

Abstract
Castration-resistant prostate cancer (CRPC) is an androgen receptor (AR)-dependent disease expected to cause the death of more than 27,000 Americans in 2015. There are only a few available treatments for CRPC, making the discovery of new drugs an urgent need. We report that CUDC-101 (an inhibitor od HER2/NEU, EGFR and HDAC) inhibits both the full length AR (flAR) and the AR variant AR-V7. This observation prompted experiments to discover which of the known activities of CUDC-101 is responsible for the inhibition of flAR/AR-V7 signaling. We used pharmacologic and genetic approaches, and found that the effect of CUDC-101 on flAR and AR-V7 was duplicated only by other HDAC inhibitors, or by silencing the HDAC isoforms HDAC5 and HDAC10. We observed that CUDC-101 treatment or AR-V7 silencing by RNAi equally reduced transcription of the AR-V7 target gene, PSA, without affecting viability of 22Rv1 cells. However, when cellular proliferation was used as an end point, CUDC-101 was more effective than AR-V7 silencing, raising the prospect that CUDC-101 has additional targets beside AR-V7. In support of this, we found that CUDC-101 increased the expression of the cyclin-dependent kinase inhibitor p21, and decreased that of the oncogene HER2/NEU. To determine if CUDC-101 reduces growth in a xenograft model of prostate cancer, this drug was given for 14 days to castrated male SCID mice inoculated with 22Rv1 cells. Compared to vehicle, CUDC-101 reduced xenograft growth in a statistically significant way, and without macroscopic side effects. These studies demonstrate that CUDC-101 inhibits wtAR and AR-V7 activity and growth of 22Rv1 cells in vitro and in vivo. These effects result from the ability of CUDC-101 to target not only HDAC signaling, which was associated with decreased flAR and AR-V7 activity, but multiple additional oncogenic pathways. These observations raise the possibility that treatment of CRPC may be achieved by using similarly multi-targeted approaches.
Electronic supplementary material
The online version of this article (doi:10.1007/s12672-016-0257-2) contains supplementary material, which is available to authorized users.
Keywords: Androgen Receptor, Prostate Specific Antigen, Gefitinib, Androgen Deprivation Therapy, HDAC Inhibitor
Introduction
Castration-resistant prostate cancer (CRPC) arises after patients with prostate cancer (PCa) fail androgen deprivation therapy (ADT) [1]. The most sensitive biochemical sign of CRPC relapse is an increase of the AR-dependent protein, prostate specific antigen (PSA). This event indicated that AR is still signaling despite patients receiving ADT usually have anorchid levels of serum androgens [2–4]. Second-generation ADT treatments to block AR activity in CRPC have been approved by the FDA and include the CYP17A1 inhibitor abiraterone acetate (AA) [5] and the AR antagonist enzalutamide [6]. Although yielding an average increase in life of a few months, AA and enzalutamide are limited in effect, as some patients manifest de novo resistance, while others relapse within a short period of time [7–9]. According to a number of recent reports [10, 11], the accumulation in the recurring tumor of the carboxyl terminal truncated and constitutively active AR variant (AR-V), AR-V7 [12–17] is an important contributor to resistance to second generation ADT. In a parallel project, we embarked on a small library screen of FDA approved drugs to establish classes of bioactive molecules that inhibit AR and AR-V7 signaling, and thus may be useful to treat CRPC [18]. One class of compounds identified with this approach (manuscript in preparation) consisted of histone deacetylase inhibitors (HDACi), which lead to testing commercially available agents with the same mechanism of action. Using this approach, we found that CUDC-101 (a commercially available combined HDAC, EGFR, and HER2/Neu inhibitor) [19] is a strong inhibitor of flAR and AR-V7. Herewith, we describe the mechanism CUDC-101 uses to inhibit flAR (full-length AR) and AR-V7 activity, and in vivo experiments showing that this compound prevents growth of CRPC xenografts in SCID mice.
Material and Methods
Cell Lines
LNCaP [20], PC-3 [21], HeLa [22], VCaP [23], and 22Rv1 [24] cells were purchased from ATCC (Rockville, MD). C4-2 cells [25] were purchased from UroCor (Oklahoma City, OK). LAPC4 cells were obtained from Dr. C. Sawyers and CWR-R1 from Dr. S. Dehm [26]. LNCaPAR-V7/pLenti (a gift of Dr. Nancy Weigel) were generated as previously reported [27, 28]. In addition to flAR containing the T877A mutation, LNCaPAR-V7/pLenti express AR-V7 upon addition of 0.25 ng/mL doxycycline for 24 h. Stably transfected PC-3-GFP-AR-V7 [29], HELA-GFP-wtAR [30], and PC-3-GFP-wtAR [31] were generated as previously described and express constitutively GFP-wtAR or GFP-AR-V7 under the control of the CMV promoter. Cell lines were cultured in 5 % CO2 at 37 °C using RPMI 1640 + 10 % FBS.
Cell lines were chosen based on the fact that they express AR’s containing the wild-type sequence (LAPC4, VCaP, HELA-GFP-wtAR, and PC-3-GFP-wtAR), the full-length sequence with point mutations (LNCaP [32], C4-2 [32], 22Rv1 [33], LNCaPAR-V7/pLenti [32], and CWR-R1) or the AR variant AR-V7 (22Rv1, PC-3-GFP-AR-V7, CWR-R1, and LNCaPAR-V7/pLenti). Some of the cell lines are known to express more than one form of AR; for instance, 22Rv1 cells express full-length (fl)AR containing mutation H784Y [34] and a duplication of exon 3 [35], AR-V7 [14] and AR-V4 [36]. CWR-R1 cells express both flAR [containing the H874Y mutation [33] and AR-V7 [14]. In this manuscript, AR’s containing the wild-type sequence or point mutations were abbreviated as flAR.
Reagents
HDAC inhibitors (HDACi) CUDC-101 [19], pracinostat [37], MGCD0103 [38], MC1568 [39], and PCI-34051 [40] were purchased from Selleck Chemicals. The specific inhibitors of EGFR and HER2/NEU, Gefitinib [41], and CP-724714 [42], were also purchased from Selleck. Sodium butyrate [43] was purchased from Sigma Aldrich. All HDACi were used at published IC50. Gefitinib and CP-724714 were used at a range of concentrations (108–540 nM and 108–864 nM, respectively). In the experiments determining IC50, dose response and time course activity against AR-V7 and flAR activity (Fig. 1), CUDC-101 was used between 0.3 nM to 1 μM from 3 to 24 h. It was used at a concentration of 300 nM for 6 or 24 h in the remaining experiments, unless stated otherwise. All reagents were dissolved in DMSO, except DHT, which was dissolved in ethanol.
Fig. 1.

a–b: LNCaPAR-V7/pLenti cells were transiently transfected with plasmid ARR2PBLuc. After 24 h in charcoal stripped-FBS (CS-FBS), cells were treated with doxycycline 0.25 μg/ml. After 24 h, CUDC was added for 24 h at concentrations ranging between 0 and 1000 nM. Panel a percent inhibition of relative luciferase activity in the presence of CUDC-101. b parallel immunoblot analysis demonstrating the induction of AR-V7 by doxycycline. Dox (−) represents a control where no dox was added, that was not used in the experiment of panel a. c Dose response experiment: 22Rv1 were seeded and grown for 24 h in CS-FBS, cells were then treated for 24 h with vehicle or increasing concentrations of CUDC-101 (0 to 0.3 μM) alone, to measure AR-V7 transcriptional activity, or DHT (2 nM) + CUDC-101 (0 to 0.3 μM) to measure flAR transcriptional activity. After 24 h, cells were harvested and aliquots obtained for determination of PSA mRNA. Bars ± SE represent PSA mRNA corrected for 18 S mRNA normalized to cells treated with vehicle. d Time course experiment: 22Rv1 cells were seeded and grown for 24 h in CS-FBS, cells were then treated with vehicle (V) or DHT (D) for additional 24 h. CUDC-101 (300 nM) was added for 3, 6, or 24 h in addition to vehicle to measure % inhibition of AR-V7 transcriptional activity (black bars). CUDC-101 (300 nM) + DHT (2 nM) were added for 3, 6, or 24 h to cells previously treated with DHT for 24 h to measure % inhibition of flAR transcriptional activity. At the indicated time points, cells were harvested and aliquots obtained for determination of PSA mRNA. Bars ± SE represent PSA mRNA corrected for 18 S mRNA normalized to cells treated with vehicle. e Parallel immunoblot analysis of flAR, AR-V7 and beta actin using aliquots of 22Rv1 cells treated with CUDC101 + DHT from panel c. f Parallel immunoblot analysis of flAR, AR-V7, and beta actin using aliquots of cells treated with CUDC-101+ DHT from panel d. **P < 0.01 compared to vehicle-treated control. ***P < 0.001 compared with vehicle + DHT- treated control. Experiments were done in triplicate a minimum of three times
Cell Line-Based Experiments
In the experiments performed with the LNCaPAR-V7/pLenti line, cells were seeded and grown in charcoal stripped-FBS (CS-FBS). After 24 h, Doxycycline (Dox) was added at 0.25 ng/ml to induce AR-V7 expression. When testing AR-V7 activity, CUDC-101 was added after 24 h of Dox stimulation at different concentrations and for different amounts of time, as indicated in the main text and legends. When testing flAR activity, 2 nM DHT, or 2 nM DHT + CUDC-101 were added simultaneously. CUDC-101 was administered at different concentrations and length of time depending on the experimental conditions, as indicated in the main text and legends. Inhibition of AR-V7 transcriptional activity was calculated as the % inhibition of luciferase or PSA mRNA expression in CUDC-101-treated cells compared with vehicle control. Inhibition of flAR activity was calculated as the % inhibition of luciferase or PSA mRNA expression in DHT + CUDC-101-treated cells compared with DHT alone.
All other cell lines were seeded and grown in CS-FBS for 24 h. When testing the activity of AR-V7, cells were treated with vehicle or CUDC-101, other HDAC inhibitors (HDACi) (pracinostat, MGCD0103, MC1568, or PCI-34051), or inhibitors of EGFR or HER2/Neu (Gefitinib and CP724714, respectively) at different concentrations and length of time depending on the experimental conditions, as indicated in the main text and legends. When testing the activity of flAR, cells were seeded and grown in CS-FBS. After 24 h 2 nM DHT was added alone or together with CUDC-101, other HDAC inhibitors (HDACi) (pracinostat, MGCD0103, MC1568, or PCI-34051), or inhibitors of EGFR or HER2/Neu (Gefitinib and CP724714, respectively) at different concentrations and length of time depending on the experimental conditions, as indicated in the main text and legends. Inhibition of AR-V7 transcriptional activity was calculated as the % inhibition of luciferase or PSA mRNA expression in CUDC-101-treated cells compared with vehicle control. Inhibition of flAR activity was calculated as the % inhibition of luciferase or PSA mRNA expression in DHT and CUDC-101-treated cells compared with DHT.
In the experiment testing AR and AR-V7 protein stability (Supplemental Fig. S5a), 22Rv1 cells were seeded in CS-FBS. After 24 h, cells were treated with cycloheximide (10 μg/mL) or cycloheximide (10 μg/mL) + CUDC-101 (300 nM) for 0, 3, 6 and 24 h. At the end of the experiment, cells were harvested and used for immunoblot analysis of flAR, AR-V7, and β-actin.
In the experiments testing if CUDC-101 decreases transcription of AR-dependent genes by directly interfering with AR transcription (Supplemental Fig. S5c), PC-3-GFP-AR-V7 were transfected with ARR2PB-Luc and grown in CS-FBS. After 24 h, cells were treated with CUDC-101 (300 nM) for 0, 6, 24 h. At the end of the experiment, aliquots of cells were used to determine luciferase activity and protein expression of GFP-AR-V7 and β-actin.
All experiments were done in triplicate and a minimum of three times.
Western Blotting
Cells were lysed in TESH buffer [10 mmol/L Tris, 1 mmol/L EDTA, 12 mmol/L thioglycerol (pH 7.7)] [19] with 0.4 M NaCl using three rounds of freeze/thaw; 30 μg of protein were resolved on a 6.5 % SDS gel. Proteins were detected using ECL reagents and antibodies raised against HDAC1 (sc-7872, rabbit polyclonal, used at 1:200), HDAC5 (sc-133225, monoclonal, used at 1:100), HDAC6 (sc-11420, rabbit polyclonal, used at 1:200), HDAC7 (sc-74563, monoclonal, used at 1:250), HDAC8 (sc11405, rabbit polyclonal, used at 1:200), HDAC9 (sc 28732, rabbit polyclonal, used at 1:100), and HDAC10 (sc134995, rabbit polyclonal, used at 1:200) all from Santa Cruz (Santa Cruz, CA), AR [monoclonal anti AR antibody (AR441), used at 1:1000] [44] (a gift of Nancy Weigel, Baylor College of Medicine) and β-actin [rabbit polyclonal antibody from Sigma-Aldrich (A-2066) (used at 1:250)]. Phospho-HER2/ErbB2 (Tyr1248) Antibody #2247 (used at 1:1000) and HER2/ErbB2 Ab #2165 (used at 1:1000) were from Life Technologies. Phospho-EGF Receptor (Tyr1068) Ab # 3777 (used at 1:1000) and EGF Receptor Antibody #2232 (used at 1:1000) were from Life Technologies.
Quantitative PCR
qPCR was performed in triplicate and a minimum of three times as described previously [29], using 18S as calibrator gene.
RNAi Silencing
Each siRNA was purchased from Applied Biosystem and transfected using siPORT™ NeoFX™ following the protocol recommended by the manufacturer. Briefly, cells were trypsinized and resuspended in medium containing 4 % CS-FBS. 7.5 μl siRNA (5 μM) was incubated with transfection reagents at room temperature. The mixture was added to a six-well plate and overlaid with 2 × 105 cells per well. After 48 h, cells were used to perform the experiment of interest (as outlined in the main text). At the end of the experiment, one aliquot was used to determine the effect of treatment of flAR or AR-V7 transcriptional activity, the second aliquot was used to verify successful silencing of the gene of interest by immunoblot analysis.
Cell Viability Assay
Cell viability was measured using the CCK-8 kit (Dojindo, Rockville MD) as per manufacturer instructions.
ChIP Analysis
In the experiment shown in Fig. 5a, 22Rv1 cells were transfected with siRNAs to silence flAR or with scrambled siRNA. Cells were then starved for 24 h in CS-FBS, treated for 6 h with CUDC101 (300 nM) and harvested.
Fig. 5.
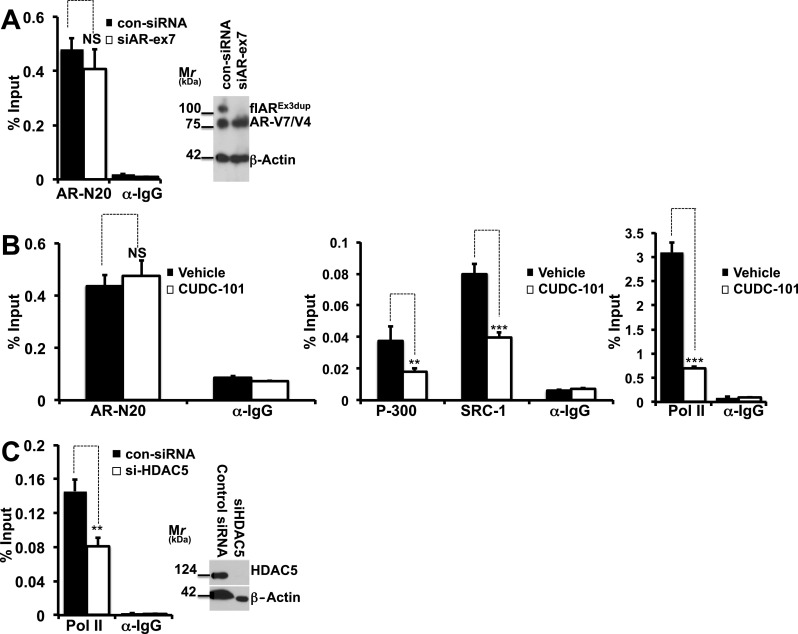
a–c CUDC-101 treatment or HDAC5 silencing prevent formation of an active AR-V7-dependent transcriptional complex at the level of the PSA enhancer. a 22Rv1 cells underwent experimental manipulation with specific siRNA’s to silence flAR or with control siRNA’s. Cells were grown in CS-FBS for 24 h, harvested and used for ChIP analysis performed with anti AR Ab-N20 (Santa Cruz) or rabbit IgG. Right panel Immunoblot analysis for flAR, AR-V7 and beta actin. Left panel ChIP analysis measuring AR-V7 bound to PSA promoter. NS: denotes statistically non-significant difference when comparing 22Rv1 cells with silenced flAR (siAR-ex7) to control cells (con-siRNA). b 22Rv1 cells were starved in CS-FBS medium. After 24 h cells were treated for 6 h with CUDC101 at the concentrations of 300 nM and harvested. AR, P-300, SRC-1 and RNA pol II binding to the PSA enhancer was measured by ChIP followed by qPCR using AR, RNA polymerase II, p300, SRC-1, and rabbit and mouse immunoglobulin G (IgG) antibodies (all from Santa Cruz). NS: statistically non-significant; **P > 0.01, ***P > 0.001 compared to vehicle treated cells. c 22Rv1 cells underwent experimental manipulation with specific siRNA’s to silence HDAC5 or with control siRNA’s. Cells were grown in CS-FBS for 24 h and harvested. RNA pol II bound to the PSA enhancer was measured by ChIP followed by qPCR (left panel) using anti Pol II or rabbit IgG antibodies. Right panel Immunoblot analysis for HDAC5 and beta actin demonstrating silencing of HDAC5. **P < 0.01 when comparing 22Rv1 cells with silenced HDAC5 (si-HDAC5) to control cells (con-siRNA)
In experiment 5b, 22Rv1 cells were starved in CS-FBS medium. After 24 h cells were treated for 6 h with CUDC101 (300 nM) and harvested.
In experiment 5c, 22Rv1 cells were transfected with siRNAs to silence HDAC5 or with scrambled siRNA. Cells were then starved for 24 h in CS-FBS, treated for 6 h with CUDC101 (300 nM) and harvested. Aliquots were used to demonstrate silencing of flAR (Fig. 5a) or HDAC5 (Fig. 5c) and for ChIP analysis. ChIP was performed as previously described [45] with minor modifications. Briefly, after culture medium removal from each dish, 1 % molecular biology-grade formaldehyde (Sigma-Aldrich) was added to the cells for 10 min at room temperature to cross-link protein complexes to DNA. Cells were pelleted and then lysed with RIPA buffer. Chromatin was sheared by sonication to less than 1 kb fragments. The soluble chromatin fraction was collected after centrifugation at 12,000 g for 5 min. Following pre-cleaning steps, 10 % of the supernatant was allocated as input for normalization. Equivalent amounts were subjected to antibodies for AR-N20, p-300, SRC-1 or Pol-II or normal rabbit IgG that served as a negative control. After incubation at 4 °C ON, antibodies were precipitated by adding protein A/G beads and agitated for 3 h at 4 °C. The pellet was collected and subjected to washing buffers. DNA was extracted from the beads, purified and eluted using a PCR purification kit (Qiagen), followed by quantitative real-time PCR as described above. Antibodies for AR, RNA polymerase II, p300, SRC-1, and rabbit and mouse immunoglobulin G (IgG) were all purchased from Santa Cruz. Data were normalized to total input DNA to calculate the percentage of input.
Primer sets for PSA and AR enhancer are listed below:
| PSA | Forward | 5′-GCCTGGATCTGAGAGAGATATCATC-3′ |
| Reverse | 5′-ACACCTTTTTTTTTCTGGATTGTG-3′ | |
| AR | Forward | 5′-CCTGCCTGTCTTTTCAGAGG-3′ |
| Reverse | 5′-TTCCCCTCCTTTTGCTCTTT-3′ |
Calculation of CUDC-101 IC50
Relative luciferase activity generated by plasmid ARR2PB-luciferase [46] was used to determine the IC50 of CUDC-101 against AR-V7 transcriptional activity. LNCaP Dox cells were transiently transfected with plasmid ARR2-PB-Luc and treated with 0.25 ng/ml doxycycline. After 24 h CUDC-101 was added at concentrations ranging from 0 to 1000 nM for 24 h. Cells were harvested. Percentage inhibition of relative luciferase activity was calculated in the presence of CUDC-101 and the results were applied to fit an antagonist curve using GraphPad Prism 5.0 (Fig. 1a), which yielded an IC50 of 88 nM. CUDC-101 was used at a final concentration of 300 nM in most of the subsequent experiments based on the calculated IC50 and on the results of the experiments shown in Fig. 1c, d.
Xenograft Experiments
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Baylor College of Medicine Institutional Animal Care and Use Committee (protocol AN-6250). All surgery was performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering. 2 × 105 22Rv1 cells natively expressing both wtAR and AR-V7 were mixed at a 1:1 volume ratio with Matrigel (BD Biosciences) and injected into the right flank of 16 male SCID mice. After 5 days, mice were castrated, and examined every 3 days for the occurrence of palpable sc xenografts. Xenograft-bearing animals were randomized to a control group (treated with IP injections of 50 μl of DMSO every 24 h) and a group receiving CUDC-101 (40 mg/Kg) in 50 μl of DMSO. We planned to carry on the experiment for four weeks, however by the second week many tumors in the control group had grown beyond 10 % of the baseline body weight, and, as per IRB specifications, the experiment had to be terminated earlier at 14 days. Tumor size was measured with a caliper using the equation m1.2 × m2 × 0.5236 (where m1 and m2 are the smallest and largest diameters) [47]. Mice were weighed at the beginning and end of the experiment. Upon sacrifice, a macroscopic autopsy was performed to rule out presence of visible abnormalities. Data were expressed as percent change from baseline; unpaired t test was used for statistical analysis with significance set at a P < 0.05.
Statistical Analysis
Each experiment was performed in triplicate and repeated a minimum of three times. One-way ANOVA was used for statistical analysis. Statistical analysis comparing xenografts was performed using an unpaired t test.
Results
CUDC 101 Inhibits AR-V7 in LNCaPAR-V7/pLenti
In a parallel project, we embarked on a small library screen of FDA-approved drugs to establish classes of bioactive molecules inhibiting AR signaling, which may be useful to treat CRPC [18]. Enrichment analysis of active compounds showed that histone deacetylase inhibitors (HDACi) were most active (manuscript in preparation). This led to testing commercially available agents with the same mechanism of action, including CUDC-101, a commercially available inhibitor of HDAC, HER2/NEU, and EGFR [19]. CUDC-101 was found to be a strong inhibitor of AR-V7 activity in LNCaPAR-V7/pLenti cells (Fig. 1a, b) transiently transfected with plasmid ARR2PB. The experiment shows that treatment with doxycycline strongly induced AR-V7 expression (Fig. 1b), and that CUDC-101 inhibited AR-V7-dependent activation of the ARR2PB-luc reporter in a dose-dependent manner (Fig. 1a). The half maximal inhibitory concentration (IC50) was calculated at 88 nM.
CUDC-101 Inhibits AR-V7 and flAR Activity in 22Rv1 Cells: Dose Response and Time Course Experiments
22Rv1 cells were used in dose-response and time-course experiments (Fig. 1c, e) to study AR-V7 or flAR inhibition by comparing PSA mRNA expression in response to vehicle vs. CUDC-101 or DHT vs. DHT + CUDC-101, respectively. In dose-response experiments cells were grown for 24 h in CS-FBS and then treated with vehicle ± concentrations of CUDC-101 ranging between 0 and 3 μM (black bars), or 2 nM DHT ± concentrations of CUDC-101 ranging from 0 to 3 μM (gray bars) for 24 h (Fig. 1c). In time-course experiments, cells were grown in CS-FBS for 24 h and then treated with vehicle ± 300 nM CUDC-101 (black bars) or 2 nM DHT ± 300 nM CUDC-101 (gray bars) for 0, 3, 6 and 24 h (Fig. 1d). CUDC-101 ≥ 30 nM significantly inhibited constitutive AR-V7 and DHT-induced flAR transcriptional activity (Fig. 1c); 300 nM of CUDC-101 significantly inhibited constitutive AR-V7 and DHT-induced flAR transcriptional activity after ≥6 h of treatment (Fig. 1d). Figure 1e, f are western blots performed with aliquots of 22Rv1 cells treated with DHT + CUDC shown in Fig. 1c, d, respectively. They show that flAR and AR-V7 protein expression was decreased after exposure to CUDC-101 ≥ 300 nM for 24 h. Figure 1c, e demonstrated that the lowest effective concentration of CUDC-101 inhibiting transcriptional activity, i.e., 30 nM, was not paralleled by simultaneous inhibition of AR-V7 and flAR protein expression (Fig. 1e).
CUDC101 Inhibits AR Signaling Without Decreasing Cell Viability
It was important to demonstrate that CUDC-101 inhibited AR signaling without affecting cell viability. At this purpose, the experiment of Supplemental Fig. S1a, b shows that viability was the same as at baseline after 24 h of CUDC-101 in LNCaPAR-V7/pLenti and 22Rv1 cells, however CUDC-101 significantly slowed cell proliferation compared to controls. That CUDC-101 does not induce systemic cellular toxicity was also inferred by unchanged levels of the calibrator mRNA 18s and housekeeping protein β-actin in every qPCR or western analysis performed, and by lack of necroscopic abnormalities in mice receiving CUDC-101 for 15 days.
Based on the experiments of Fig. 1 and Supplemental Fig. S1, CUDC-101 was used at 300 nM in every remaining experiment because this concentration strongly inhibited AR-V7 and flAR activity (Fig. 1a, c, d), and did not affect cell viability (Supplemental Fig. S1).
CUDC-101 is a Pan-AR Inhibitor
Supplemental Fig. S2 demonstrated that CUDC-101 inhibits AR-V7 and flAR transcriptional activity in every cell line tested. When given at 300 nM for 6 h, CUDC-101 inhibited AR-V7 in PC-3-GFP-AR-V7, 22Rv1, CWR1 and LNCaPAR-V7/pLenti cells [Supplemental Fig. S2 panels a–d] and flAR in CWR-R1, LNCaPAR-V7/pLenti, 22Rv1, C4-2, LAPC4, VCaP, and PC-3-GFP-wtAR cells [Supplemental Fig. S2c–i].
The AR forms expressed in these cell lines include flAR with wild type sequence in LAPC, VCaP, and PC3-GFP-AR, flAR with point mutations T877A in LNCaP and C4-2 cells [48] and H874Y in 22Rv1 and CWR1 cells [33], or the AR variant AR-V7 (22Rv1, CWR1, PC-3-GFP-AR-V7, and LNCaPAR-V7/pLenti). Thus, these experiments suggested that CUDC-101 is a pan AR inhibitor.
CUDC-101 Inhibits AR-V7 and flAR by Inhibiting HDAC Signaling
We dissected the three known inhibitory functions of CUDC-101 and determined how AR-V7 (in 22Rv1 cells) and flAR (in LNCaP cells) responded to specific inhibitors of EGFR (Gefitinib), Her-2/Neu (CP-724714) or of various HDAC isoforms (Fig. 2). Figure 2g shows that the lowest concentrations of the EGFR and Her-2/Neu inhibitors used to test flAR/AR-V7 activity in Fig. 2a were inhibiting EGFR and Her2/Neu signaling. Of the tested compounds, only the pan HDACi, CUDC-101 (Fig. 2c lane 2, Fig. 2d, e lanes 3), or the class II (i.e., HDAC 4, 5, 6, 7, 9 and 10) HDACi, MC1568 (Fig. 2c lane 4, Fig. 2d and e lanes 7), inhibited AR-V7 and flAR activities. In contrast Gefitinib and CP-724714 were ineffective (Fig. 2a, d, e lanes 4 and 5), while class I (i.e., HDAC 1, 2, 3) and IV (i.e., HDAC 11) HDAC inhibitor MGCD0103 induced AR-V7 (Fig. 2c lane 3) and had no effect on flAR activity (Fig. 2d, e lanes 5). In other experiments we used 22Rv1 cells to test two additional pan-HDACi (i.e., sodium butyrate and pracinostat) and HDAC8i PCI34051 (Supplemental Fig. S3). The common denominator of these additional experiments confirmed that the two pan-HDACi effectively decreased AR-V7 or flAR activity (Supplemental Fig. S3c, d), while PCI34051 did not have any measurable effect (Supplemental Fig. S3a, b). These experiments suggested that CUDC-101 inhibits flAR and AR-V7 activity by inhibiting class II HDAC isoforms.
Fig. 2.
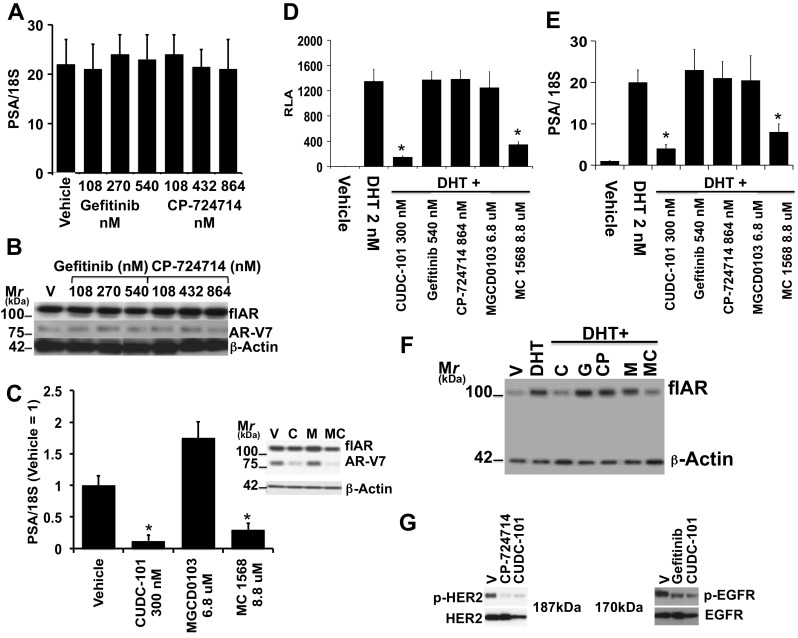
a, b, c Testing AR-V7 activity in 22Rv1 cells. a Cells were seeded in CS-FBS. After 24 h they were treated for 24 h with Vehicle (V), Gefitinib (G) at concentration ranging between 108 and 540 nM, CP-724714 at concentration ranging between 108 and 864 nM. b Parallel immunoblot analysis of the experiment of panel a showing flAR, AR-V7 and beta actin protein expression. c Cells were seeded in CS-FBS. After 24 h, they were treated for 24 with Vehicle (V), CUDC-101 (C) (300 nM), MGCD0103 (M) (6.8 μM) and MC1568 (MC) (8.8 μM). Cells were harvested; one aliquot was used to measure PSA mRNA transcriptional output, the other for the immunodetection of flAR, AR-V7 and beta actin (inset). d, e, f: LNCaP cells were transiently transfected with plasmid ARR2PB-Luc and grown in CS-FBS. After 24 h, cells were treated with vehicle, DHT 2 nM, or DHT + CUDC-101 (C) (300 nM), Gefitinib (G) (540 nM), CP-724714 (CP) (864 nM), MGCD0103 (M) (6.8 μM) or MC1568 (MC) (8.8 μM). After 24 h, cells were harvested and aliquots used for the determination of relative luciferase activity (d) PSA mRNA transcriptional output (e), or for the immunodetection of flAR and beta actin (f). Bars ± SE indicate PSA/18S mRNA (a, e) or RLA activity (d). CUDC-101 (*) and MGCD0103 (*) inhibited AR-V7 (c) and flAR (d, e) transcriptional activity in a statistically significant way (i.e., P < 0.0001). CUDC-101 and MGCD0103 decreased the expression of AR-V7 and to a lesser degree flAR (panel c inset), and of flAR (panel f). g 22Rv1 cells were seeded and grown in CS-FBS. After 24 h cells were treated with CP-724714 or Gefitinib both at 108 nM for 60 min. Cells were then harvested and lysates subjected to western analysis with antibodies for phospho-(Tyr-1248) Her2 or Her2 (left panel) or phospho-(Tyr1068) EGFR or EGFR (right panel)
HDAC5 and 10 are Involved in Mediating the Effect of CUDC-101 on AR-V7 and flAR Transcriptional Activity
Based on these observations, we used 22Rv1 cells and silenced class II HDAC’s (i.e., HDAC4, 5, 6, 7, 9, and 10) individually or in pairs. After silencing, cells were grown for 24 h. in CS-FBS. AR-V7 activity (black bars) was determined as a function of PSA/18S mRNA measured after 6 additional h of vehicle vs. CUDC-101 (300 nM). flAR activity (gray bars) was determined as a function of PSA/18S mRNA measured after 6 additional hours of DHT (2 nM) vs. DHT (2 nM) + CUDC-101 (300 nM). Parallel aliquots were used to verify successful silencing of the various HDAC’s by western analysis (insets).
Silencing HDAC4 (Fig. 3a inset) increased AR-V7 and (to a lesser degree) flAR activity (Fig. 3a compare lanes 1 and 5, 3 and 7). Under conditions of HDAC4 silencing, CUDC101 exerted similar inhibition of AR-V7 and flAR activity.
Fig. 3.

22Rv1 cells underwent manipulation with specific siRNA’s to silence class II HDAC’s or with control siRNA’s. Cells were then seeded in CS-FBS. After 24 h cells were treated with vehicle ± CUDC-101 (300 nM) or DHT (2 nM) ± CUDC-101 (300 nM) for 6 h to test the activities of AR-V7 or flAR, respectively. Bars ± SE indicate PSA/18S mRNA measured after 6 h with activity of cells treated with vehicle and control siRNA located on the far left of the graph set at 1. Parallel aliquots were used to control for successful silencing of the various HDAC’s by western analysis (insets). Asterisks denote statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001. Experiments were performed in triplicate and a minimum of three times
Figure 3b demonstrates that AR-V7 and flAR activities were not affected when HDAC7 and 9 were silenced (Fig. 3b, inset), however HDAC6 silencing (Fig. 3b, insets) led to a decreased transcriptional activity of flAR (dotted line), but not AR-V7 (dashed line). Under conditions of HDAC6, 7 or 9 silencing, CUDC-101 exerted similar inhibition of AR-V7 and flAR activity (Fig. 3b).
In cells where expression of HDAC5 or 10 was individually silenced (Fig. 3c), there was inhibition of AR-V7 and flAR transcriptional activity ranging between 25 and 30 % (dotted lines). The degree of inhibition of AR-V7 reached 65–70 % when HDAC5 and 10 were silenced simultaneously (Fig. 3d, dotted line), and was comparable to cells treated with CUDC-101 (Fig. 3d). These results lead us to the conclusion that AR-V7 and flAR transcriptional activities are, at least in part, modulated by HDAC5 and 10. HDAC1 (a class I HDAC) is known to modulate AR activity by direct interaction [49] and to be recruited to the PSA promoter by androgen stimulation [50]. To determine if HDAC1 played a relevant role in our model, we performed the experiment presented in Supplemental Fig. S4, and showed that under conditions of HDAC1 silencing there were no changes in AR-V7 or flAR transcriptional activity.
CUDC-101 Interferes With flAR and AR-V7 Transcriptional Activity
CUDC-101 (Fig. 1c, d, e, f) and the specific class II inhibitor MC1568 (Fig. 2c, d, e, f) decreased AR-V7 and flAR protein expression and transcriptional activity. To explore if the mechanism used by CUDC-101 to inhibit flAR and AR-V7 consists in decreased stability of these proteins or direct inhibition of their transcriptional activity, we used 22Rv1 and PC-3-GFP-AR-V7 cells. In 22Rv1 cells treated with the protein synthesis inhibitor cycloheximide, stability of flAR and AR-V7 proteins was unaffected by CUDC101 (Supplemental Fig. S5a). This suggested that CUDC-101 does not decrease flAR and AR-V7 transcriptional activity by increasing their rate of degradation. PC-3-GFP-AR-V7 is a model where the constitutively active CMV promoter controls AR-V7. Unlike 22Rv1, where AR and AR-V7 expression is under their native promoter, AR-V7 protein expression does not change when PC-3-GFP-AR-V7 are treated with CUDC-101 for 6 and 24 h (Supplemental Fig. S5b). However, despite constant levels of AR-V7 expression, induction of the luciferase reporter was still inhibited by CUDC-101 in this cell line (Fig. 5c), suggesting that this drug exerts a direct inhibitory effect on AR-V7 transcriptional activity. Similar results and conclusions were obtained using other cell lines, such as PC-3-GFP-AR-V7 (Supplemental Fig. S2a), PC-3-GFP-AR (Supplemental Fig. S2i) or LNCaPAR-V7/pLenti (Fig. 1a, b, Supplemental Fig. S2d), where the expression of flAR and AR-V7 is driven by the constitutively active CMV (Supplemental Fig. S2a, i) or dox-inducible TREG3 (Supplemental Figs. S1a, b, and S2d) promoters. Supplemental Figs. S2a, d, i and S1a, b show that in these cell lines CUDC-101 inhibited AR-V7 and flAR transcriptional activity without decreasing the level of protein expression.
Effects of AR-V7 Silencing Compared to CUDC-101 Treatment
Experiments were conducted to determine if AR-V7 silencing or treatment with CUDC-101 had equivalent effects on transcription of PSA mRNA expression and cell proliferation. We used 22Rv1 cells and silenced AR-V7, flAR or both (Fig. 4a). Cells were then grown in CS-FBS for 24 h, and treated with vehicle or CUDC-101 (300 nM) for an additional 6 h. PSA mRNA expression decreased significantly in cells with silenced AR-V7 [Fig. 4b compare lane 1 (cells treated with control siRNA and thus expressing both flAR and AR-V7) with lane 2 (cells treated with si-AR-V7, causing AR-V7 silencing)]. Addition of CUDC-101 did not change the degree of PSA mRNA suppression in cells with silenced AR-V7 (Fig. 4b, compare lanes 2 and 5), suggesting that AR-V7 silencing and CUDC-101 had equal effects on PSA mRNA transcriptional output. In contrast, PSA mRNA expression did not change under conditions of flAR silencing (Fig. 4b compare lanes 1 and 3) but decreased significantly after addition of CUDC-101 (Fig. 4b compare lane 3 and 6). Casodex and MDV3100 are AR antagonists that interact with the AR ligand-binding domain (LBD), and are not expected to inhibit AR-V7, where the LBD is deleted. In agreement with this, MDV3100 and Casodex inhibited PSA mRNA expression in 22Rv1 cells only when they were stimulated with DHT [Fig. 4c compare lane 1 (DHT alone) with lanes 3 (DHT + MDV3100) and 4 (DHT + Casodex)], but not under control conditions (Fig. 4c; compare lane 2 (CS-FBS + vehicle) with lanes 4 (CS-FBS + MDV3100) and 6 (CS-FBS + Casodex)). In contrast, the dual flAR and AR-V7 inhibitor CUDC-101 decreased PSA mRNA expression to a larger degree than MDV3100 and Casodex both in the presence (Fig. 4c compare lanes 3 and 5 with lane 7) or absence of DHT (compare lanes 2, 4 and 6 with lane 8).
Fig. 4.

a 22Rv1 cells were transfected with control si-RNA’s (si-Control), si-AR-V7 (to silence AR-V7), si-ex-7 (to silence flAR) and si-ex-2 (to silence both AR-V7 and flAR). Aliquots were used for western analysis of flAR, AR-V7 and beta actin. b Aliquots of cells transfected with si-Control (1 and 4), si-AR-V7 (2 and 5), si-ex-7 (3 and 6) were seeded, grown in CS-FBS for 24 h and treated with vehicle (1, 2, and 3) or CUDC-101 (4, 5 and 6) for 6 h. Bars ± SE indicate PSA/18S mRNA, with siControl treated with vehicle (lane 1) set at 1. Dotted lined emphasize the difference between 1 (si-Control) and 2 (si-AR-V7), and the lack of difference between 2 (si-AR-V7 treated with vehicle) and 5 (si-AR-V7 treated with CUDC-101). c 22Rv1 cells were seeded and grown for 24 h in CS-FBS. Cells were treated with vehicle, DHT (2 nM), MDV3100 (10 μM), Casodex (10 μM) or CUDC-101 (300 nM) for 24 h. Bars ± SE indicate PSA/18S mRNA with cells treated with DHT set at 1. d Aliquots of the experiment presented in A were seeded and grown in the presence of vehicle or CUDC-101 (300 nM) for 72 h. Bars ± SE indicate OD450 as a marker of cell proliferation. Data are normalized to cells grown in the presence of vehicle and transfected with control siRNA. e. 22Rv1 cells were seeded and grown in CS-FBS for 24 h, and treated with vehicle, MDV3100 (10 μM) or Casodex (10 μM) for 72 h. Bars ± SE indicate OD450. Data are normalized to cells grown under control (vehicle) conditions to 100 %. f. Immunoblot analysis for p21, HER2/NEU, Acetyl-H3, Acetyl alpha tubulin and alpha tubulin. 22Rv1 cells were seeded in CS-FBS for 24 h, and treated with vehicle or CUDC-101 for 6 h. Asterisks denote statistical significance: *P < 0.05, ** P < 0.01, *** P < 0.001
In parallel experiments, we used an aliquot of the cells shown in Fig. 4a to determine how AR-V7 silencing affected cell proliferation compared to CUDC-101. AR-V7 silencing (Fig. 4d compare lanes 1 and 3 or 1 and 7) but not first- or second-generation anti-androgens (Fig. 4e) decreased proliferation of 22Rv1 cells in CS-FBS. Interestingly, CUDC-101 was more effective than AR-V7 silencing in decreasing proliferation of 22Rv1 (Fig. 4d compare lanes 3 vs. 4 or 7 vs. 8), indicating that this compound has other activities, besides inhibiting AR-V7, that result in additional cell growth inhibition. It is known that HDAC inhibitors affect expression of the p21WAF and other cell cycle regulators [51]. In addition, HDAC6 inhibition (achieved by CUDC-101, a pan HDAC inhibitor) causes HSP90 hyperacetylation and inactivation, which is followed by loss of maturation, stability, and activity of the several client proteins of this chaperone [52]. Based on this, we treated 22Rv1 cells for 6 h with CUDC101 and showed that at a concentration in which this compound is active (as shown by induction of H3 and tubulin acetylation), it also induces expression of the cell cycle regulator, p21WAF1, and repressed expression of the HSP90 client kinase HER2/NEU (Fig. 4f).
Taken together these experiments suggest that: (1) CUDC-101 is equally effective as AR-V7 silencing in inhibiting PSA transcriptional output (Fig. 4b). (2) CUDC-101 is more effective than AR-V7 silencing in decreasing cell proliferation (Fig. 4d), implying that it has additional activities affecting cell proliferation besides inhibition of AR-V7 activity. (3) It is likely that these differences are related to induction of cell cycle regulators such a p21 WAF1 and in part to inhibition of HSP90 client proteins by CUDC-101 (Fig. 4f).
Mechanism of Transcriptional Inhibition of AR-V7
The experiments described argue that CUDC-101 inhibits transcription of PSA, the prototypical target mRNA of AR-V7 and flAR. To determine the mechanism involved, we studied by ChIP if CUDC-101 affects assembly of the AR-V7 transcription complex at the level of the PSA enhancer, and investigated 22Rv1 cells grown in CSS using a N-terminal AR antibody. N-terminal AR antibodies do not discriminate between flAR and AR-V7, however one would expect that under conditions of growth in CS-FBS only AR-V7 is recruited at the level of the PSA enhancer. In support of this hypothesis, we performed the experiments shown in Fig. 5a where flAR was silenced (Fig. 5a, right panel). Under experimental conditions in which DHT was not administered, the amount of AR recovered from the PSA enhancer did not change whether or not flAR had been silenced (Fig. 5a, left panel). This implied that under control conditions only a carboxy-terminally truncated AR reaches the PSA enhancer. 22Rv1 cells contain both AR-V7 and AR-V4 [36], however the expected AR-V4 band migrating at ∼ 75 kDa was not visualized when we silenced AR-V7 (Fig. 4a lane 2). This suggested that AR-V4 is either not expressed at a detectable level (Fig. 4a lane 2) or it is expressed at the mRNA level but not translated to a protein in the 22Rv1 cell line used in these studies, and argues that AR-V7 is the predominant AR form recovered from the PSA enhancer in the experiment of Fig. 5a. Based on this, in additional experiments, we studied 22Rv1 cells cultured in CSS in the presence of vehicle or CUDC-101 using the same N-terminal antibody. Figure 5b shows that under control conditions AR-V7 was recruited by the PSA enhancer and formed a complex together with SRC-1, P300 and RNA Polymerase II. Treatment with CUDC-101 did not alter the recruitment of AR-V7, but recruitment of SRC-1, P300 and RNA Polymerase II was significantly impaired. Because the experiments of Fig. 3c demonstrated that HDAC5 is one of the HDAC isoforms that is associated with AR-V7 transcriptional activity, we investigated by ChIP the consequences of silencing HDAC5 (Fig. 5c, right panel) and found that under these experimental conditions binding of RNA polymerase II to the PSA enhancer was decreased by 43 % (Fig. 5c, left panel). Together, these results indicated that addition of CUDC-101 prevents recruitment of p-300, SRC-1, and RNA polymerase II to AR-V7 at the level of the PSA enhancer, and that this process is partially HDAC-5 dependent.
Treatment with CUDC-101 Decreases Growth of 22Rv1 Xenografts
22Rv1 cells mixed with Matrigel (2 × 105) were injected in the R flank of 16 SCID mice. After 5 days, mice were castrated. Sc tumors became palpable within 2 weeks in 14 animals, of which 6 were randomized to receive vehicle and 8 CUDC-101. Animals were injected daily by IP with DMSO 50 μl, or 50 μg/kg of CUDC-101 in 50 μl of DMSO for 2 weeks. Two mice in the CUDC-101 group and 1 in the control group died during treatment. There was no difference in weight between the two groups at the outset or at the end of the study (Fig. 6a), suggesting that the drug did not induce cachexia. A statistically significant reduction in tumor growth was achieved in mice treated with CUDC-101 (p < 0.05) (Fig. 6b).
Fig. 6.

Treatment with CUDC-101 reduces growth of 22Rv1 xenografts in SCID mice. Right panel change in weight of treated and control mice at baseline and after receiving CUDC-101 or DMSO for 14 days. Left panel 2 × 105 22Rv1 cells mixed with Matrigel were injected in the R flank of SCID mice. A sc tumor became palpable within 2 weeks. SCID mice were treated with CUDC-101 (50 μg/kg/day) or DMSO for 14 days. Tumors were measured with a caliper at the outset and after 14 days of treatment. Bars represent change in tumor size from baseline (mean ± SE) of 6 (CUDC-101) and 5 (DMSO) treated mice. *P < 0.05 when comparing xenografts treated with CUDC-101 and DMSO
Discussion
The discovery that AR reactivation is the main mechanism associated with failure of first generation ADT and progression of PCa to the CRPC phenotype lead to the discovery and subsequent FDA approval of the second-generation drugs abiraterone acetate [5] and enzalutamide [6]. The rise in the PSA level, which may signal AR reactivation, occurs after a median of 8–10 months of treatment with abiraterone or enzalutamide [7–9], and has been attributed to a variety of mechanisms including AR or steroidogenic enzyme overexpression [53], selection of constitutively active AR variants [10, 11, 17, 54] or AR mutations [55], gain of function mutations of steroidogenic enzymes [56] or activation of pathways shared between GR and AR [57]. The common AR target of first and second-generation drugs is the carboxyl terminal ligand-binding domain (LBD). However, medications targeting the LBD are not expected to antagonize C-terminally truncated AR variants, and only EPI001, an experimental bisphenol A (BPA)-metabolite with promising therapeutic effects, has shown activity against AR-V7 [58, 59]. We report on the ability of CUDC-101 (a commercially available inhibitor of EGFR, HER2/NEU, and of all HDAC isoforms) to inhibit AR-V7, wild-type flAR, and flAR’s carrying a variety of point mutations. We identified three mechanisms through which CUDC-101 interferes with flAR and AR-V7 signaling, and they include: (1) Inhibition of HDAC5 and 10 (Fig. 3c, d). (2) Direct inhibition of flAR and AR-V7 transcriptional activity (Supplemental Fig S5). (3) Preventing ARV7 from forming an active transcriptional complex on the PSA enhancer (Fig. 5b).
Known Efficacy of CUDC-101; In Vitro, In Vivo, and Human Data
This is the first study describing the ability of CUDC-101 to block at the same time flAR and AR-V7 in a prostate cancer cell line with a phenotype of castration-independence and to inhibit its growth, however this agent has already been the focal point of previous important investigations in cancer research. Following, the initial paper describing its mechanism of action and ability to inhibit growth of several human cancer cell lines and xenografts [19], additional reports have demonstrated that this agent is effective in suppressing tumor growth. The study of Wang et al. demonstrated that, thanks to its multitargeted mechanism of action, CUDC-101 remains effective against cell lines that have developed resistance to single target EGFR inhibitors [60]. Zhang et al. performed quantitative high-throughput screening of 3282 clinically approved drugs and drugs candidates using in vitro and in vivo experimental models of anaplastic thyroid cancer, and found that CUDC-101 is the most active suppressor of tumor growth and metastasis of this lethal human malignancy [61]. Phase I studies have been conducted with CUDC-101 alone or in combination with chemoradiation. The first study reported that a dose of 275 mg/m2 in 25 patients with solid tumors was generally well tolerated and showed some antitumor activity [62]. The second study enrolled 12 patients with intermediate or high risk head and neck squamous cell carcinoma (HNSCC) in a trial of CUDC-101 given with chemoradiation, and resulted in a high rate (41 %) of dose-limiting toxicity-independent discontinuation of the drug [63], suggesting the need for alternate protocols, doses and routes of administration.
The Effect of CUDC-101 on flAR and AR-V7 is Mediated by Histone Deacetylase Activity
Known mechanisms of action of CUDC-101 consist in inhibition of EGFR, HER2/NEU and HDAC signaling [19]. To discover which of these three activities modulates AR-V7 and flAR transcriptional activity, we used pharmacologic and genetic experiments. Using a variety of drugs targeting the EGFR, HER2/NEU, and HDAC signaling pathways, we found that CUDC-101 decreases the transcriptional activity of flAR and AR-V7 by inhibiting class II HDAC’s. This did not come as a surprise because molecules involved in AR transcription such as histones [64], the chaperone HSP90 [65], and AR itself [66, 67] are known substrates for acetylation/deacetylation. The interaction between AR and HDAC signaling is complex, as demonstrated by contrasting reports suggesting that HDAC inhibition can increase [66, 67], or decrease [68–71] AR transcriptional activity or AR-mediated prostate cancer cell growth. This complexity was confirmed in our experiments, where we observed that AR-V7 and flAR activities were inhibited by pan- (CUDC-101, sodium butyrate and pracinostat) or class II (MC 1568) HDACi but were not affected by the specific HDAC8i, PCI34051, or responded differently to the class I HDACi MGCD0103, which induced AR-V7 activity but did not affect flAR. To better understand this complexity, we silenced each class II HDAC and observed a variety of changes on flAR and AR-V7 activation. First: most of the effects of CUDC-101 on flAR and AR-V7 activity were recapitulated by knocking down the expression of HDAC5 and 10. Second: AR-V7 and flAR transcriptional activities were increased in cells with silenced HDAC4. Third: HDAC6 silencing was associated with decreased flAR (but not AR-V7) transcriptional activity. The observation that HDAC5 and 10 modulate AR-V7 and flAR activity is novel. The changes observed in cells with silenced HDAC4 or HDAC6 can be explained based on previous observations: (1) It is known that HDAC4 suppresses flAR transcriptional activity through SUMOylation [72], hence, when HDAC4 is silenced AR does not undergo SUMOylation and its activity is expected to increase. (2) HDAC6 is a specific HSP90 deacetylase. HDAC6 silencing is expected to lead to HSP90 hyperacetylation, loss of its chaperone activity [52] and expedited proteasome degradation with loss of function of all HSP-90 client proteins, including AR [65]. Unlike flAR, AR-V7 activity was not impaired by HDAC6 silencing (Fig. 3b). This difference is explainable based on the observation that the C-terminally truncated AR-V7 remains active despite HSP90 inhibition [27, 36].
Effects of CUDC 101 on Cell Proliferation: AR-Dependent and Independent Mechanisms
CUDC-101 has a complex antineoplastic mechanism of action. As shown in Fig. 4b (compare lanes 2 and 5), AR-V7 silencing and CUDC-101 treatment were equivalent in their ability to inhibit AR-V7 transcription. In contrast, CUDC-101 treatment affected cell proliferation more than AR-V7 silencing (Fig. 4d, compare lanes 3 and 4, or 7 and 8). The non-AR-dependent mechanisms responsible for slower cellular proliferation have not been thoroughly investigated, however they can be inferred from known activities of CUDC-101 and in part from experiments reported in these studies, which include: (1) Direct inhibition of the receptor tyrosine kinases, HER2 (Fig. 2g and [19]) and EGFR [19], which are both strong inducers of cell proliferation and anti-apoptosis. (2) Inhibition of class I and II HDAC molecules [73]. This effect, which can be achieved by numerous drugs including the pan HDAC inhibitor CUDC-101, leads to increased expression/activity of the cell cycle inhibitors p21WAF1 (Fig. 4f and [74, 75] and p27KIP [76]). (3) Inhibition of HDC6. Inhibition of this HDAC isoform leads to HSP90 hyperacetylation, loss of its chaperone activity [52] and accelerated degradation with loss of function of the large family of HSP-90 client proteins (http://www.picard.ch/downloads/Hsp90interactors.pdf), of which HER2/NEU (Fig. 4f) is a member.
Effects of CUDC-101 on Xenograft Growth
22Rv1 formed aggressive xenografts in castrate SCID mice necessitating euthanasia of the control group after only 14 days post-implantation, rather than 4 weeks as originally planned. Despite this, there was an overall delay of xenograft growth (P < 0.05) compared to DMSO-treated animals in mice receiving a daily dose of CUDC-101 (40 μg/kg). The absence of macroscopic abnormalities in animals receiving the drug and the uniformity of weight between groups at the end of the experiment argue that CUDC-101 did not exert systemic toxicity at the concentrations used.
Conclusions
A gene program mediated by the reactivation of flAR, AR-Vs or alternative steroid receptors such as GR puts in motion a downstream signaling pathways leading to CRPC survival and proliferation. This AR-dependent program is paralleled by activation of other oncogenic pathways contributing disease progression by determining growth and survival of the disease [77]. These studies described in this paper indicate that an effective way to defeat prostate cancer consists in developing multi-targeted therapeutic modalities inhibiting not only AR signaling but also other non-AR-dependent oncogenic pathways [29, 78]. Our data are a proof of concept that by virtue of a complex mechanism of action targeting AR and non-AR-dependent signaling pathways, CUDC-101 successfully inhibits in vitro and in vivo growth of a castration resistant prostate cancer cell line.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
(DOCX 109 kb)
(PPTX 755 kb)
Acknowledgments
We thank Drs. Scott Dehm and Nancy Weigel for sharing cell lines.
Supported by DAMD W81XWH-10-1-0390 (MM) and the VA Merit Review Program (MM). Research conducted in joint participation with Diana Helis Henry Medical Research Foundation (M.A.M., M.M.) through its direct engagement in the continuous active conduct of medical research in conjunction with Baylor College of Medicine and the Cancer Program. A.T.S. is a K12 scholar supported by NIH grant K12DK0083014.
Compliance with Ethical Standards
Conflict of Interest
The authors have no conflict of interest to declare.
Footnotes
Huiying Sun, Sanjay N. Mediwala and Adam T. Szafran contributed equally to this work.
References
- 1.Pienta KJ, Bradley D. Mechanisms underlying the development of androgen-independent prostate cancer. Clin Cancer Res. 2006;12(6):1665–1671. doi: 10.1158/1078-0432.CCR-06-0067. [DOI] [PubMed] [Google Scholar]
- 2.Taplin ME. Drug insight: role of the androgen receptor in the development and progression of prostate cancer. Nat Clin Pract Oncol. 2007;4(4):236–244. doi: 10.1038/ncponc0765. [DOI] [PubMed] [Google Scholar]
- 3.Chen Y, Clegg NJ, Scher HI. Anti-androgens and androgen-depleting therapies in prostate cancer: new agents for an established target. Lancet Oncol. 2009;10(10):981–991. doi: 10.1016/S1470-2045(09)70229-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Attard G, Richards J, de Bono JS. New strategies in metastatic prostate cancer: targeting the androgen receptor signaling pathway. Clin Cancer Res. 2011;17(7):1649–1657. doi: 10.1158/1078-0432.CCR-10-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Potter GA, Barrie SE, Jarman M, Rowlands MG. Novel steroidal inhibitors of human cytochrome P45017 alpha (17 alpha-hydroxylase-C17,20-lyase): potential agents for the treatment of prostatic cancer. J Med Chem. 1995;38(13):2463–2471. doi: 10.1021/jm00013a022. [DOI] [PubMed] [Google Scholar]
- 6.Jung ME, Ouk S, Yoo D, Sawyers CL, Chen C, Tran C, Wongvipat J. Structure-activity relationship for thiohydantoin androgen receptor antagonists for castration-resistant prostate cancer (CRPC) J Med Chem. 2010;53(7):2779–2796. doi: 10.1021/jm901488g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB, Jr, Saad F, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364(21):1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, Fizazi K, Mainwaring P, Piulats JM, Ng S, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368(2):138–148. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367(13):1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 10.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371(11):1028–1038. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steinestel J, Luedeke M, Arndt A, Schnoeller TJ, Lennerz JK, Wurm C, Maier C, Cronauer MV, Steinestel K, Schrader AJ (2015) Detecting predictive androgen receptor modifications in circulating prostate cancer cells. Oncotarget [DOI] [PMC free article] [PubMed]
- 12.Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68(13):5469–5477. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69(1):16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, Kong X, Melamed J, Tepper CG, Kung HJ, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69(6):2305–2313. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, Kim K, Sawyers CL. Inaugural Article: constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci U S A. 2010;107:16759–16765. doi: 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, Page ST, Coleman IM, Nguyen HM, Sun H, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120(8):2715–2730. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu Z, Chen S, Sowalsky AG, Voznesensky OS, Mostaghel EA, Nelson PS, Cai C, Balk SP (2014) Rapid induction of androgen receptor splice variants by androgen deprivation in prostate cancer. Clin Cancer Res [DOI] [PMC free article] [PubMed]
- 18.Szafran AT, Mancini MA. The myImageAnalysis project: a web-based application for high-content screening. Assay Drug Dev Technol. 2014;12(1):87–99. doi: 10.1089/adt.2013.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lai CJ, Bao R, Tao X, Wang J, Atoyan R, Qu H, Wang DG, Yin L, Samson M, Forrester J, et al. CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity. Cancer Res. 2010;70(9):3647–3656. doi: 10.1158/0008-5472.CAN-09-3360. [DOI] [PubMed] [Google Scholar]
- 20.Horoszewicz JS, Leong SS, Chu TM, Wajsman ZL, Friedman M, Papsidero L, Kim U, Chai LS, Kakati S, Arya SK, et al. The LNCaP cell line--a new model for studies on human prostatic carcinoma. Prog Clin Biol Res. 1980;37:115–132. [PubMed] [Google Scholar]
- 21.Kaighn ME, Narayan KS, Ohnuki Y, Lechner JF, Jones LW. Establishment and characterization of a human prostatic carcinoma cell line (PC-3) Investig Urol. 1979;17:16–23. [PubMed] [Google Scholar]
- 22.Scherer WF, Hoogasian AF. Preservation at subzero temperatures of mouse fibroblasts (strain L) and human epithelial cells (strain HeLa) Proc Soc Exp Biol Med. 1954;87(2):480–487. doi: 10.3181/00379727-87-21419. [DOI] [PubMed] [Google Scholar]
- 23.Korenchuk S, Lehr JE, Mclean L, Lee YG, Whitney S, Vessella R, Lin DL, Pienta KJ. VCaP, a cell-based model system of human prostate cancer. Vivo. 2001;15(2):163–168. [PubMed] [Google Scholar]
- 24.Sramkoski RM, Pretlow TG, 2nd, Giaconia JM, Pretlow TP, Schwartz S, Sy MS, Marengo SR, Rhim JS, Zhang D, Jacobberger JW. A new human prostate carcinoma cell line, 22Rv1. In Vitro Cell Dev Biol Anim. 1999;35(7):403–409. doi: 10.1007/s11626-999-0115-4. [DOI] [PubMed] [Google Scholar]
- 25.Thalmann GN, Anezinis PE, Chang SM, Zhau HE, Kim EE, Hopwood VL, Pathak S, von Eschenbach AC, Chung LW. Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer [published erratum appears in Cancer Res 1994 Jul 15;54(14):3953] Cancer Res. 1994;54(10):2577–2581. [PubMed] [Google Scholar]
- 26.Li Y, Hwang TH, Oseth LA, Hauge A, Vessella RL, Schmechel SC, Hirsch B, Beckman KB, Silverstein KA, Dehm SM. AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression. Oncogene. 2012;31(45):4759–4767. doi: 10.1038/onc.2011.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shafi AA, Cox MB, Weigel NL. Androgen receptor splice variants are resistant to inhibitors of Hsp90 and FKBP52, which alter androgen receptor activity and expression. Steroids. 2013;78(6):548–554. doi: 10.1016/j.steroids.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krause WC, Shafi AA, Nakka M, Weigel NL (2014) Androgen receptor and its splice variant, AR-V7, differentially regulate FOXA1 sensitive genes in LNCaP prostate cancer cells. Int J Biochem Cell Biol 54:49–59 [DOI] [PMC free article] [PubMed]
- 29.Mediwala SN, Sun H, Szafran AT, Hartig SM, Sonpavde G, Hayes TG, Thiagarajan P, Mancini MA, Marcelli M. The activity of the androgen receptor variant AR-V7 is regulated by FOXO1 in a PTEN-PI3K-AKT-dependent way. Prostate. 2013;73(3):267–277. doi: 10.1002/pros.22566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szafran AT, Szwarc M, Marcelli M, Mancini MA. Androgen receptor functional analyses by high throughput imaging: determination of ligand, cell cycle, and mutation-specific effects. PLoS ONE. 2008;3(11):e3605. doi: 10.1371/journal.pone.0003605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murthy S, Marcelli M, Weigel NL. Stable expression of full length human androgen receptor in PC-3 prostate cancer cells enhances sensitivity to retinoic acid but not to 1alpha,25-dihydroxyvitamin D3. Prostate. 2003;56(4):293–304. doi: 10.1002/pros.10261. [DOI] [PubMed] [Google Scholar]
- 32.Veldscholdte J, Ris-Stalpers C, Kuiper GGJM, Jentser G, Berrevoets C, Claassen E, Rooij HCJV, Trapman J, Brinkmann AO, Mulder E. A mutation in the ligand binding domain of the androgen receptor of LnCAP cells affects steroid binding characteristics and response to anti-androgens. Biochem Biophys Res Commun. 1990;173:534–540. doi: 10.1016/S0006-291X(05)80067-1. [DOI] [PubMed] [Google Scholar]
- 33.Tan J, Sharief Y, Hamil KG, Gregory CW, Zang DY, Sar M, Gumerlock PH, deVere WRW, Pretlow TG, Harris SE, et al. Dehydroepiandrosterone activates mutant androgen receptors expressed in the androgen-dependent human prostate cancer xenograft CWR22 and LNCaP cells. Mol Endocrinol. 1997;11(4):450–459. doi: 10.1210/mend.11.4.9906. [DOI] [PubMed] [Google Scholar]
- 34.van Bokhoven A, Varella-Garcia M, Korch C, Johannes WU, Smith EE, Miller HL, Nordeen SK, Miller GJ, Lucia MS. Molecular characterization of human prostate carcinoma cell lines. Prostate. 2003;57(3):205–225. doi: 10.1002/pros.10290. [DOI] [PubMed] [Google Scholar]
- 35.Chlenski A, Nakashiro K, Ketels KV, Korovaitseva GI, Oyasu R. Androgen receptor expression in androgen-independent prostate cancer cell lines. Prostate. 2001;47(1):66–75. doi: 10.1002/pros.1048. [DOI] [PubMed] [Google Scholar]
- 36.Chan SC, Li Y, Dehm SM. Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. J Biol Chem. 2012;287(23):19736–19749. doi: 10.1074/jbc.M112.352930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Novotny-Diermayr V, Sangthongpitag K, Hu CY, Wu X, Sausgruber N, Yeo P, Greicius G, Pettersson S, Liang AL, Loh YK, et al. SB939, a novel potent and orally active histone deacetylase inhibitor with high tumor exposure and efficacy in mouse models of colorectal cancer. Mol Cancer Ther. 2010;9(3):642–652. doi: 10.1158/1535-7163.MCT-09-0689. [DOI] [PubMed] [Google Scholar]
- 38.Fournel M, Bonfils C, Hou Y, Yan PT, Trachy-Bourget MC, Kalita A, Liu J, Lu AH, Zhou NZ, Robert MF, et al. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol Cancer Ther. 2008;7(4):759–768. doi: 10.1158/1535-7163.MCT-07-2026. [DOI] [PubMed] [Google Scholar]
- 39.Nebbioso A, Manzo F, Miceli M, Conte M, Manente L, Baldi A, De Luca A, Rotili D, Valente S, Mai A, et al. Selective class II HDAC inhibitors impair myogenesis by modulating the stability and activity of HDAC-MEF2 complexes. EMBO Rep. 2009;10(7):776–782. doi: 10.1038/embor.2009.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Balasubramanian S, Ramos J, Luo W, Sirisawad M, Verner E, Buggy JJ. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia. 2008;22(5):1026–1034. doi: 10.1038/leu.2008.9. [DOI] [PubMed] [Google Scholar]
- 41.Angelucci A, Gravina GL, Rucci N, Millimaggi D, Festuccia C, Muzi P, Teti A, Vicentini C, Bologna M. Suppression of EGF-R signaling reduces the incidence of prostate cancer metastasis in nude mice. Endocr Relat Cancer. 2006;13(1):197–210. doi: 10.1677/erc.1.01100. [DOI] [PubMed] [Google Scholar]
- 42.Jani JP, Finn RS, Campbell M, Coleman KG, Connell RD, Currier N, Emerson EO, Floyd E, Harriman S, Kath JC, et al. Discovery and pharmacologic characterization of CP-724,714, a selective ErbB2 tyrosine kinase inhibitor. Cancer Res. 2007;67(20):9887–9893. doi: 10.1158/0008-5472.CAN-06-3559. [DOI] [PubMed] [Google Scholar]
- 43.Candido EP, Reeves R, Davie JR. Sodium butyrate inhibits histone deacetylation in cultured cells. Cell. 1978;14(1):105–113. doi: 10.1016/0092-8674(78)90305-7. [DOI] [PubMed] [Google Scholar]
- 44.Nazareth LV, Stenoien DL, Bingman WE, 3rd, James AJ, Wu C, Zhang Y, Edwards DP, Mancini M, Marcelli M, Lamb DJ, et al. A C619Y mutation in the human androgen receptor causes inactivation and mislocalization of the receptor with concomitant sequestration of SRC-1 (steroid receptor coactivator 1) Mol Endocrinol. 1999;13(12):2065–2075. doi: 10.1210/mend.13.12.0382. [DOI] [PubMed] [Google Scholar]
- 45.Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, Gannon F. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115(6):751–763. doi: 10.1016/S0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- 46.Zhang JF, Thomas TZ, Kasper S, Matusik RJ. A small composite probasin promoter confers high levels of prostate-specific gene expression through regulation by androgens and glucocorticoids in vitro and in vivo . Endocrinology. 2000;141(12):4698–4710. doi: 10.1210/endo.141.12.7837. [DOI] [PubMed] [Google Scholar]
- 47.Janik P, Briand P, Hartmann NR. The effect of estrone-progesterone treatment on cell proliferation kinetics of hormone-dependent GR mouse mammary tumors. Cancer Res. 1975;35(12):3698–3704. [PubMed] [Google Scholar]
- 48.Veldscholte J, Berrevoets CA, Ris-Stalpers C, Kuiper GG, Jenster G, Trapman J, Brinkmann AO, Mulder E. The androgen receptor in LNCaP cells contains a mutation in the ligand binding domain which affects steroid binding characteristics and response to antiandrogens. J Steroid Biochem Mol Biol. 1992;41:665–669. doi: 10.1016/0960-0760(92)90401-4. [DOI] [PubMed] [Google Scholar]
- 49.Gaughan L, Logan IR, Cook S, Neal DE, Robson CN. Tip60 and histone deacetylase 1 regulate androgen receptor activity through changes to the acetylation status of the receptor. J Biol Chem. 2002;277(29):25904–25913. doi: 10.1074/jbc.M203423200. [DOI] [PubMed] [Google Scholar]
- 50.Shang Y, Myers M, Brown M. Formation of the androgen receptor transcription complex. Mol Cell. 2002;9(3):601–610. doi: 10.1016/S1097-2765(02)00471-9. [DOI] [PubMed] [Google Scholar]
- 51.Lagger G, O’Carroll D, Rembold M, Khier H, Tischler J, Weitzer G, Schuettengruber B, Hauser C, Brunmeir R, Jenuwein T, et al. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 2002;21(11):2672–2681. doi: 10.1093/emboj/21.11.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18(5):601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 53.Mostaghel EA, Marck BT, Plymate SR, Vessella RL, Balk S, Matsumoto AM, Nelson PS, Montgomery RB. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res. 2011;17(18):5913–5925. doi: 10.1158/1078-0432.CCR-11-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hornberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, Bergh A, Wikstrom P. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS One. 2011;6(4):e19059. doi: 10.1371/journal.pone.0019059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, Moon M, Maneval EC, Chen I, Darimont B, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013;3(9):1020–1029. doi: 10.1158/2159-8290.CD-13-0226. [DOI] [PubMed] [Google Scholar]
- 56.Chang KH, Li R, Kuri B, Lotan Y, Roehrborn CG, Liu J, Vessella R, Nelson PS, Kapur P, Guo X, et al. A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell. 2013;154(5):1074–1084. doi: 10.1016/j.cell.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, Shah N, Cai L, Efstathiou E, Logothetis C, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013;155(6):1309–1322. doi: 10.1016/j.cell.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Myung JK, Banuelos CA, Fernandez JG, Mawji NR, Wang J, Tien AH, Yang YC, Tavakoli I, Haile S, Watt K, et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J Clin Invest. 2013;123(7):2948–2960. doi: 10.1172/JCI66398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, Watt K, Tam T, Yang YC, Banuelos CA, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17(6):535–546. doi: 10.1016/j.ccr.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 60.Wang J, Pursell NW, Samson ME, Atoyan R, Ma AW, Selmi A, Xu W, Cai X, Voi M, Savagner P, et al. Potential advantages of CUDC-101, a multitargeted HDAC, EGFR, and HER2 inhibitor, in treating drug resistance and preventing cancer cell migration and invasion. Mol Cancer Ther. 2013;12(6):925–936. doi: 10.1158/1535-7163.MCT-12-1045. [DOI] [PubMed] [Google Scholar]
- 61.Zhang L, Zhang Y, Mehta A, Boufraqech M, Davis S, Wang J, Tian Z, Yu Z, Boxer MB, Kiefer JA et al (2015) Dual inhibition of HDAC and EGFR signaling with CUDC-101 induces potent suppression of tumor growth and metastasis in anaplastic thyroid cancer. Oncotarget [DOI] [PMC free article] [PubMed]
- 62.Shimizu T, LoRusso PM, Papadopoulos KP, Patnaik A, Beeram M, Smith LS, Rasco DW, Mays TA, Chambers G, Ma A, et al. Phase I first-in-human study of CUDC-101, a multitargeted inhibitor of HDACs, EGFR, and HER2 in patients with advanced solid tumors. Clin Cancer Res. 2014;20(19):5032–5040. doi: 10.1158/1078-0432.CCR-14-0570. [DOI] [PubMed] [Google Scholar]
- 63.Galloway TJ, Wirth LJ, Colevas AD, Gilbert J, Bauman JE, Saba NF, Raben D, Mehra R, Ma AW, Atoyan R, et al. A phase I study of CUDC-101, a multitarget inhibitor of HDACs, EGFR, and HER2, in combination with chemoradiation in patients with head and neck squamous cell carcinoma. Clin Cancer Res. 2015;21(7):1566–1573. doi: 10.1158/1078-0432.CCR-14-2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Durrin LK, Mann RK, Kayne PS, Grunstein M. Yeast histone H4 N-terminal sequence is required for promoter activation in vivo. Cell. 1991;65(6):1023–1031. doi: 10.1016/0092-8674(91)90554-C. [DOI] [PubMed] [Google Scholar]
- 65.Ai J, Wang Y, Dar JA, Liu J, Liu L, Nelson JB, Wang Z. HDAC6 regulates androgen receptor hypersensitivity and nuclear localization via modulating Hsp90 acetylation in castration-resistant prostate cancer. Mol Endocrinol. 2009;23(12):1963–1972. doi: 10.1210/me.2009-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fu M, Rao M, Wang C, Sakamaki T, Wang J, Di Vizio D, Zhang X, Albanese C, Balk S, Chang C, et al. Acetylation of androgen receptor enhances coactivator binding and promotes prostate cancer cell growth. Mol Cell Biol. 2003;23(23):8563–8575. doi: 10.1128/MCB.23.23.8563-8575.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fu M, Wang C, Reutens AT, Wang J, Angeletti RH, Siconolfi-Baez L, Ogryzko V, Avantaggiati ML, Pestell RG. p300 and p300/cAMP-response element-binding protein-associated factor acetylate the androgen receptor at sites governing hormone-dependent transactivation. J Biol Chem. 2000;275(27):20853–20860. doi: 10.1074/jbc.M000660200. [DOI] [PubMed] [Google Scholar]
- 68.Welsbie DS, Xu J, Chen Y, Borsu L, Scher HI, Rosen N, Sawyers CL. Histone deacetylases are required for androgen receptor function in hormone-sensitive and castrate-resistant prostate cancer. Cancer Res. 2009;69(3):958–966. doi: 10.1158/0008-5472.CAN-08-2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ellis L, Lehet K, Ramakrishnan S, Adelaiye R, Miles KM, Wang D, Liu S, Atadja P, Carducci MA, Pili R. Concurrent HDAC and mTORC1 inhibition attenuate androgen receptor and hypoxia signaling associated with alterations in microRNA expression. PLoS One. 2011;6(11):e27178. doi: 10.1371/journal.pone.0027178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gryder BE, Akbashev MJ, Rood MK, Raftery ED, Meyers WM, Dillard P, Khan S, Oyelere AK. Selectively targeting prostate cancer with antiandrogen equipped histone deacetylase inhibitors. ACS Chem Biol. 2013;8(11):2550–2560. doi: 10.1021/cb400542w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gravina GL, Marampon F, Muzi P, Mancini A, Piccolella M, Negri-Cesi P, Motta M, Lenzi A, Di Cesare E, Tombolini V, et al. PXD101 potentiates hormonal therapy and prevents the onset of castration-resistant phenotype modulating androgen receptor, HSP90, and CRM1 in preclinical models of prostate cancer. Endocr Relat Cancer. 2013;20(3):321–337. doi: 10.1530/ERC-12-0240. [DOI] [PubMed] [Google Scholar]
- 72.Yang Y, Tse AK, Li P, Ma Q, Xiang S, Nicosia SV, Seto E, Zhang X, Bai W. Inhibition of androgen receptor activity by histone deacetylase 4 through receptor SUMOylation. Oncogene. 2011;30(19):2207–2218. doi: 10.1038/onc.2010.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bellucci L, Dalvai M, Kocanova S, Moutahir F, Bystricky K. Activation of p21 by HDAC inhibitors requires acetylation of H2A.Z. PLoS One. 2013;8(1):e54102. doi: 10.1371/journal.pone.0054102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xiao H, Hasegawa T, Miyaishi O, Ohkusu K, Isobe K. Sodium butyrate induces NIH3T3 cells to senescence-like state and enhances promoter activity of p21WAF/CIP1 in p53-independent manner. Biochem Biophys Res Commun. 1997;237(2):457–460. doi: 10.1006/bbrc.1997.7158. [DOI] [PubMed] [Google Scholar]
- 75.Sambucetti LC, Fischer DD, Zabludoff S, Kwon PO, Chamberlin H, Trogani N, Xu H, Cohen D. Histone deacetylase inhibition selectively alters the activity and expression of cell cycle proteins leading to specific chromatin acetylation and antiproliferative effects. J Biol Chem. 1999;274(49):34940–34947. doi: 10.1074/jbc.274.49.34940. [DOI] [PubMed] [Google Scholar]
- 76.Park WH, Jung CW, Park JO, Kim K, Kim WS, Im YH, Lee MH, Kang WK, Park K. Trichostatin inhibits the growth of ACHN renal cell carcinoma cells via cell cycle arrest in association with p27, or apoptosis. Int J Oncol. 2003;22(5):1129–1134. [PubMed] [Google Scholar]
- 77.Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene. 2013;32(49):5501–5511. doi: 10.1038/onc.2013.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Reuveni H, Flashner-Abramson E, Steiner L, Makedonski K, Song R, Shir A, Herlyn M, Bar-Eli M, Levitzki A. Therapeutic destruction of insulin receptor substrates for cancer treatment. Cancer Res. 2013;73(14):4383–4394. doi: 10.1158/0008-5472.CAN-12-3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 109 kb)
(PPTX 755 kb)