges to understanding the diversity of human responses, given the limitations on human experimentation. This is one reason that inbred mouse has been so ideal in that many of the variables that affect the immune system, such as MHC polymorphism, confounding diseases, and genetic diversity, can be easily controlled for. But these are also limitations for the study of a normal immune system, because it typically has to deal with both of these factors in abundance. There is also the problem of the very diverse phenotypes and functional characteristics of T cells and other immune cells. Thus, the developments of cellular analysis technologies that can handle this diversity and make the most of limited human samples are crucial (Newell and Davis 2014). One such very important tool in immunology has been the development of fluorescence-activated cell sorting (FACS) and analysis, first developed by the Herzenbergs in the 1970s. This has allowed the detailed characterization of immune cells on a single-cell basis and has been a mainstay of immunological research ever since. Through development of new fluorophores and more sophisticated instrumentation, the number of parameters that can be simultaneously measured by this approach has been steadily increasing over the years (Bendall et al. 2011). However, because it relies on different fluorescent labels, the effects of excitation and emission spectral overlap become increasingly difficult to manage because the numbers of parameters are increased. In the last decade, this has put an effective cap on the number of parameters that can be simultaneously measured at around 12, severely limiting the amount of information that can be obtained from a precious human blood or tissue sample. An alternate approach developed by Tanner and colleagues (Bandura et al. 2009) and recently introduced by the DVS Sciences company is a remarkable new approach that uses high-throughput mass spectrometry to analyze metal isotope labels on single cells. The method is called “mass cytometry” or cytometry by time of flight (CyTOF) and has the advantage that isotopes can be resolved at a single atomic mass unit (AMU) resolution. That is, an isotope that is 150 AMU is easily distinguished from one that is 151 AMU with little overlap. In addition, because there are a large number of isotopically purified isotopes amenable to this approach, we currently have 40 different labels that can be used as antibody labels. The remarkable power of this instrument was nicely shown in the work of Nolan and colleagues (Bendall et al. 2011), where they were able to survey the entire hematopoietic lineage in humans in one panel, revealing complexity in subsets that was not visible before. This work was followed by Newell et al. (2012) who took a deeper dive into CD8+ T cells with 35 labels, and found that these cells were much more complex than previously known. Specifically, Newell and colleagues (2012) found that CD8+ T cells were far more complex than previously appreciated, expressing 242 different combinations of nine functional markers (mostly cytokines) of 512 possible, showing an essentially combinatorial pattern. Similar results have been seen with CD4+ T cells (Wen et al. in prep.) and in human natural killer (NK) cells, where Blish and colleagues (Horowitz et al. 2013) found an estimated 100,000 or so distinct phenotypes of NK cells. Thus, this technology is quickly redefining every lymphocyte subset. It is also particularly important in human work in that it provides a much more information-rich way to characterize white cells in the blood, approaching gene expression (previously the undisputed champ in this regard), and unlike gene expression, it is closer to functional activity than RNA, and is, by nature, a single-cell assay, so there is no doubt as to what cells you are looking at any particular time.
Another technical problem we have addressed in our work is how to assess diverse T-cell responses to pathogens and more efficiently discover new epitopes. Intact pathogens have hundreds or easily thousands of T-cell epitopes that can be displayed by similar numbers of MHC alleles. Initially, Newell et al. (2009) addressed this problem by introducing combinatorial tetramer labeling that enables four different fluorophores to be the basis for 15 different peptide–MHC tetramers. In this technique, T cells are labeled with up to 15 different tetramers, and the combinations of labeling each cell are assessed with a standard FACS instrument. Routinely, 15 different pMHC specificities can be resolved using different labeled streptavidins. Similarly, Schumacher and colleagues used combinations of tetramers coupled to different quantum dots to create 25 different labels (Hadrup et al. 2009; Hadrup and Schumacher 2010).
But recently, we have expanded the range dramatically using mass cytometry, and one currently has up to 40 different labels. Here, Newell et al. used 10 of these labels to create “bar codes” of three different labels to make a tetramer panel of up to 109 different labels (120 are possible) (Newell et al. 2013). In this method, each peptide–MHC combination would be separately labeled with one of three different metal isotopes and then T cells that bind to all three of those tetramers are identified as specific for that particular ligand, which is done automatically by a program. In this way, we have surveyed blood and gut lymphocyte samples in a search for novel rotavirus epitopes. We found six or more, including the most dominant epitope yet found, out of 72 candidates screened with HLA-A0201 as the restricting element (Fig. 5). These data also showed that for both rotavirus and the EBV epitopes included in the survey, two very distinct phenotypes were evident, depending on the antigen specificity of the labeled T cell (Fig. 5).
Figure 5.
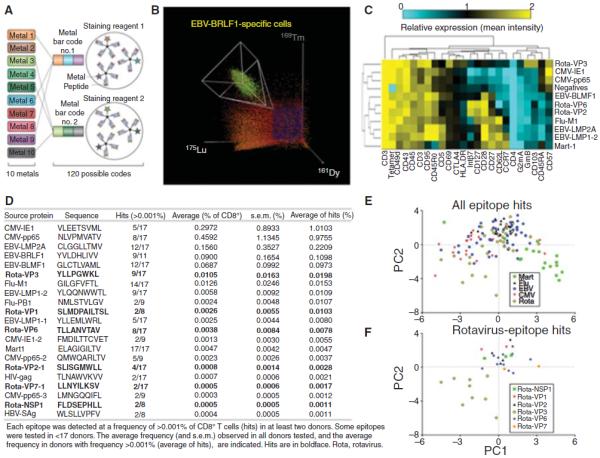
Combinatorial pMHC tetramer staining analyzed by flow cytometry and CyTOF. (A) For this experiment, each T-cell antigen specificity is coded by a unique combination of three out of 10 CyTOF labels. There are 120 possible unique combinations. (B) Cells positive for one of these unique combinations of three were identified as specific for the corresponding antigen as shown for an epitope of EBV complexed to HLA-A0201. Each donor sample was staining with a panel of 109 epitopes including both previously defined T-cell epitopes and candidate rotavirus epitopes, together with an additional 23 surface/intracellular markers that were used to phenotypically characterize the antigen-specific cells. (C) The average expression of each phenotypic marker (listed at bottom) on T cells specific for the indicated epitopes (listed at right) from one representative blood donor are plotted as a clustered heatmap. (D) Single blood samples from each of 17 donors were analyzed and the frequencies of hits identified in two or more donors were tabulated. (E and F). To compare the phenotypes of cells derived from all donors, the first two components obtained from principal component analysis (PCA) of z-score normalized average expression levels for each of these markers are plotted for each hit (each point is a single epitope–donor combination, as listed in D for Mart1, flu-, EBV-, CMV- and rotavirus-specific cells (E) and T cells specific for indicated rotavirus peptides (F). (A, Reprinted, with permission, from Harvey and Wucherpfennig 2013; B, reprinted from Newell and Davis 2014; C–F reprinted from Newell et al. 2013.)
We have recently extended this approach to create panels of Class II MHC tetramers for influenza and rotavirus, in particular using a large number (HM McGuire, in prep.) of different influenza peptide antigens restricted to DR1501 to peripheral blood mononuclear cell (PBMC) samples from influenza vaccine recipients. Although these studies are still ongoing, the results are very promising and indicate that this approach is quite broad and will be able to considerably enrich our understanding of T-cell responses to vaccination or infection and their relationship to outcomes.
CONCLUDING REMARKS
Although many challenges remain, our experience in working on human immunity shows that there is much to be learned about basic immunology in this area, as well as the likely benefits to practical issues such as vaccine development.
REFERENCES
- Aaby P, Martins CL, Garly ML, Bale C, Andersen A, Rodrigues A, Ravn H, Lisse IM, Benn CS, Whittle HC. Non-specific effects of standard measles vaccine at 4.5 and 9 months of age on childhood mortality: Randomised controlled trial. BMJ. 2010;341:c6495. doi: 10.1136/bmj.c6495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akue AD, Lee JY, Jameson SC. Derivation and maintenance of virtual memory CD8 T cells. J Immunol. 2012;188:2516–2523. doi: 10.4049/jimmunol.1102213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amur S, Parekh A, Mummaneni P. Sex differences and genomics in autoimmune diseases. J Autoimmun. 2012;38:J254–J265. doi: 10.1016/j.jaut.2011.12.001. [DOI] [PubMed] [Google Scholar]
- Bandura DR, Baranov VI, Ornatsky OI, Antonov A, Kinach R, Lou X, Pavlov S, Vorobiev S, Dick JE, Tanner SD. Mass cytometry: Technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal Chem. 2009;81:6813–6822. doi: 10.1021/ac901049w. [DOI] [PubMed] [Google Scholar]
- Bendall SC, Simonds EF, Qiu P, Amir el-AD, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–696. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm MA, Pinto AK, Daniels KA, Schneck JP, Welsh RM, Selin LK. T cell immunodominance and maintenance of memory regulated by unexpectedly cross-reactive pathogens. Nat Immunol. 2002;3:627–634. doi: 10.1038/ni806. [DOI] [PubMed] [Google Scholar]
- Casanova JL, Abel L. Inborn errors of immunity to infection: The rule rather than the exception. J Exp Med. 2005;202:197–201. doi: 10.1084/jem.20050854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu HH, Moon JJ, Kruse AC, Pepper M, Jenkins MK. Negative selection and peptide chemistry determine the size of naive foreign peptide-MHC class II-specific CD4+ T cell populations. J Immunol. 2010;185:4705–4713. doi: 10.4049/jimmunol.1002276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppieters KT, Dotta F, Amirian N, Campbell PD, Kay TW, Atkinson MA, Roep BO, von Herrath MG. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med. 2012;209:51–60. doi: 10.1084/jem.20111187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MM. A prescription for human immunology. Immunity. 2008;29:835–838. doi: 10.1016/j.immuni.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decman V, Laidlaw BJ, Doering TA, Leng J, Ertl HC, Goldstein DR, Wherry EJ. Defective CD8 T cell responses in aged mice are due to quantitative and qualitative changes in virus-specific precursors. J Immunol. 2012;188:1933–1941. doi: 10.4049/jimmunol.1101098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kauwe AL, Chen Z, Anderson RP, Keech CL, Price JD, Wijburg O, Jackson DC, Ladhams J, Allison J, McCluskey J. Resistance to celiac disease in humanized HLA-DR3-DQ2-transgenic mice expressing specific anti-gliadin CD4+ T cells. J Immunol. 2009;182:7440–7450. doi: 10.4049/jimmunol.0900233. [DOI] [PubMed] [Google Scholar]
- Du Pre MF, Kozijn AE, van Berkel LA, ter Borg MN, Lindenbergh-Kortleve D, Jensen LT, Kooy-Winkelaar Y, Koning F, Boon L, Nieuwenhuis EE, et al. Tolerance to ingested deamidated gliadin in mice is maintained by splenic, type 1 regulatory T cells. Gastroenterology. 2011;141:610–620. 620, e611–e612. doi: 10.1053/j.gastro.2011.04.048. [DOI] [PubMed] [Google Scholar]
- Fallang LE, Bergseng E, Hotta K, Berg-Larsen A, Kim CY, Sollid LM. Differences in the risk of celiac disease associated with HLA-DQ2.5 or HLA-DQ2.2 are related to sustained gluten antigen presentation. Nat Immunol. 2009;10:1096–1101. doi: 10.1038/ni.1780. [DOI] [PubMed] [Google Scholar]
- Feldmann M, Maini RN. Anti-TNF alpha therapy of rheumatoid arthritis: What have we learned? Annu Rev Immunol. 2001;19:163–196. doi: 10.1146/annurev.immunol.19.1.163. [DOI] [PubMed] [Google Scholar]
- Furman D, Jojic V, Kidd B, Shen-Orr S, Price J, Jarrell J, Tse T, Huang H, Lund P, Maecker HT, et al. Apoptosis and other immune biomarkers predict influenza vaccine responsiveness. Mol Syst Biol. 2013a;9:659. doi: 10.1038/msb.2013.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman D, Hejblum BP, Simon N, Jojic V, Dekker CL, Thiebaut R, Tibshirani RJ, Davis MM. Systems analysis of sex differences reveals an immunosuppressive role for testosterone in the response to influenza vaccination. Proc Natl Acad Sci. 2013b;111:869–874. doi: 10.1073/pnas.1321060111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaucher D, Therrien R, Kettaf N, Angermann BR, Boucher G, Filali-Mouhim A, Moser JM, Mehta RS, Drake DR, III, Castro E, et al. Yellow fever vaccine induces integrated multilineage and polyfunctional immune responses. J Exp Med. 2008;205:3119–3131. doi: 10.1084/jem.20082292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green PH, Cellier C. Celiac disease. New Engl J Med. 2007;357:1731–1743. doi: 10.1056/NEJMra071600. [DOI] [PubMed] [Google Scholar]
- Grosso JF, Jure-Kunkel MN. CTLA-4 blockade in tumor models: An overview of preclinical and translational research. Cancer Immun. 2013;13:5. [PMC free article] [PubMed] [Google Scholar]
- Hadrup SR, Schumacher TN. MHC-based detection of antigen-specific CD8+ T cell responses. Cancer Immunol Immunother. 2010;59:1425–1433. doi: 10.1007/s00262-010-0824-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadrup SR, Bakker AH, Shu CJ, Andersen RS, van Veluw J, Hombrink P, Castermans E, Thor Straten P, Blank C, Haanen JB, et al. Parallel detection of antigen-specific T-cell responses by multidimensional encoding of MHC multimers. Nat Methods. 2009;6:520–526. doi: 10.1038/nmeth.1345. [DOI] [PubMed] [Google Scholar]
- Haluszczak C, Akue AD, Hamilton SE, Johnson LD, Pujanauski L, Teodorovic L, Jameson SC, Kedl RM. The antigen-specific CD8+ T cell repertoire in unimmunized mice includes memory phenotype cells bearing markers of homeostatic expansion. J Exp Med. 2009;206:435–448. doi: 10.1084/jem.20081829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han A, Newell EW, Glanville J, Fernandez-Becker N, Khosla C, Chien YH, Davis MM. Dietary gluten triggers concomitant activation of CD4+ and CD8+ alphabeta T cells and gammadelta T cells in celiac disease. Proc Natl Acad Sci. 2013;110:13073–13078. doi: 10.1073/pnas.1311861110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CJ, Wucherpfennig KW. Cracking the code of human T-cell immunity. Nat Biotechnol. 2013;31:609–610. doi: 10.1038/nbt.2626. [DOI] [PubMed] [Google Scholar]
- Horowitz A, Strauss-Albee DM, Leipold M, Kubo J, Nemat-Gorgani N, Dogan OC, Dekker CL, Mackey S, Maecker H, Swan GE, et al. Genetic and environmental determinants of human NK cell diversity revealed by mass cytometry. Sci Transl Med. 2013;5:208ra145. doi: 10.1126/scitranslmed.3006702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabri B, Sollid LM. Tissue-mediated control of immunopathology in coeliac disease. Nat Rev Immunol. 2009;9:858–870. doi: 10.1038/nri2670. [DOI] [PubMed] [Google Scholar]
- Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421:852–856. doi: 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- Klein SL, Poland GA. Personalized vaccinology: One size and dose might not fit both sexes. Vaccine. 2013;31:2599–2600. doi: 10.1016/j.vaccine.2013.02.070. [DOI] [PubMed] [Google Scholar]
- Lackritz EM, Satten GA, Aberle-Grasse J, Dodd RY, Raimondi VP, Janssen RS, Lewis WF, Notari EP, IV, Petersen LR. Estimated risk of transmission of the human immunodeficiency virus by screened blood in the United States. New Engl J Med. 1995;333:1721–1725. doi: 10.1056/NEJM199512283332601. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol. 2012;28:137–161. doi: 10.1146/annurev-cellbio-101011-155745. [DOI] [PubMed] [Google Scholar]
- Laubach VE, Foley PL, Shockey KS, Tribble CG, Kron IL. Protective roles of nitric oxide and testosterone in endotoxemia: Evidence from NOS-2-deficient mice. Am J Physiol. 1998;275:H2211–H2218. doi: 10.1152/ajpheart.1998.275.6.H2211. [DOI] [PubMed] [Google Scholar]
- Meresse B, Malamut G, Cerf-Bensussan N. Celiac disease: An immunological jigsaw. Immunity. 2012;36:907–919. doi: 10.1016/j.immuni.2012.06.006. [DOI] [PubMed] [Google Scholar]
- Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl RM, Jenkins MK. Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity. 2007;27:203–213. doi: 10.1016/j.immuni.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi Y, Lu B, Gerard C, Iwasaki A. CD8(+) T lymphocyte mobilization to virus-infected tissue requires CD4(+) T-cell help. Nature. 2009;462:510–513. doi: 10.1038/nature08511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakaya HI, Wrammert J, Lee EK, Racioppi L, Marie-Kunze S, Haining WN, Means AR, Kasturi SP, Khan N, Li GM, et al. Systems biology of vaccination for seasonal influenza in humans. Nat Immunol. 2011;12:786–795. doi: 10.1038/ni.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell EW, Davis MM. Beyond model antigens: Highly dimensional analysis of antigen-specific T cells. Nat Biotechnol. 2014 doi: 10.1038/nbt.2783. doi: 10.1038/nbt.2783. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell EW, Klein LO, Yu W, Davis MM. Simultaneous detection of many T-cell specificities using combinatorial tetramer staining. Nat Methods. 2009;6:497–499. doi: 10.1038/nmeth.1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell EW, Ely LK, Kruse AC, Reay PA, Rodriguez SN, Lin AE, Kuhns MS, Garcia KC, Davis MM. Structural basis of specificity and cross-reactivity in T cell receptors specific for cytochrome c-I-E(k) J Immunol. 2011;186:5823–5832. doi: 10.4049/jimmunol.1100197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell EW, Sigal N, Bendall SC, Nolan GP, Davis MM. Cytometry by time-of-flight shows combinatorial cytokine expression and virus-specific cell niches within a continuum of CD8+ T cell phenotypes. Immunity. 2012;36:142–152. doi: 10.1016/j.immuni.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell EW, Sigal N, Nair N, Kidd BA, Greenberg HB, Davis MM. Combinatorial tetramer staining and mass cytometry analysis facilitate T-cell epitope mapping and characterization. Nat Biotechnol. 2013;31:623–629. doi: 10.1038/nbt.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentcheva-Hoang T, Corse E, Allison JP. Negative regulators of T-cell activation: Potential targets for therapeutic intervention in cancer, autoimmune disease, and persistent infections. Immunol Rev. 2009;229:67–87. doi: 10.1111/j.1600-065X.2009.00763.x. [DOI] [PubMed] [Google Scholar]
- Price JV, Jarrell JA, Furman D, Kattah NH, Newell E, Dekker CL, Davis MM, Utz PJ. Characterization of influenza vaccine immunogenicity using influenza antigen microarrays. PLoS One. 2013;8:e64555. doi: 10.1371/journal.pone.0064555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querec T, Bennouna S, Alkan S, Laouar Y, Gorden K, Flavell R, Akira S, Ahmed R, Pulendran B. Yellow fever vaccine YF-17D activates multiple dendritic cell subsets via TLR2, 7, 8, and 9 to stimulate polyvalent immunity. J Exp Med. 2006;203:413–424. doi: 10.1084/jem.20051720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raki M, Fallang LE, Brottveit M, Bergseng E, Quarsten H, Lundin KE, Sollid LM. Tetramer visualization of gut-homing gluten-specific T cells in the peripheral blood of celiac disease patients. Proc Natl Acad Sci. 2007;104:2831–2836. doi: 10.1073/pnas.0608610104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinherz EL, Tan K, Tang L, Kern P, Liu J, Xiong Y, Hussey RE, Smolyar A, Hare B, Zhang R, et al. The crystal structure of a T cell receptor in complex with peptide and MHC class II. Science. 1999;286:1913–1921. doi: 10.1126/science.286.5446.1913. [DOI] [PubMed] [Google Scholar]
- Reiser JB, Darnault C, Gregoire C, Mosser T, Mazza G, Kearney A, van der Merwe PA, Fontecilla-Camps JC, Housset D, Malissen B. CDR3 loop flexibility contributes to the degeneracy of TCR recognition. Nat Immunol. 2003;4:241–247. doi: 10.1038/ni891. [DOI] [PubMed] [Google Scholar]
- Rudd BD, Venturi V, Li G, Samadder P, Ertelt JM, Way SS, Davenport MP, Nikolich-Zugich J. Nonrandom attrition of the naive CD8+ T-cell pool with aging governed by T-cell receptor:pMHC interactions. Proc Natl Acad Sci. 2011;108:13694–13699. doi: 10.1073/pnas.1107594108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg-Bennich S, Dahlquist G, Kallen B. Coeliac disease is associated with intrauterine growth and neonatal infections. Acta Paediatr. 2002;91:30–33. doi: 10.1080/080352502753457905. [DOI] [PubMed] [Google Scholar]
- Selin LK, Nahill SR, Welsh RM. Cross-reactivities in memory cytotoxic T lymphocyte recognition of heterologous viruses. J Exp Med. 1994;179:1933–1943. doi: 10.1084/jem.179.6.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci. 2013;110:3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300:337–339. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- Sollid LM, Jabri B. Triggers and drivers of autoimmunity: Lessons from coeliac disease. Nat Rev Immunol. 2013;13:294–302. doi: 10.1038/nri3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollid LM, Qiao SW, Anderson RP, Gianfrani C, Koning F. Nomenclature and listing of celiac disease relevant gluten T-cell epitopes restricted by HLA-DQ molecules. Immunogenetics. 2012;64:455–460. doi: 10.1007/s00251-012-0599-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorup S, Villumsen M, Ravn H, Benn CS, Sorensen TI, Aaby P, Jess T, Roth A. Smallpox vaccination and all-cause infectious disease hospitalization: A Danish register-based cohort study. Int J Epidemiol. 2011;40:955–963. doi: 10.1093/ije/dyr063. [DOI] [PubMed] [Google Scholar]
- Su LF, Kidd BA, Han A, Kotzin JJ, Davis MM. Virus-specific CD4(+) memory-phenotype T cells are abundant in unexposed adults. Immunity. 2013;38:373–383. doi: 10.1016/j.immuni.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300:339–342. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trowsdale J. The MHC, disease and selection. Immunol Lett. 2011;137:1–8. doi: 10.1016/j.imlet.2011.01.002. [DOI] [PubMed] [Google Scholar]
- Wang B, Gonzalez A, Benoist C, Mathis D. The role of CD8+ T cells in the initiation of insulin-dependent diabetes mellitus. Eur J Immunol. 1996;26:1762–1769. doi: 10.1002/eji.1830260815. [DOI] [PubMed] [Google Scholar]
- Weber S, Tian H, van Rooijen N, Pirofski LA. A serotype 3 pneumococcal capsular polysaccharide-specific monoclonal antibody requires Fcgamma receptor III and macrophages to mediate protection against pneumococcal pneumonia in mice. Infect Immun. 2012;80:1314–1322. doi: 10.1128/IAI.06081-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong FS, Visintin I, Wen L, Flavell RA, Janeway CA., Jr CD8 T cell clones from young nonobese diabetic (NOD) islets can transfer rapid onset of diabetes in NOD mice in the absence of CD4 cells. J Exp Med. 1996;183:67–76. doi: 10.1084/jem.183.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xian L, Wu X, Pang L, Lou M, Rosen CJ, Qiu T, Crane J, Frassica F, Zhang L, Rodriguez JP, et al. Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nat Med. 2012;18:1095–1101. doi: 10.1038/nm.2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HY, Dundon PL, Nahill SR, Welsh RM. Virus-induced polyclonal cytotoxic T lymphocyte stimulation. J Immunol. 1989;142:1710–1718. [PubMed] [Google Scholar]