

Abstract
The human neutrophil peptides (HNP) bind to vascular smooth muscle cells and regulate vascular tone. We hypothesized that HNP act on endothelial cells to modulate the expression of vasoactive byproducts. We observed a time- and dose-dependent increase in the expression of cyclooxygenase-2 (COX-2) by human umbilical vein endothelial cells (HUVEC) in response to HNP stimulation, while COX-1 levels remained unchanged. Despite an upregulated expression of COX-2, HNP did not significantly enhance the production of the COX-2-derived prostaglandins PGI2 and PGE2. HNP significantly induced the release of endothelin-1 (ET-1) as well as the formation of nitrotyrosine. The HNP-induced COX-2 and ET-1 production was attenuated by the treatment with the oxygen free radical scavenger N-acetyl-L-cysteine, and the inhibitors of p38 MAPK and NF-κB, respectively. We conclude that HNP may play an important role in the regulation of the course of cardiovascular diseases by activating endothelial cells to produce vasoactive byproducts.
Keywords: Inflammation, immunity, leukocyte
INTRODUCTION
Human neutrophil peptides (HNP), mainly released from neutrophils during inflammation, are key players in innate immunity by upregulation of co-stimulatory molecules (1), stimulation of cell adhesion (1,2), release of chemokines (3–5), induction of reactive oxygen species (ROS) (6), and T cell chemotaxis (7,8). In addition, HNP bind to vascular smooth muscle cells and regulate vascular tone (9,10). Elevated circulating levels of HNP are found in patients with inflammatory diseases such as acute respiratory distress syndrome and sepsis (11–13), where pulmonary vasoconstriction and systemic vasodilation are frequently implicated.
The vascular endothelium actively participates in maintaining vascular homeostasis by balancing vasoactive compounds between endothelium-derived relaxing and contracting factors. The major vasoactive by-products include prostaglandins, nitric oxide (NO) and Endothelin-1 (ET-1).
Prostaglandins are produced following the sequential oxidation of arachidonic acid by cyclooxygenases (COX-1 and COX-2) and terminal prostaglandin synthases. COX-1 is responsible for the constitutive levels of prostaglandins, while COX-2 produces inducible prostaglandins in scenarios of inflammation through stimulation of endothelial cells by various growth factors and cytokines (14). The major vasodilatory prostaglandin E2 (PGE2) is generated by catalyzing prostaglandin E synthases on prostaglandin H2 (PGH2). Similarly, prostacyclin (PGI2) synthase (PGIS) converts PGH2 into PGI2. PGE2 and PGI2 also exert anti-inflammatory properties (15).
Endothelin-1 (ET-1), produced mainly by endothelial cells is a potent vasoconstrictor by interaction with two key receptor types, ETA and ETB. ETA receptors are found on the smooth muscle cells of blood vessels, and ETB is primarily located on the endothelial cells. Binding of ET-1 to the receptors increases vasoconstriction and the retention of sodium (16). ET-1 is reportedly associated with the pathogenesis of atherosclerosis (17).
It has been recently established that inflammation plays a crucial role in mediating all stages of atherosclerosis from initiation through progression (18). During leukocyte dominated inflammatory responses, neutrophils release large amounts of HNP into the extracellular milieu. In turn, HNP stimulate cells to produce ROS (6). The latter is known to modulate the expression of COX-2 and ET-1 in endothelial cells (19,20). In the present study, we tested and proved the hypothesis that HNP enhanced expression of COX-2 and ET-1 in endothelial cells through ROS-dependent mechanisms.
METHODS
Reagents
Anti-COX-1 and COX-2 polyclonal antibodies were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Monoclonal anti-β-actin antibodies, N-Acetyl-L-cysteine (NAC) were obtained from Sigma (St. Louis, MO). Phospho- and total-p38 MAPK antibodies were from Cell Signaling Technology (Danvers, MA), and HRP-conjugated anti-goat and anti-rabbit antibodies were from Jackson ImmunoResearch Laboratories (West Grove, PA). The selective inhibitors U0126, SB203580, PG-490 and JSH-23 against MEK1/2, p38 MAPK and NF-κB, respectively were purchased from Calbiochem (La Jolla, CA). Enhanced chemiluminesence kit was from Perkin Elmer LAS, Inc (Boston, MA). The purification of HNP has been previously described (1,3).
Cell Culture
Human umbilical vein endothelial cells (HUVEC, Cell Applications, Inc., San Diego, CA) were cultured in HUVEC medium (Cell Applications, Inc) at 37°C in a 5% CO2. The cells were used on passage 2.
Western Blotting
Confluent HUVEC in 30 mm2 dishes (Corning Costar, Cambridge, MA) were deprived of serum for 12–16 h, and subjected to HNP stimulation. The cells were then lysed in a buffer containing63.5 mM Tris-HCl (pH 6.8), 10% (vol/vol) glycerol, 2% (wt/vol) SDS, 1 mM Na3VO4, 1 mM 4-(2-aminoethyl)-benzene sulfonyl fluoride, and 50 μg/ml leupeptin. Cell extracts were subject to SDS-PAGE and transferred to Nitrocellulose membrane (Bio-Rad Laboratories, Inc., Hercules, CA). Nonspecific antibody binding was blocked by incubation of membranes in 50 mM Tris, 150 M NaCl, and 0.02% (vol/vol) Tween 20, pH7.4 (TBST) containing 5% milk for 2 h. Membranes were subsequently incubated with the appropriate primary antibody in TBST/10% BSA. After extensive washing with TBST, membranes were incubated with horseradish peroxidase–conjugated secondary antibodies. Membranes were washed and signals were visualized with an enhanced chemiluminescence detection system (ECL Kit; Perkin Elmer LAS, Inc., Boston, MA). The band density was determined by a Kodak image station 2000MM (Mandel Scientific, Guelph, ON, Canada).
Measurement of PGI2, PGE2 and ET-1
Upon completion of the experiments, the cell culture medium was collected and centrifuged at 112g for 5 min. The cell culture supernatants were assayed for 6-keto-PGF1α, a stable intermediate of PGI2, using an ELISA kit (GE Healthcare, Buckinghamshire, UK). Levels of PGE2 (GE Healthcare) and ET-1 were measured by ELISA kits (R & D Systems, Minneapolis, MN).
Nitrotyrosine assay
Nitro-tyrosine levels were detected in cell culture supernatants by using a commercial kit (Bioxytech Nitrotyrosine-EIA, Oxis International, Inc., Portland, OR).
Cytotoxicity measurement
To confirm excellent cell viability in the conditions where HNP and/or the inhibitors were present, lactate dehydrogenase (LDH) activity was measured at 490 nm (Cytotoxicity detection kit, Roche Applied Science, Penzberg, Germany) in all experiments.
Statistical Analysis
Data are presented as mean ± SD. Data were analyzed in non-parametric tests by using Prism Graphpad 4.0 software package (Prism, San Diego, CA). Comparison among groups was performed by ANOVA using Kruskal-Wallis test. A P < 0.05 was considered as significant.
RESULTS
HNP increased expression of COX-2 by activation of p38 MAPK, ERK1/2 and NF-κB
HUVEC (1×106 cells) were stimulated with HNP at 5 μg/mL and 25 μg/mL for 4h, 6h and 8h, respectively. A time- and dose-dependent increase in COX-2 expression was observed, while the expression of COX-1 remained unchanged in response to HNP stimulation (Fig. 1A).
Figure 1. HNP increased expression of COX-2 by activation of p38 MAPK, ERK1/2 and NF-κB. A.

Confluent HUVEC monolayers were incubated overnight with serum-free medium, and exposed to either 0.01% acetic acid as vehicle control or HNP. B and C. Cells were treated with either vehicle (DMSO alone), SB203580 (10 μM), U0126 (10 μM), JSH23 (10 μM), or PG490 (70 nM) for 30 min prior to stimulation with HNP (25 μg/mL) for additional 6h. Cells were lysed for western blotting. Representative blots are illustrated from three independent experiments. The bar graphs present densitometric analysis of three experiments. * p < 0.05 vs. vehicle controls, respectively; † p < 0.05 vs. the immediate previous group; and ‡ p < 0.05 vs. HNP alone at identical conditions, respectively.
We next showed that the HNP-induced COX-2 expression was completely blocked by inhibition of p38 MAPK, and largely attenuated by inhibition of MEK1/2 at the dose used (Fig. 1B). Since COX-2 expression is possibly mediated by NF-κB (21), and we have previously demonstrated that stimulation of epithelial and CD4+ T cells with HNP induced NF-κB translocation (1), we thus examined the role of NF-κB on the HNP-induced COX2 expression by using the inhibitors JSH-23 and PG-490 (Triptolide) in HUVEC. The two compounds have been widely used as NF-κB inhibitors in in vivo and in vitro conditions (21–24). We observed a significant attenuation of the HNP-induced COX-2 expression by using the NF-κB inhibitors (Fig. 1C).
HNP had no significant effects on PGI2 and PGE2
PGI2 and PGE2 are known major byproducts derived from COX-2. However, the expression of the two prostaglandins was not significantly altered although the level of PGI2 tended to increase in the cell culture medium after HNP stimulation (Figs. 2A and 2B). We also stimulated the cells with IL-1α as a positive control (25) to ensure that the cells were able to produce PGI2 and PGE2 (Fig. 2A and 2B).
Figure 2. HNP had no significant effects on PGI2 and PGE2.

Confluent HUVEC were exposed to HNP for 6h, concentrations of PGI2 and PGE2 were measured in cell supernatants. IL-1α was used as a positive control. N = 3. * p < 0.05 vs. vehicle control at identical conditions, respectively.
HNP increased ET-1 release by activation of p38 MAPK, ERK1/2 and NF-κB
Since ET-1 is a major vasoconstrictor produced by endothelium, we measured the concentration of ET-1 which is produced independent of the COX-2 pathway. We observed a significant increase in ET-1 release as early as 30 min, followed by a time-dependent increase in response to HNP stimulation at a dose of 25 μg/mL (Fig 3A). Stimulation with HNP at a dose lower than 25 μg/mL for 8 h had no effects on ET-1 production (Fig. 3B). The HNP-induced ET-1 expression was significantly reduced by the use of inhibitors against p38 MAPK, ERK1/2 and NF-κB, respectively (Fig. 3C).
Figure 3. HNP increased ET-1 release by activation of p38 MAPK, ERK1/2 and NF-κB.

A. Time-dependent increase in ET-1. HUVEC were stimulated with HNP (25 μg/mL) or vehicle control (0.01% acetic acid) for the indicated times. Concentration of ET-1 was measured in the cell supernatants. B. Dose-dependent increase in ET-1. HUVEC were treated with indicated doses of HNP for 6h. C. Cells were pretreated with either vehicle (DMSO alone), U0126 (10 μM), SB203580 (10 μM), PG-490 (70nM) or JSH-23 (10μM) for 30min prior to stimulation with HNP (25 μg/mL) for additional 6h. N = 3. * p < 0.05 vs vehicle control at identical conditions, respectively. † p < 0.05 vs. HNP alone.
HNP induced COX-2 and ET-1 through oxidative stress
ROS have been reported as key mediators to induce production of COX-2 (19, 20) and ET-1 (19, 26). We have previously reported that HNP stimulate lung tissue to produce hydrogen peroxide (6). We thus measured the concentration of nitrotyrosine in HUVEC culture medium after HNP stimulation. Nitrotyrosine is a marker of peroxynitrite formation as a result of generation of NO and superoxide (27). We observed that the stimulation of HUVEC with HNP at 25 μg/mL resulted in a 4-fold and 2.5-fold increase in nitrotyrosine level at 30 min and 4h, respectively (Fig. 4). When the antioxidant N-acetyl-L-cysteine (NAC) was used to inhibit the HNP-induced oxidative stress, we observed a decrease in the expression of both COX-2 and ET-1 (Fig. 5A and Fig. 5B) associated with an attenuation of p38 MAPK phosphorylation (Fig. 5C). Taken together, these results suggest that the HNP-induced increase in the expression of COX-2 and ET-1 through oxidative stress.
Figure 4. HNP induced nitrotyrosine formation in HUVEC.
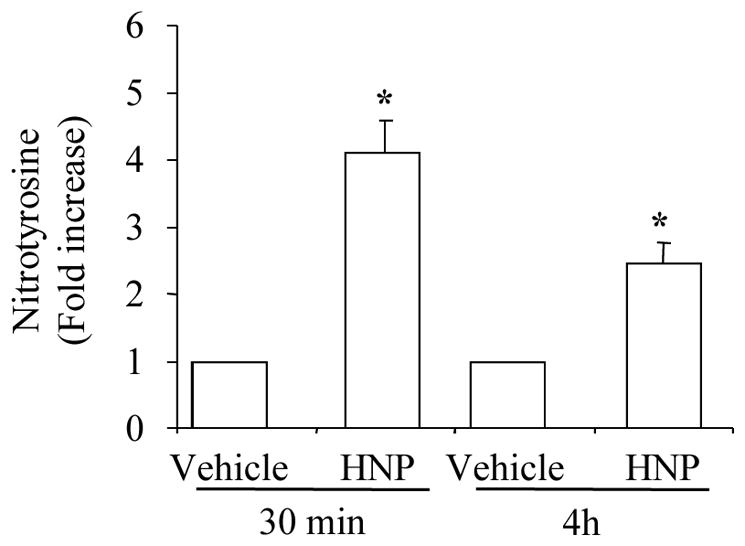
Cells were treated with HNP (25 μg/mL) for the indicated times. Nitrotyrosine levels in cell supernatants were measured by ELISA. N = 3. * p < 0.05 vs. vehicle control.
Figure 5. HNP upregulated COX-2 and ET-1 through oxidative stress.

HUVEC were pretreated with NAC 30 min prior to exposure to HNP for additional 6h. Cell lysates were analyzed by Western blotting for COX-2 protein. A. Representative immunoblot is shown from three separate experiments. B. ET-1 released in cell supernatants was assayed by ELISA. C. HUVEC were pretreated with NAC 30 min prior to exposure to HNP for the indicated times. The cell lysates were analyzed for phosphorylated and total levels of p38 by Western blotting. N = 3. * p < 0.05 vs. vehicle control; † p < 0.05 vs. HNP alone, respectively. ‡ p < 0.05 vs. HNP alone at 30 min, and 60 min, respectively.
HNP had no effects on Angiotensin-II to modulate COX-2 and ET-1
A large body of evidence supports a role of Angiotensin-II in modulation of the expression of COX-2 and ET-1 (19, 20, 28). We thus examined the Angiotensin-II pathway in the HNP-induced expression of COX-2 and ET-1 by using the dicarboxylate-containing angiotensin converting enzyme inhibitor Enalapril. Our results showed no effects on the HNP-induced expression of COX-2 or ET-1 by using Enalapril at a dose of 100 μM (Fig. 6A and Fig. 6B).
Figure 6. HNP had no effects on Angiotensin-II to modulate COX-2 and ET-1.

HUVEC were incubated with Enalapril for 30 min prior to stimulation with HNP for additional 6h. COX-2 expression was detected by Western blotting. B. Concentration of ET-1 was measured in cell supernatant by ELISA. N = 2. * p < 0.05 vs. vehicle (0.01% acetic acid) control, respectively.
DISCUSSION
A main finding of our study is that HNP can stimulate human endothelial cells to increase the expression of COX-2 and ET-1, and ROS play an important role in mediating the HNP-induced activation of endothelial cells. The treatment of endothelial cells with the ROS scavenger NAC attenuated the elevated expression of COX-2 and ET-1 in response to HNP stimulation.
Previous in vivo and in vitro studies have described the role of ROS and peroxynitrite in the regulation of COX-2 expression (29, 30). We have demonstrated that exposure of lung explants to HNP resulted in an increase in the hydrogen peroxide production (6). We now observe an increased formation of nitrotyrosine that reflects the interaction between NO and ROS by human endothelial cells, in response to HNP stimulation. Further, the HNP-induced COX-2 expression was attenuated by the treatment with NAC. This observation indicates that HNP increased COX-2 expression through oxidative stress. Previous studies have reported that an upregulation of COX-2 after stimulation with cytokines, thrombin and growth factors usually led to an increased production of PGI2 in HUVEC (24, 31). However, production of the prostaglandins PGI2 and PGE2 was not significantly altered despite an elevated COX-2 expression after HNP stimulation in our study. Interestingly, previous studies have also reported an absence of prostaglandin release in spite of an elevated COX-2 expression following stimulation with NO donors (32, 33). A couple of mechanisms may explain the paradox of an increased COX-2 expression with lack of prostaglandin production. Peroxynitrite can cause tyrosine nitration of COX leading to inactivation of COX activity (34). Peroxynitrite can also act as a potent inhibitor of PGI synthase, a terminal enzyme in PGI2 biosynthesis (35). It is noteworthy that we examined only PGI2 and PGE2 because they are the most characterized prostaglandins. The effects of HNP on other prostaglandins remain yet to be determined.
ET-1 is a dominant vasoconstrictor produced by endothelial cells (36, 37). We observed a rapid increase in ET-1 release as early as 30 min after HNP stimulation in HUVEC. The early and sustained expression of ET-1 could be due to autocrine regulatory properties by which ET-1 acts on cell surface receptors that in turn activate cells releasing more ET-1 (36). In addition, the production of ET-1 was blocked by the treatment of the cells with NAC, suggesting a role of ROS in mediating the HNP-induced ET-1 production. Our results are consistent with other studies reporting that ROS play an important role to stimulate smooth muscle cells (26) and endothelial cells (37) producing ET-1 in in vitro and in vivo conditions (38,39).
To understand the intracellular signaling mechanisms by which HNP upregulated the expression of COX-2 and ET-1, we examined several signaling pathways including p38 MAPK, ERK1/2 and NF-κB since they we have previously demonstrated that the kinases and NF-κB are involved in the modulation of adhesion molecules and IL-8 by monocytes and CD4+ cells in response to HNP stimulation (1,4). By employing the specific inhibitors, we observed that p38 MAPK, ERK1/2 and NF-κB signaling pathways are required to modulate the HNP-induced expression of COX-2 and ET-1 in HUVEC. Other studies have demonstrated that the kinases and NF-κB signaling are required in the regulation of COX-2 and ET-1 in response to a variety of stimuli (40, 41).
It has been suggested that angiotensin-II modulates COX-2 and ET-1 expression (19, 20). We did not observe any significant effects of the angiotensin-II pathway on the HNP-induced COX-2 or ET-1 expression in HUVEC by using Enalapril, an inhibitor of angiotensin converting enzyme. Unlike other angiotensin converting enzyme inhibitors, Enalapril does not interfere with the NO and ROS pathways (42). Taken together, these studies further support the central role of ROS by which HNP activate endothelial cells.
In summary, we demonstrate that HNP increased COX-2 and ET-1 expression in endothelial cells via ROS-dependent mechanisms that require the activation of p38 MAPK, ERK1/2 and NF-κB signaling pathways. The effects of HNP in the modulation of the course of cardiovascular diseases are yet to be elucidated in in vivo conditions.
Acknowledgments
Supported by Canadian Institutes of Health Research (CIHR) operating grants to AS and HZ and Ontario Thoracic Society (OTS) Grant-in-Aid to HZ. HZ is a recipient of Ontario Premier’s Research Excellence Award.
References
- 1.Vaschetto R, Grinstein J, Del Sorbo L, Khine AA, Voglis S, Tullis E, Slutsky AS, Zhang H. Role of human neutrophil peptides in the initial interaction between lung epithelial cells and CD4+ lymphocytes. J Leukoc Biol. 2007;81:1022–1031. doi: 10.1189/jlb.0706435. [DOI] [PubMed] [Google Scholar]
- 2.Chaly YV, Paleolog EM, Kolesnikova TS, Tikhonov, Petratchenko EV, Voitenok NN. Neutrophil alpha-defensin human neutrophil peptide modulates cytokine production in human monocytes and adhesion molecule expression in endothelial cells. Eur Cytokine Netw. 2000;11:257–66. [PubMed] [Google Scholar]
- 3.Khine AA, Del Sorbo L, Vaschetto R, Voglis S, Tullis E, Slutsky AS, Downey GP, Zhang H. Human neutrophil peptides induce interleukin-8 production through the P2Y6 signaling pathway. Blood. 2006;107:2936–2942. doi: 10.1182/blood-2005-06-2314. [DOI] [PubMed] [Google Scholar]
- 4.Syeda F, Liu HY, Tullis E, Liu M, Slutsky AS, Zhang H. Differential signaling mechanisms of HNP-induced IL-8 production in human lung epithelial cells and monocytes. J Cell Physiol. 2007 Sep 4; doi: 10.1002/jcp.21279. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Wetering S, Mannesse-Lazeroms SP, Van Sterkenburg MA, Daha MR, Dijkman JH, Hiemstra PS. Effect of defensins on interleukin-8 synthesis in airway epithelial cells. Am J Physiol. 1997;(272):L888–96. doi: 10.1152/ajplung.1997.272.5.L888. [DOI] [PubMed] [Google Scholar]
- 6.Porro GA, Lee JH, de Azavedo J, Crandall I, Whitehead T, Tullis E, Ganz T, Liu M, Slutsky AS, Zhang H. Direct and indirect bacterial killing functions of neutrophil defensins in lung explants. Am J Physiol Lung Cell Mol Physio. 2001;281:L1240–7. doi: 10.1152/ajplung.2001.281.5.L1240. [DOI] [PubMed] [Google Scholar]
- 7.Yang D, Chen Q, Chertov O, Oppenheim JJ. Human neutrophil defensins selectively chemoattract naive T and immature dendritic cells. J Leukoc Biol. 2000;68:9–14. [PubMed] [Google Scholar]
- 8.Chertov O, Michiel DF, Xu L, Wang JM, Tani K, Murphy WJ, Longo DL, Taub DD, Oppenheim JJ. Identification of defensin-1, defensin-2, and CAP37/azurocidin as T-cell chemoattractant proteins released from interleukin-8-stimulated neutrophils. J Biol Chem. 1996;271:2935–40. doi: 10.1074/jbc.271.6.2935. [DOI] [PubMed] [Google Scholar]
- 9.Nassar T, Akkawi S, Bar-Shavit R, Haj-Yehia A, Bdeir K, Al-Mehdi AB, Tarshis M, Higazi AA. Human alpha-defensin regulates smooth muscle cell contraction: a role for low-density lipoprotein receptor-related protein/alpha 2-macroglobulin receptor. Blood. 2002;100:4026–32. doi: 10.1182/blood-2002-04-1080. [DOI] [PubMed] [Google Scholar]
- 10.Lopez-Bermejo A, Chico-Julia B, Castro A, Recasens M, Esteve E, Biarnes J, Casamitjana R, Ricart W, Fernandez-Real JM. Alpha defensins 1, 2, and 3: potential roles in dyslipidemia and vascular dysfunction in humans. Arterioscler Thromb Vasc Biol. 2007;27:1166–71. doi: 10.1161/ATVBAHA.106.138594. [DOI] [PubMed] [Google Scholar]
- 11.Ashitani J, Matsumoto N, Nakazato M. Elevated levels of antimicrobial peptides in bronchoalveolar lavage fluid in patients with chronic eosinophilic pneumonia. Respiration. 2007;74:69–75. doi: 10.1159/000090199. [DOI] [PubMed] [Google Scholar]
- 12.Ashitani J, Mukae H, Arimura Y, Sano A, Tokojima M, Nakazato M. High concentrations of alpha-defensins in plasma and bronchoalveolar lavage fluid of patients with acute respiratory distress syndrome. Life Sci. 2004;75:1123–34. doi: 10.1016/j.lfs.2004.01.028. [DOI] [PubMed] [Google Scholar]
- 13.Panyutich AV, Panyutich EA, Krapivin VA, Baturevich EA, Ganz T. Plasma defensin concentrations are elevated in patients with septicemia or bacterial meningitis. J Lab Clin Med. 1993;122:202–207. [PubMed] [Google Scholar]
- 14.Tsatsanis C, Androulidaki A, Venihaki M, Margioris AN. Signalling networks regulating cyclooxygenase-2. Int J Biochem Cell Biol. 2006;38:1654–61. doi: 10.1016/j.biocel.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 15.Yin H, Cheng L, Langenbach R, Ju C. Prostaglandin I(2) and E(2) mediate the protective effects of cyclooxygenase-2 in a mouse model of immune-mediated liver injury. Hepatology. 2007;45:159–69. doi: 10.1002/hep.21493. [DOI] [PubMed] [Google Scholar]
- 16.Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351:1425–36. doi: 10.1056/NEJMra040291. [DOI] [PubMed] [Google Scholar]
- 17.Fan J, Unoki H, Iwasa S, Watanabe T. Role of Endothelin-1 in atherosclerosis. Ann NY Acad Sci. 2000;902:84–94. doi: 10.1111/j.1749-6632.2000.tb06303.x. [DOI] [PubMed] [Google Scholar]
- 18.Libby P, Aikawa M, Jain MK. Vascular endothelium and atherosclerosis. Handb Exp Pharmacol. 2006;176:285–306. doi: 10.1007/3-540-36028-x_9. [DOI] [PubMed] [Google Scholar]
- 19.Cingolani HE, Villa-Abrille MC, Cornelli M, Nolly A, Ennis IL, Garciarena C, Suburo AM, Torbidoni V, Correa MV, Camilionde Hurtado MC, Aiello EA. The positive inotropic effect of angiotensin II: role of endothelin-1 and reactive oxygen species. Hypertension. 2006;47:727–34. doi: 10.1161/01.HYP.0000208302.62399.68. [DOI] [PubMed] [Google Scholar]
- 20.Jaimes EA, Tian RX, Pearse D, Raij L. Up-regulation of glomerular COX-2 by angiotensin II: role of reactive oxygen species. Kidney Int. 2005;68:2143–53. doi: 10.1111/j.1523-1755.2005.00670.x. [DOI] [PubMed] [Google Scholar]
- 21.Dai Y-Q, Jin D-Z, Zhu X-Z, Lei D-L. Triptolide inhibits COX-2 expression via NF-kappa B pathway in astrocytes. Neuroscience Research. 2006;55:154–160. doi: 10.1016/j.neures.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 22.Corson TW, Crews CM. Molecular understanding and modern application of traditional medicines: triumphs and trials. Cell. 2007;130:769–74. doi: 10.1016/j.cell.2007.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shin HM, Kim MH, Kim BH, Jung SH, Kim YS, Park HJ, Hong JT, Min KR, Kim Y. Inhibitory action of novel aromatic diamine compound on lipopolysaccharide-induced nuclear translocation of NF-kappaB without affecting IkappaB degradation. FEBS Lett. 2004;571:50–4. doi: 10.1016/j.febslet.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 24.Syeda F, Grosjean J, Houliston RA, Keogh RJ, Carter TD, Paleolog E, Wheeler-Jones CP. Cyclooxygenase-2 induction and prostacyclin release by protease-activated receptors in endothelial cells require cooperation between mitogen-activated protein kinase and NF-kappaB pathways. J Biol Chem. 2006;281:11792–804. doi: 10.1074/jbc.M509292200. [DOI] [PubMed] [Google Scholar]
- 25.Dianzani C, Collino M, Gallicchio M, Di Braccio M, Roma G, Fantozzi R. Effects of anti-inflammatory [1, 2, 4]triazolo[4, 3-a] [1, 8]naphthyridine derivatives on human stimulated PMN and endothelial cells: an in vitro study. J Inflamm (Lond) 2006;3:4. doi: 10.1186/1476-9255-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kahler J, Ewert A, Weckmuller J, Stobbe S, Mittmann C, Koster R, Paul M, Meinertz T, Munzel T. Oxidative stress increases endothelin-1 synthesis in human coronary artery smooth muscle cells. J Cardiovasc Pharmacol. 2001;38:49–57. doi: 10.1097/00005344-200107000-00006. [DOI] [PubMed] [Google Scholar]
- 27.Hurst JK. Whence nitrotyrosine? J Clin Invest. 2002;109:1287–9. doi: 10.1172/JCI15816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ushio-Fukai M, Alexander RW, Akers M, Griendling KK. p38 Mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin II. Role in vascular smooth muscle cell hypertrophy. J Biol Chem. 1998;273:15022–9. doi: 10.1074/jbc.273.24.15022. [DOI] [PubMed] [Google Scholar]
- 29.Kim SF, Huri DA, Snyder SH. Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science. 2005;310:1966–70. doi: 10.1126/science.1119407. [DOI] [PubMed] [Google Scholar]
- 30.Landino LM, Crews BC, Timmons MD, Morrow JD, Marnett LJ. Peroxynitrite, the coupling product of nitric oxide and superoxide, activates prostaglandin biosynthesis. Proc Natl Acad Sci U S A. 1996;93:15069–74. doi: 10.1073/pnas.93.26.15069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olszanecki R, Gebska A, Korbut R. Production of prostacyclin and prostaglandin E2 in resting and IL-1beta-stimulated A549, HUVEC and hybrid EA.HY 926 cells. J Physiol Pharmacol. 2006;57:649–60. [PubMed] [Google Scholar]
- 32.Eligini S, Habib A, Lebret M, Creminon C, Levy-Toledano S, Maclouf J. Induction of cyclo-oxygenase-2 in human endothelial cells by SIN-1 in the absence of prostaglandin production. Br J Pharmacol. 2001;133:1163–71. doi: 10.1038/sj.bjp.0704163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reddy ST, Herschman HR. Prostaglandin synthase-1 and prostaglandin synthase-2 are coupled to distinct phospholipases for the generation of prostaglandin D2 in activated mast cells. J Biol Chem. 1997;272:3231–7. doi: 10.1074/jbc.272.6.3231. [DOI] [PubMed] [Google Scholar]
- 34.Upmacis RK, Deeb RS, Hajjar DP. Oxidative alterations of cyclooxygenase during atherogenesis. Prostaglandins Other Lipid Mediat. 2006;80:1–14. doi: 10.1016/j.prostaglandins.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 35.Cosentino F, Eto M, De Paolis P, van der Loo B, Bachschmid M, Ullrich V, Kouroedov A, Delli Gatti C, Joch H, Volpe M, Luscher TF. High glucose causes upregulation of cyclooxygenase-2 and alters prostanoid profile in human endothelial cells: role of protein kinase C and reactive oxygen species. Circulation. 2003;107:1017–23. doi: 10.1161/01.cir.0000051367.92927.07. [DOI] [PubMed] [Google Scholar]
- 36.Murakoshi N, Miyauchi T, Kakinuma Y, Ohuchi T, Goto K, Yanagisawa M, Yamaguchi I. Vascular endothelin-B receptor system in vivo plays a favorable inhibitory role in vascular remodeling after injury revealed by endothelin-B receptor-knockout mice. Circulation. 2002;106:1991–8. doi: 10.1161/01.cir.0000032004.56585.2a. [DOI] [PubMed] [Google Scholar]
- 37.Kahler J, Mendel S, Weckmuller J, Orzechowski HD, Mittmann C, Koster R, Paul M, Meinertz T, Munzel T. Oxidative stress increases synthesis of big endothelin-1 by activation of the endothelin-1 promoter. J Mol Cell Cardiol. 2000;32:1429–37. doi: 10.1006/jmcc.2000.1178. [DOI] [PubMed] [Google Scholar]
- 38.Zhou Y, Mitra S, Varadharaj S, Parinandi N, Zweier JL, Flavahan NA. Increased expression of cyclooxygenase-2 mediates enhanced contraction to endothelin ETA receptor stimulation in endothelial nitric oxide synthase knockout mice. Circ Res. 2006;98:1439–45. doi: 10.1161/01.RES.0000224120.52792.10. [DOI] [PubMed] [Google Scholar]
- 39.Hink U, Munzel T. COX-2, another important player in the nitric oxide-endothelin cross-talk: good news for COX-2 inhibitors? Circ Res. 2006;98:1344–6. doi: 10.1161/01.RES.0000228471.38761.93. [DOI] [PubMed] [Google Scholar]
- 40.Lee HM, Won KJ, Kim J, Park HJ, Kim HJ, Roh HY, Lee SH, Lee CK, Kim B. Endothelin-1 induces contraction via a Syk-mediated p38 mitogen-activated protein kinase pathway in rat aortic smooth muscle. J Pharmacol Sci. 2007;103:427–33. doi: 10.1254/jphs.fp0070039. [DOI] [PubMed] [Google Scholar]
- 41.Song HJ, Min YS, Shin CY, Jeong JH, Sohn UD. Activation of p38 MAPK is involved in endothelin-1-stimulated COX-2 expression in cultured Feline esophageal smooth muscle cells. Mol Cells. 2006;22:44–50. [PubMed] [Google Scholar]
- 42.Scribner AW, Loscalzo J, Napoli C. The effect of angiotensin-converting enzyme inhibition on endothelial function and oxidant stress. Eur J Pharmacol. 2003;482:95–9. doi: 10.1016/j.ejphar.2003.10.002. [DOI] [PubMed] [Google Scholar]