

Abstract
PI3K/AKT and RAS/MAPK pathway co-activation in the prostate epithelium promotes both epithelial-mesenchymal transition (EMT) and metastatic castration-resistant prostate cancer (mCRPC), which is currently incurable. To study the dynamic regulation of the EMT process, we developed novel genetically-defined cellular and in vivo model systems from which epithelial, EMT, and mesenchymal-like tumor cells with Pten deletion and Kras activation can be isolated. When cultured individually, each population has the capacity to regenerate all three tumor cell populations, indicative of epithelial-mesenchymal plasticity. Despite harboring the same genetic alterations, mesenchymal-like tumor cells are resistant to PI3K and MAPK pathway inhibitors, suggesting that epigenetic mechanisms may regulate the EMT process, as well as dictate the heterogeneous responses of cancer cells to therapy. Among differentially expressed epigenetic regulators, the chromatin remodeling protein HMGA2 is significantly upregulated in EMT and mesenchymal-like tumors cells, as well as in human mCRPC. Knockdown of HMGA2, or suppressing HMGA2 expression with the histone deacetylase (HDAC) inhibitor LBH589, inhibits epithelial-mesenchymal plasticity and stemness activities in vitro and dramatically reduces tumor growth and metastasis in vivo through successful targeting of EMT and mesenchymal-like tumor cells. Importantly, LBH589 treatment in combination with castration prevents mCRPC development and significantly prolongs survival following castration by enhancing p53 and AR acetylation and in turn sensitizing castration-resistant mesenchymal-like tumor cells to ADT. Taken together, these findings demonstrate that cellular plasticity is regulated epigenetically, and that mesenchymal-like tumor cell populations in mCRPC that are resistant to conventional and targeted therapies can be effectively treated with the epigenetic inhibitor LBH589.
Keywords: epithelial-mesenchymal transition (EMT), castration-resistant prostate cancer (CRPC), metastasis, epigenetics, HDAC inhibitor
Introduction
Prostate cancer is the most prevalent malignancy in men and a leading cause of cancer-related death worldwide.1 Nearly all prostate cancer-associated mortality is caused by distant metastasis. The most common treatment for advanced prostate cancer is androgen deprivation therapy (ADT), owing to the central role of androgens and androgen receptor (AR) signaling in normal prostate development and prostate tumor growth. While most men initially respond to ADT, the therapeutic benefits are short-lived, and patients usually succumb to castration-resistant prostate cancer (CRPC) within 18-24 months.2 Treatment of CRPC with new generation androgen signaling inhibitors such as enzalutamide and abiraterone acetate has improved survival outcomes;3, 4 however, CRPC remains incurable, and patients generally die within 2 years.5 Therefore, novel therapies for CRPC, including those that would prevent distant metastasis, are desperately needed.
Genetic and phenotypic heterogeneity within the same prostate tumor is frequently observed despite common underlying pathway alterations,6-10 a finding that suggests a degree of cellular plasticity at the level of RNA and protein expression within a given patient that is uncoupled from mutations and chromosomal abnormalities. There is accumulating evidence that epithelial-mesenchymal plasticity, referring to the reversible processes of the epithelial-mesenchymal transition (EMT) and the mesenchymal-epithelial transition (MET), is induced by ADT and other therapies and plays a role in both treatment resistance and metastatic progression through the acquisition of stemness and invasion programs.11-16 Therefore, co-targeting regulators of epithelial-mesenchymal plasticity may increase the therapeutic efficacy of ADT. However, the molecular mechanisms regulating epithelial-mesenchymal plasticity are poorly understood, and validated biomarkers of epithelial-mesenchymal plasticity are still required.
We and others have previously shown that PI3K/AKT and RAS/MAPK pathway activation is highly associated with metastatic CRPC (mCRPC), and that activation of both pathways in the Pb-Cre+/-;PtenL/L;KrasG12D/+ (CPK) mouse model is sufficient to induce an EMT and distant metastasis.7, 16 To study the direct role of EMT in prostate cancer stem cell formation and distant metastasis in vivo, we crossed CPK mice with Vim-GFP reporter mice, as vimentin is one of the earliest expressed genes during EMT, and generated the Pb-Cre+/-;PtenL/L;KrasG12D/+;Vim-GFP (CPKV) mouse model.17 We demonstrated that epithelial, EMT, and mesenchymal-like (MES-like) prostate tumor cell populations could be isolated from murine prostate tumors of CPKV mice using EpCAM and Vim-GFP as markers.17 EMT tumor cells, which co-express both epithelial and mesenchymal markers, and mesenchymal-like tumor cells, which are derived from an EMT but have fully lost epithelial marker expression, have enhanced stemness qualities and tumor-initiating capacity compared to epithelial tumor cells.17 Fascinatingly, we observed that prostate tumors initiated by EMT and MES-like tumor cells isolated from CPKV prostates contained regenerated epithelial glandular structures, indicative of MET in vivo.17 In the present report, we studied the dynamic regulation of epithelial-mesenchymal plasticity using this genetically-defined system. We find that epithelial-mesenchymal plasticity is regulated epigenetically through the activity of the chromatin remodeling protein HMGA2, which is highly upregulated in EMT and MES-like tumor cells, as well as in tumors from men with mCRPC. Importantly, inhibition of HMGA2 activity with the histone deacetylase inhibitor (HDACi) LBH589 is able to eliminate castration-resistant MES-like tumor cells and prevent mCRPC in vivo.
Results
Prostate tumor cells with PI3K/AKT and RAS/MAPK co-activation display epithelial-mesenchymal plasticity
To explore whether prostate tumor cells with PI3K/AKT and RAS/MAPK co-activation have an inherent plasticity to switch between epithelial and mesenchymal states, we FACS sorted EpCAM+GFP- epithelial tumor cells from 10-week old Pb-Cre+/-;PtenL/L;KrasG12D/+;Vim-GFP (CPKV) prostates and cultured them in vitro (Figure 1a). After 14 days in culture, epithelial tumor cells that were originally sorted and plated as GFP- cells began to transition into GFP+ cells (Figure 1b). FACS analysis conducted on this cell line (hereafter referred to as the PKV cell line) revealed the existence of the same epithelial (EpCAM+GFP-), EMT (EpCAM+GFP+), and mesenchymal-like (MES-like) (EpCAM-GFP+) tumor cell populations that could be identified and isolated from primary CPKV prostates in vivo (Figure 1c).17 Similar to EMT and MES-like tumor cells isolated from CPKV prostates, EMT and MES-like tumor cells within the PKV cell line were also initially derived from epithelial tumor cells that underwent Cre recombination and harbor Pten deletion and Kras activation (Supplementary Figure 1a), as well as exhibit enhanced EMT signature gene expression and invasive capacity compared to epithelial tumor cells (Figures 1d and e).
Figure 1. Prostate tumor cells with PI3K/AKT and RAS/MAPK co-activation display epithelial-mesenchymal plasticity in vitro.
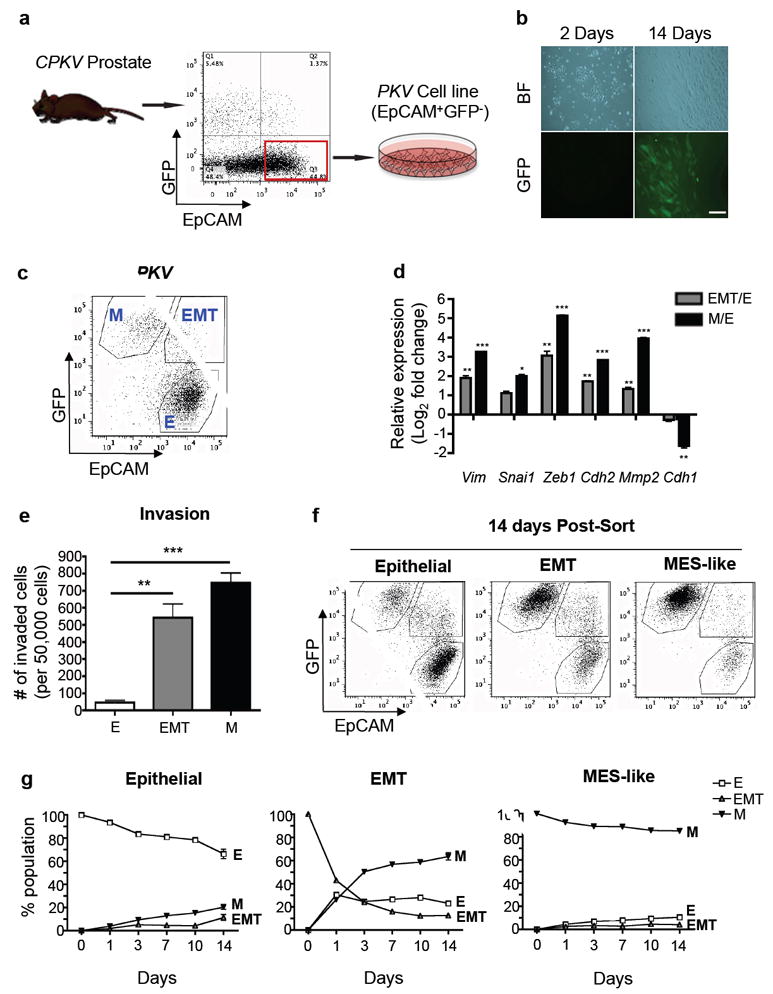
(a) Schematic outlining the generation of the PKV cell line from EpCAM+/GFP- epithelial cells FACS sorted from 10-12 week old CPKV prostates. (b) EpCAM+/GFP- epithelial cells plated in culture spontaneously undergo EMT and express GFP. Scale bar, 50 μm; BF, brightfield. (c) The PKV cell line contains heterogenous epithelial (E), EMT, and MES-like (M) tumor cell populations as assessed by FACS analysis. (d) qPCR analysis confirms that EMT and MES-like (M) tumor cells from the PKV cell line have upregulated EMT signature gene expression compared to epithelial tumor cells. Expression is relative to gene expression values found in epithelial (E) tumor cells. (e) Matrigel invasion assay reveals that EMT and MES-like (M) tumor cells are significantly more invasive than epithelial (E) tumor cells isolated from the PKV cell line. (f) Each tumor cell population within the PKV cell line was isolated by FACS and cultured separately in vitro. Representative FACS plots of each cell population 14 days after plating are shown. Each tumor cell population has the plasticity to generate all 3 tumor cell populations. (g) Each tumor cell population within the PKV cell line was isolated by FACS and cultured separately in vitro. The percentage of each tumor cell population (E, EMT, M) within each individually plated cell type (Epithelial, EMT, MES-like) was assessed by FACS 1, 3, 7, 10, and 14 days post-sort. Data in d, e, and g are represented as mean ± SEM from 2-3 independent experiments done in triplicate. *, p<0.05; **, p<0.01; ***, p<0.001.
Having shown that epithelial tumor cells have the plasticity to transition into EMT and MES-like tumor cells, we next wanted to determine if EMT and MES-like tumor cells also had the capacity to generate each of the three tumor cell populations. Epithelial, EMT, and MES-like tumor cell populations were isolated by FACS from the PKV line (Figure 1c) and cultured separately. Fourteen days after plating, each population was able to give rise to all three tumor cell populations as determined by FACS analysis and fluorescent imaging (Figure 1f and Supplementary Figure 1b). Interestingly, while the majority of sorted epithelial and MES-like tumor cells remained in their initial cell state, with small subsets of the other cell populations arising, the majority of EMT tumor cells had transitioned into fully epithelial or MES-like states as early as 24 hours after plating (Figure 1g). Moreover, each sorted cell population maintained a similar percentage of EMT tumor cells 14 days after plating, demonstrating that EMT tumor cells exist in a plastic, transitory state (Figure 1g). Overall, these results demonstrate that prostate tumor cells with PI3K/AKT and RAS/MAPK co-activation have the plasticity to readily transition between epithelial and mesenchymal states through both an EMT and MET.
Epithelial-mesenchymal transition states dictate response to PI3K and MAPK pathway inhibition and differential gene expression profile
The dynamic epithelial-mesenchymal plasticity observed in our genetically defined system raised the issue as to whether such plasticity contributes to the heterogeneous response of prostate cancer cells to targeted therapies, including PI3K and MAPK pathway inhibitors. To address this issue, PKV cells were treated with the dual PI3K/mTOR inhibitor PKI-587, the MEK inhibitor PD0325901, or both for 7 days, and the total number of each tumor cell subpopulation remaining after treatment was assessed by FACS and presented as the percentage of each subpopulation compared with vehicle-treated control cells. While the total number of the epithelial and EMT tumor cells was drastically reduced by treatment with PKI587, PD0325901, or both, the MES-like tumor cell population was relatively unaffected by PI3K and MAPK pathway inhibition (Figure 2a).
Figure 2. Epithelial-mesenchymal transition states dictate response to PI3K and MAPK pathway inhibition and differential gene expression profile.
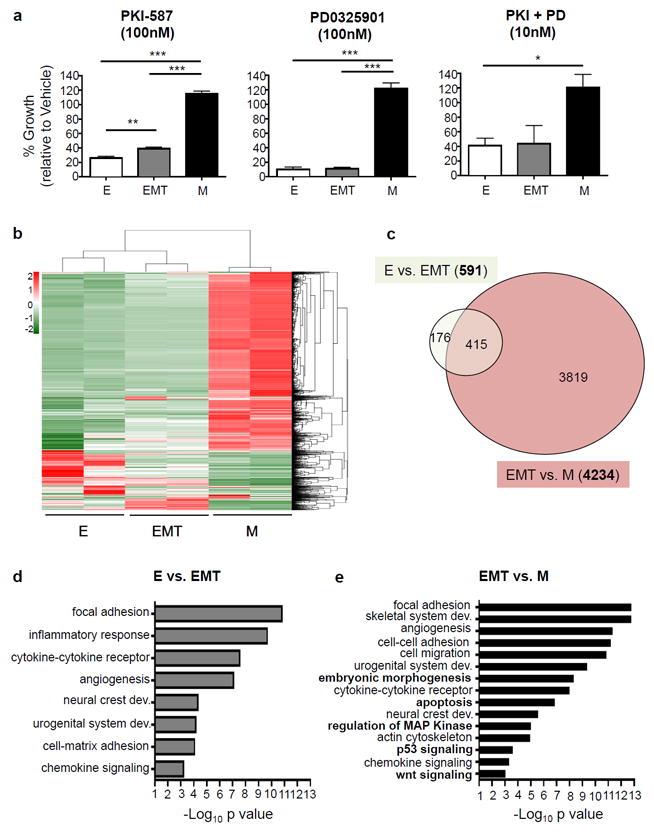
(a) PKV cells were treated with vehicle alone (DMSO), the PI3K/mTOR inhibitor PKI-587 (100nM), the MEK inhibitor PD0325901 (100nM), or both (10nM for each) for 7 days. MES-like (M) tumor cell growth is unaffected by treatment with PI3K and MAPK pathway inhibitors. % growth is relative to vehicle-treated cells (DMSO). (b) The gene transcription profiles of epithelial (E), EMT, and MES-like (M) tumor cells isolated from the prostates of 10-12 week old CPKV mice as assessed by RNA-seq. Heatmap displays mean-centered gene transcription levels of 2190 genes with average FPKM no less than 0.01 and coefficients of variation higher than 0.5. (c) Venn diagram showing the overlap of differentially expressed genes (DEGs) between epithelial vs. EMT (E vs. EMT) and EMT vs. MES-like populations (EMT vs. M). The MES-like tumor cell population has a large number of DEGs compared to the EMT (4234) tumor cell population. (d) Significantly enriched Gene Ontology (GO) items for differentially transcribed genes between epithelial (E) and EMT tumor cell populations. (e) Significantly enriched GO items for differentially transcribed genes between EMT and MES-like (M) tumor cell populations. Items in bold are solely enriched in the EMT-M transition. Data in a is represented as mean ± SEM from 3 independent experiments done in triplicate. *, p<0.05; **, p<0.01; ***, p<0.001.
As epithelial, EMT, and MES-like tumor cells were all initially derived from Cre+ prostate epithelial cells harboring Pten deletion and Kras activation and are in principle genetically identical, we next wanted to ascertain what additional pathways may be altered during the EMT process to account for their differential phenotypes and responses to PI3K/AKT and RAS/MAPK pathway inhibition, especially during the transition of EMT cells to a fully mesenchymal state (EMT-M transition). To this end, we profiled the transcriptomes of epithelial, EMT, and MES-like tumor cells isolated from the prostates of 10-12 week old CPKV mice through RNA-sequencing (RNA-seq) analysis. RNA-seq analysis revealed that the epithelial and EMT tumor cell populations (E-EMT transition) had a relatively similar gene expression profile with only 591 differentially expressed genes (DEGs) between the two states; the EMT-M transition, on the other hand, had a dramatically different gene expression profile with 4234 DEGs (Figures 2b and c, Supplementary Table 1). Gene Ontology (GO) analysis revealed shared pathway alterations between the E-EMT and EMT-M transitions, including changes in focal adhesion, actin cytoskeleton regulation, developmental pathways (urogenital system development, neural crest development) and inflammatory pathways (inflammatory response, cytokine-cytokine receptor, chemokine signaling) (Figure 2d and e). However, additional pathway alterations were found during the EMT-M transition, including changes in embryonic morphogenesis, Wnt signaling, apoptosis, and p53 signaling pathways (Figure 2e), which may explain the enhanced in vivo tumorigenic potential and stemness activity of mesenchymal-like tumor cells as was reported in our previous publication.17 Overall, while the early EMT process (E-EMT transition) has relatively few changes in gene expression, the late EMT process (EMT-M transition) is accompanied by a widespread shift in gene expression, including changes in key stem cell, developmental, and cell survival pathways that may ultimately lead to resistance PI3K/AKT and RAS/MAPK pathway inhibition.
EMT signature genes highly expressed in EMT and mesenchymal-like tumor cells are also highly expressed in human metastatic prostate cancer
To determine if EMT-related genes are also expressed in human metastatic prostate cancer samples, we used rank-rank hypergeometric overlap (RRHO) analysis to find those genes that are highly expressed in metastatic tumors in two separate human prostate cancer datasets: 1) the Taylor et al. dataset, which has 29 normal, 131 primary tumor, and 19 metastatic tumor samples,7 and 2) the Grasso et al. dataset, which contains 28 benign, 59 localized cancer, and 34 metastatic CRPC samples.18 As shown in Figure 3a, the two human datasets are very similar to each other. Importantly, a set of EMT signature genes is upregulated in human metastatic prostate tumor samples from both datasets (red circle in Figure 3a). We then analyzed the expression of the EMT signature genes found to be upregulated in human metastatic prostate cancer in two different murine gene expression datasets derived from 1) microarray analysis of laser capture microdissected well-differentiated (epithelial) and poorly differentiated tumor tissue from CPK prostates (Supplementary Table S2) and 2) RNA-seq analysis of epithelial, EMT, and MES-like tumor cells FACS sorted from CPKV prostates (Supplementary Table S1). We determined that these EMT signature genes, which include Snail, Twist1, Foxc2, Fn1, Mmp3, Mmp9, Lox, and Loxl2, were also upregulated in the poorly differentiated murine prostate cancer tissue samples and cells with EMT and MES-like characteristics (Figures 3a and b). These data provide evidence that EMT signature gene expression, as defined in our EMT and MES-like tumor cell populations, is associated with metastatic disease and CRPC in the human setting and is therefore likely to have clinical relevance.
Figure 3. HMGA2 is highly expressed in human mCRPC and in murine EMT and mesenchymal-like tumor cells.
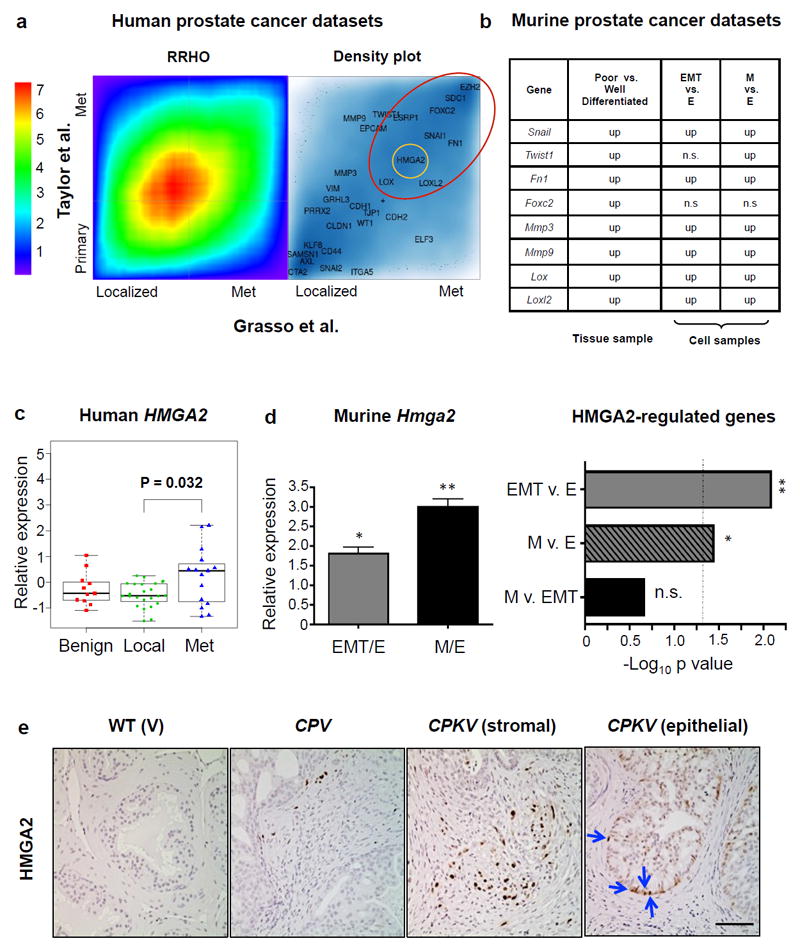
(a) Heat map comparing the overlap of differentially expressed genes in primary/localized and metastatic (met) prostate tumor specimens from the Taylor et al.7 and Grasso et al.18 human prostate cancer datasets based on rank-rank hypergeometric overlap (RRHO) analysis used to measure and visualize the degree of statistically significant overlap between two expression datasets (Left Panel). The color bar indicates the transformed log10 hypergeometric enrichment p-value between two ordered gene sets. The high heat area along the diagonal indicates significant overlap in differentially expressed genes between the two human prostate cancer datasets. Right Panel, selected epithelial and EMT signature genes were plotted corresponding to their signed and logged p-value ranks based on differential expression between metastatic and primary cancer samples. The depth of the blue color represents the density of all genes. A number of EMT signature genes (large red circle) were consistently ranked at similar positions in both human prostate cancer datasets. HMGA2 (small yellow circle) was among the group of genes differentially expressed in metastatic prostate cancer in both human datasets. (b) EMT genes upregulated in human metastatic prostate cancer were differentially expressed in murine datasets obtained from 1) laser capture microdissection microarray analysis of prostate tissue samples (p<0.05) and 2) RNA-seq analysis of FACS sorted cell populations (FDR<0.05). EMT genes were often upregulated in poorly differentiated prostate tissue and in EMT and MES-like (M) tumor cells. Up, upregulated; n.s., not significant. (c) HMGA2 expression levels in benign, localized (local), and metastatic CRPC (met) patient samples from Grasso et al.18 reveals that HMGA2 expression is significantly upregulated in mCRPC compared to localized disease. (d) Hmga2 expression is significantly upregulated in EMT and MES-like (M) tumor cells compared to epithelial tumor cells (Left Panel). Data was combined from 3 independent experiments done in triplicate. Right Panel, Fisher’s exact test was used to assess enrichment of HMGA2-regulated genes27 in differentially expressed genes between the EMT vs. epithelial (EMT v. E) and MES-like vs. epithelial (M v. E) tumor cell populations. Dotted line, p-value= 0.05; n.s, not significant. (e) HMGA2 protein expression is highly induced in both the stroma and epithelium (arrows) of CPKV prostates compared to CPV and V prostates. Scale bar, 50 μm. Data in c and d are represented as mean ± SEM. *, p<0.05; **, p<0.01.
The epigenetic regulator HMGA2 is highly expressed in human mCRPC and in murine EMT and mesenchymal-like tumor cells
Recent studies suggest that the master transcriptional regulators of the EMT process depend on epigenetic regulatory mechanisms, particularly those involved in chromatin remodeling, in order to achieve widespread changes in gene expression observed during EMT.19 The massive shift in gene expression between EMT and MES-like tumor cell populations (Figures 2b and c) prompted us to hypothesize that epigenetic alterations may play a key role in both initiating and maintaining the mesenchymal state. By surveying those genes that are highly expressed in metastatic tumors in both human datasets based on the RRHO analysis, we identified the epigenetic regulator HMGA2 (yellow circle in 3A), a non-histone chromatin remodeling protein, as a gene strongly associated with human metastatic prostate cancer. HMGA2, through its ability to bind to the minor groove of AT-rich DNA sequences and introduce structural alterations in chromatin that either promote or inhibit the actions of transcriptional enhancers, can alter global gene expression.20 HMGA2 is also known to be associated with embryonic and adult stem-cell states,21-23 and has been previously implicated in 1) modulating the microenvironment to promote prostate tumorigenesis through regulation of Wnt/β-catentin signaling, 2) regulating Snail expression through TGFβ/SMAD2, and 3) maintaining Ras-induced EMT.24-26 The expression of HMGA2 is significantly upregulated in human mCRPC compared to localized prostate cancer (Figure 3c).18 Similarly, Hmga2 expression is also significantly upregulated in both EMT and MES-like tumor cells compared to epithelial tumor cells (Figure 3d, left panel). In addition to Hmga2 expression, the HMGA2-regulated transcriptome27 is also significantly differentially expressed between EMT vs. epithelial and MES-like vs. epithelial tumor cells (Figure 3d, right panel). Compared to WT (V) and Pten null (CPV) prostates, CPKV prostates have dramatic induction of HMGA2 protein expression in both the stroma and in a small population of cells within epithelial glandular structures (arrows) (Figure 3e). Hence, these findings suggests that HMGA2 may be an essential factor in modulating epithelial-mesenchymal plasticity, and that HMGA2 expression could help to stratify human prostate cancer patients that are likely to progress to mCRPC.
HMGA2 regulates stemness and epithelial-mesenchymal plasticity in prostate tumor cells with PI3K/AKT and RAS/MAPK co-activation
To explore the functional role of HMGA2 in our model, we stably knocked down HMGA2 expression in the PKV cell line using a short hairpin RNA (shRNA) targeting HMGA2 (Figure 4a). HMGA2 knockdown had no effect on cell proliferation, as PKV-shHmga2 cells had a similar growth rate compared to PKV cells that were stably transduced with a control shScramble construct (Figure 4b). In order to investigate the effect of HMGA2 knockdown on the stemness attributes of PKV cells, we performed a matrigel sphere formation assay. Compared to control PKV-shScramble cells, PKV-shHmga2 cells had significantly reduced sphere-forming capacity, indicative of a reduced capacity for anchorage-independent and clonal growth (Figure 4c). Moreover, HMGA2 knockdown significantly reduced the expression of a number of pluripotency factors, including Oct4, Sox2, and Klf4, as well as other genes known to regulate self-renewal (Figure 4d). These findings demonstrate that HMGA2 activity is important for the maintenance of stemness in PKV cells.
Figure 4. HMGA2 regulates stemness and epithelial-mesenchymal plasticity in prostate tumor cells with PI3K/AKT and RAS/MAPK co-activation.
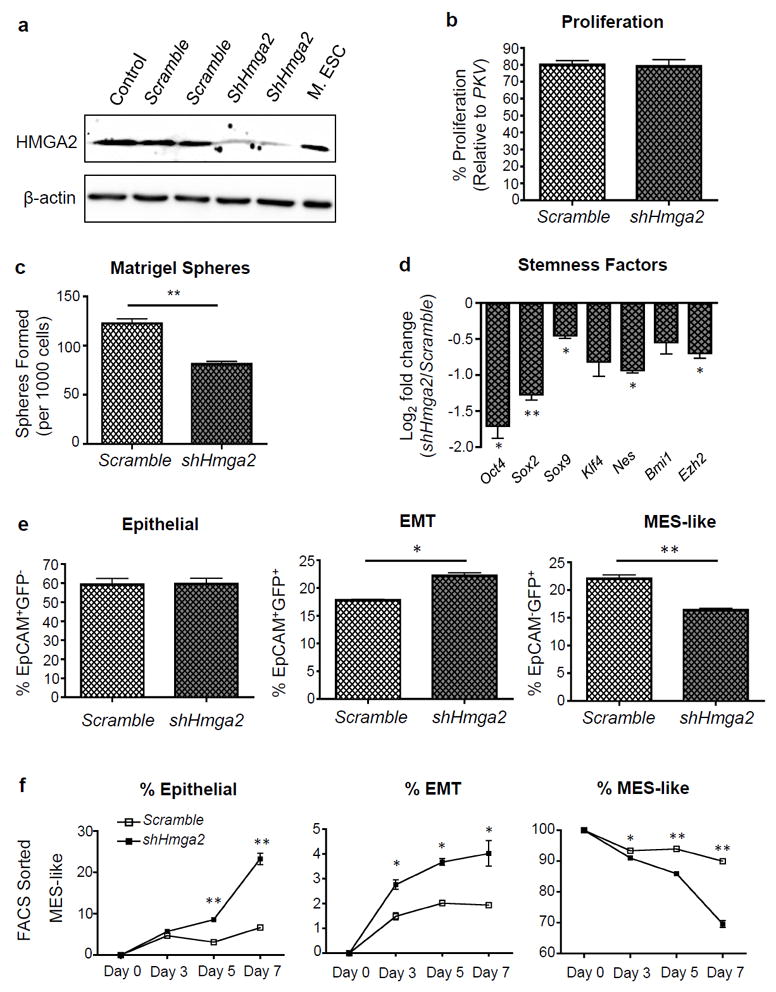
(a) ShRNA-targeted knockdown of HMGA2 protein expression in PKV cells. Mouse embryonic stem cells (M. ESC) were used as a positive control for HMGA2 expression. β-actin was used as a loading control. Control, PKV cells. Scramble, shScramble. (b) The proliferation of the PKV-shScramble (Scramble) and PKV-shHmga2 (shHmga2) cell lines was measured by MTT assay and is presented as % growth compared to control PKV cells. (c) PKV cells stably expressing shHmga2 have significantly reduced matrigel sphere-forming capacity compared to control PKV-shScramble (Scramble) cells. (d) HMGA2 knockdown reduces the expression of a number pluripotency and self-renewal factors. Expression is relative to gene expression values found in PKV-shScramble (Scramble) cells. (e) FACS analysis of the PKV-shScramble (Scramble) and PKV-shHmga2 cell lines revealed that throughout passaging, PKV-shHmga2 cells maintained a lower percentage of MES-like and higher percentage of EMT tumor cells compared to PKV-shScramble cells, indicative of a blockade in the transition of EMT tumor cells into fully MES-like tumor cells. (f) FACS sorted MES-like tumor cell populations from PKV-shHmga2 cells have reduced mesenchymal content and increased epithelial and EMT tumor cell numbers compared to control PKV-shScramble (Scramble) cells after 7 days in culture. Data in b-f are represented as mean ± SEM from 2-3 independent experiments done in triplicate. *, p<0.05; **, p<0.01.
Interestingly, compared to the PKV-shScramble cell line, the PKV-shHmga2 line maintains a relatively reduced percentage of MES-like tumor cells and an increased percentage of EMT tumor cells, suggesting that HMGA2 activity may be required for the EMT-M transition (Figure 4e). To further investigate how HMGA2 regulates epithelial-mesenchymal plasticity, we FACS sorted epithelial, EMT, and MES-like tumor cell populations from PKV-shScramble and PKV-shHmga2 cells and plated them separately in culture for 7 days. Epithelial, EMT, and MES-like tumor cell populations isolated from PKV-ShScramble cells all have the capacity to transition into all three tumor cell states (Figure 4f and Supplementary Figure 2) with similar kinetics to parental PKV cells (Figure 1g). However, in PKV-shHmga2 cells, sorted epithelial and EMT subpopulations maintained a relatively higher percentage of epithelial cells and a lower percentage of MES-like tumor cells after 7 days in culture compared with control PKV-shScramble cells, indicative of a stall in epithelial-mesenchymal plasticity (Supplementary Figure 2a). Importantly, HMGA2 knockdown significantly destabilized the MES-like tumor cell state, as a significantly higher percentage of MES-like tumor cells sorted from the PKV-shHmga2 line transitioned to both epithelial and EMT states compared to those isolated from control PKV-shScramble cells (Figure 4f). Since HMGA2 knockdown effectively destabilized the MES-like state, we were curious whether HMGA2 knockdown would also sensitize treatment-resistant MES-like cells to PI3K and MAPK pathway inhibitors. While PKI-587 treatment in the context of HMGA2 knockdown had little additive inhibitory effect on the growth of any tumor cell population compared to treatment of control PKV-ShScramble cells, HMGA2 knockdown further decreased the growth of all three tumor cell populations treated with PD0325901, including PD0325901-resistant MES-like tumor cells, as compared to the same treatment in PKV-ShScramble cells (Supplementary Figure 2b). This finding suggests that increased HMGA2 expression in MES-like tumor cells may indeed contribute to their lack of sensitivity to MAPK inhibition (see Figure 2a). From these results, we can conclude that HMGA2 knockdown 1) reduces stemness activity, 2) influences the overall plasticity of epithelial and EMT tumor cells, 3) destabilizes the MES-like state and enables MES-like tumor cells to more readily undergo an MET, and 4) sensitizes MES-like tumor cells to MAPK pathway inhibition. Therefore, HMGA2 plays an essential role in both the maintenance of stemness qualities, as well as in regulating epithelial-mesenchymal plasticity and the mesenchymal state in our model.
HDACi treatment effectively targets EMT and mesenchymal-like tumor cells through inhibition of HMGA2 activity and induction of p53-mediated apoptosis
As changes in expression of HMGA2 target genes alone cannot account for the more than 4000 DEGs found altered during the EMT-M transition (Figure 2c),27, 28 we hypothesized that other epigenetic regulators may also be important in regulating such a transition. To examine this hypothesis, we looked at differential expression of various epigenetic regulatory genes among our three tumor cell populations using a recently characterized list of genes associated with epigenetic regulation.29 While there were only 3 epigenetic regulatory genes whose expression were altered between epithelial and EMT tumor cells, 97 epigenetic regulatory genes were differentially expressed between the EMT and MES-like tumor cell states (Supplementary Figure 3a and Supplementary Table 3). Since previous studies have demonstrated that histone deacetylase inhibitors (HDACi) can effectively inhibit HMGA2 expression at the transcriptional level,30, 31 we tested whether HMGA2 expression could be modulated by HDAC activity. When we treated the PKV cell line with LBH589 (Panobinostat), a pan-HDACi, LBH589 significantly reduced Hmga2 gene expression while increasing H3K27 acetylation levels in a dose-dependent manner (Figures 5a and f). While low doses of LBH589 (1nM), which did not affect cell proliferation (data not shown) or apoptosis (Figure 5c), had little effect on epithelial and EMT tumor cells, it significantly decreased MES-like tumor cell numbers (Figure 5b) and reduced the overall sphere-forming capacity and stem cell-related gene expression of PKV cells (Figures 5d and e), mirroring the effects observed upon shRNA-mediated HMGA2 knockdown (Figure 4). This suggests that HDACi treatment can indeed inhibit HMGA2 activity and in turn suppress HMGA2-mediated epithelial-mesenchymal plasticity and stemness activity, and, importantly, target a MES-like tumor subpopulation that is relatively insensitive to PI3K and MAPK pathway inhibition (Figure 2).
Figure 5. HDACi treatment effectively targets EMT and mesenchymal-like tumor cells through inhibition of HMGA2 activity and induction of p53-mediated apoptosis.
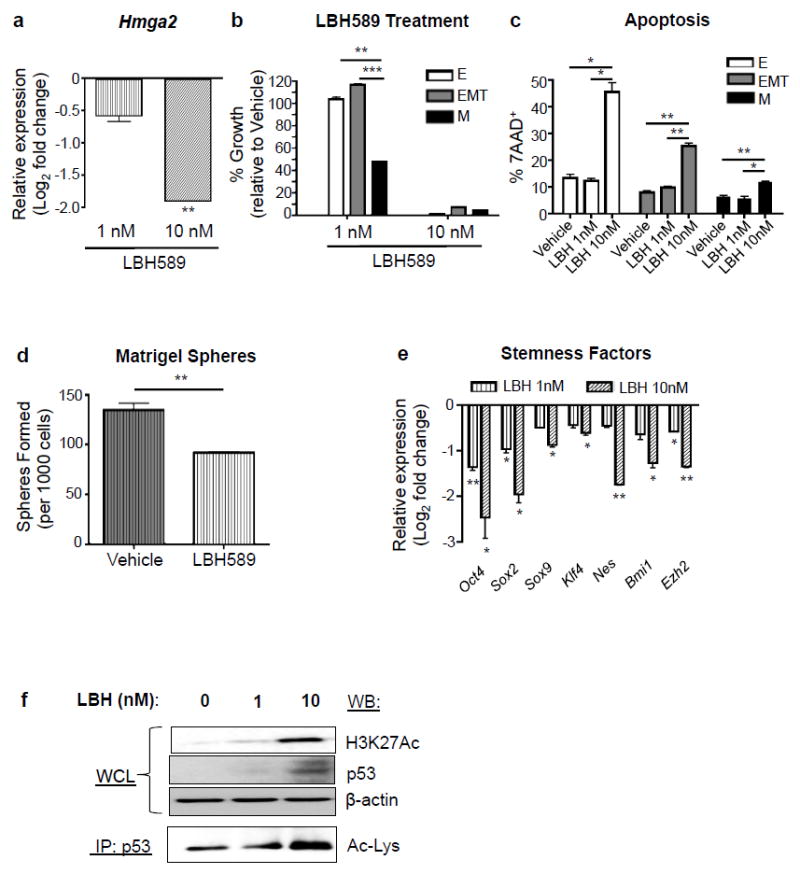
(a) LBH589 treatment (24 hr) of PKV cells reduces Hmga2 expression in a dose-dependent manner. Expression is relative to gene expression values found in vehicle-treated cells (DMSO). (b) LBH589 treatment (7 day) preferentially reduces MES-like (M) tumor cell numbers at low doses (1nM), and successfully targets all tumor cell populations at higher doses (10nM). % growth is relative to vehicle-treated cells (DMSO). (c) 7 day treatment of the PKV cell line with 10nM LBH589 induces significantly increased levels of apoptosis, as measured by the percentage of 7AAD+ cells, in all epithelial (E), EMT, and MES-like (M) tumor cell populations. (d) Low doses of LBH589 (1nM) significantly reduce the sphere-forming capacity of PKV cells after 7 days in Matrigel culture, similar to effects of HMGA2 knockdown (See Fig. 4C). (e) LBH589 treatment (24 hr) reduces the expression of various stemness factors in PKV cells compared to vehicle alone (DMSO), similar to the effects of HMGA2 knockdown (See Fig. 4D). (f) LBH589 treatment (6 hr) induces p53 expression and increases H3K27 and p53 acetylation levels in PKV cells. β-actin was used as a loading control. IP, immunoprecipitation; WCL, whole cell lystate; WB, Western Blot; Ac-Lys, acetylated lysine antibody. Data in a-f are represented as mean ± SEM. Data in b-e were collected from 2-3 independent experiments done in triplicate. *, p<0.05; **, p<0.01; ***, p<0.001.
Higher doses of LBH589 (10nM), on the other hand, were able to induce significant apoptosis and successfully inhibit the growth of all three tumor cell populations (Figures 5b and c), suggesting that LBH589 treatment may modulate other factors in addition to its effects on HMGA2 expression. We previously demonstrated that p53 protein levels are greatly reduced in the CPK murine prostate cancer model compared to Pten null and WT mice.16 We found in our current study that higher concentrations of LBH589 (10nM) significantly increased p53 protein levels (Figure 5f), while p53 and Mdm2 mRNA levels remained relatively unchanged (Supplementary Figure 3b). As p53 acetylation has been shown to enhance its stability, DNA binding affinity, and transcriptional activity associated with cell cycle arrest and apoptosis,32-34 we looked directly at LBH589-induced p53 acetylation, and found that p53 acetylation levels were significantly elevated with increasing concentrations of LBH589 (Figure 5f). P-p53 (Ser15) levels were unaffected by LBH589 treatment (data not shown), suggesting that increased p53 levels in LBH589-treated samples were not the consequence of an induced DNA damage response. Therefore, LBH589-induced p53 acetylation is one mechanism by which LBH589 is able to induce apoptosis in all PKV cell populations, including MES-like tumor cells.
HDACi treatment dramatically reduces the primary prostate tumor burden in vivo
Given the efficacy of LBH589 in targeting EMT and MES-like tumor cells in vitro, we next wanted to assess the impact of HDACi treatment on primary tumor growth in vivo. CPKV mice were treated with vehicle alone or LBH589 starting at 10 weeks of age, a time point when animals have already developed aggressive prostate tumors with poorly differentiated EMT features.16 After only 2 weeks of treatment with LBH589, there was a dramatic decrease in primary tumor size, most notably in the anterior lobes (Figure 6a, arrows in upper panel). Further histological examination revealed large glandular cysts in the anterior lobes and degenerated, scar-like tissue in the dorsolateral lobes of LBH589-treated mice (Supplementary Figure 4a), likely resulting from massive cell death. Additionally, there was also a dramatic decrease in the Ki67 proliferation index in both the epithelial and stromal compartments of LBH589-treated mice compared to mice receiving vehicle alone (Figure 6a, lower panel). H3K27 acetylation levels were increased in the prostate epithelium and stroma of CPKV mice treated with LBH589 compared to vehicle alone (Supplementary Figure 4b), verifying that LBH589 was effectively hitting its target. Importantly, FACS analysis revealed a statistically significant decrease in the EMT and MES-like tumor cell populations in the primary tumor site of LBH589-treated mice compared to those treated with vehicle alone, thus confirming the effectiveness of HDACi treatment at inhibiting these populations in vivo (Figure 6b). Mirroring our in vitro findings, the prostates of LBH589-treated CPKV mice also had substantially reduced HMGA2 expression and induction of strong p53 nuclear staining compared vehicle-treated mice (Figure 6c). Therefore, LBH589 treatment effectively reduces primary tumor growth and targets EMT and MES-like tumor cells in CPKV mice by modulating HMGA2 and p53 levels.
Figure 6. HDACi treatment inhibits prostate tumor growth, tumor cell dissemination, and metastasis in vivo.
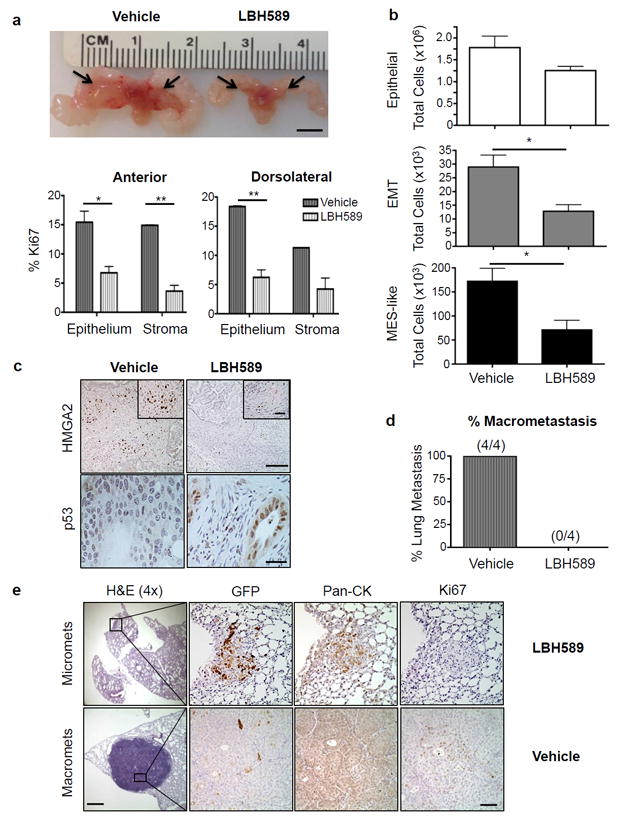
(a) 10 wk old CPKV mice treated with LBH589 for 2 weeks had dramatically reduced tumor burden, particularly in the anterior lobes (arrows), compared to vehicle-treated mice (Top Panel). Scale Bar, 5 mm. Bottom Panel, the Ki67 proliferation index is significantly reduced in LBH589-treated CPKV mice (n=4) compared to CPKV mice receiving vehicle alone (n=3) in both the anterior and dorsolateral lobes of the prostate. (b) LBH589-treated CPKV mice (n=9) have a significant reduction in the EMT and MES-like tumor cell populations compared to vehicle-treated mice (n=6). Data were combined from 2 independent experiments. (c) LBH589 treatment significantly reduces HMGA2 expression (Top Panel) and induces p53 expression (Bottom Panel) in the prostates of CPKV mice. Scale bar, top panel, low magnification, 100 μm; top panel, high magnification, 25 μm; bottom panel, 25 μm. (d) LBH589 treatment inhibits the formation of lung macrometastases in NSG mice transplanted with PKV cells by tail vein injection. (e) Representative lung histology of NOD/SCID/IL2Rγ-null (NSG) mice 8 weeks after transplantation of PKV cells by tail vein injection. While LBH589-treated mice did not develop macrometases (0/4), they did develop small micrometastases that were non-proliferative (Ki67-) and GFP+ (Top Panel). Bottom Panel, vehicle-treated mice, on the other hand, formed large, proliferating (Ki67+) macrometastases that are GFP- and have strong expression of Pan-Cytokeratin (Pan-CK), indicative of an epithelial phenotype. Scale bar, low magnification, 500 μm; Scale bar, high magnification, 50 μm. Data in a and b are represented as mean ± SEM. *, p<0.05; **, p<0.01.
HDACi treatment inhibits tumor cell dissemination and distant metastasis in vivo
Since CPKV mice already have significant tumor cell dissemination into the blood stream at 10 weeks of age,17 we wanted to investigate the impact of LBH589 therapy on circulating tumor cell (CTC) numbers and distant metastasis. Peripheral blood was collected from LBH589 and vehicle-treated CPKV mice following 2 weeks of treatment. While LBH589 treatment had little effect on the number of EpCAM+GFP- epithelial circulating tumor cells (CTCs) in the blood, treatment dramatically reduced the number of EpCAM-GFP+ CTCs with mesenchymal-like features (Supplementary Figure 4c), further validating the specificity of LBH589 treatment at targeting MES-like tumor cell populations. To determine the effect of LBH589 treatment on metastasis, we developed an in vivo dissemination model by transplanting 500 000 PKV cells by tail vein injection into NOD/SCID/IL2Rγ-null (NSG) mice. While 100% of mice treated with vehicle alone (4/4) developed lung macrometastases 8 weeks post-transplantation, LBH589 treatment completely blocked the formation of macrometastases (0/4) (Figure 6d). Although mice receiving LBH589 treatment still developed small, non-proliferative micrometastases in the lungs that maintained GFP expression, they did not develop the proliferative, GFP- macrometastases found in vehicle-treated mice (Figure 6e). Therefore, LBH589 treatment seems to block a rate-limiting step in metastasis: the transition of non-proliferative micrometastases with mesenchymal/EMT features into proliferative macrometastases with epithelial features. Overall, these data demonstrate that HDACi treatment with LBH589 is effective at suppressing primary tumor growth, tumor cell dissemination, and metastasis in vivo by inhibiting HMGA2 expression, inducing P53 dependent apoptosis, and subsequently targeting EMT and MES-like tumor cells.
Mesenchymal-like tumor cells are castration-resistant and contribute to early lethality in CPKV mice
Recent studies have shown that androgen deprivation therapy (ADT) can induce an EMT in benign and neoplastic prostate epithelium,12 and that EMT contributes to the development of CRPC.11 We therefore wanted to determine if the EMT and MES-like tumor cell populations in our model are further enriched by ADT and could potentially accelerate CRPC development. To test this hypothesis, we castrated CPKV mice at 6 weeks of age, and subsequently analyzed the effects of castration on tumor growth and overall animal survival. While CPKV prostates initially experience tumor regression, particularly in the anterior lobes at 1 week post-castration, by 2 weeks post-castration, castration-resistant tumors have completely grown back (Supplementary Figure 5a, black circles), inducing early lethality in CPKV mice with a median survival of ~3 weeks post-castration (Figure 7a). Indeed, although the Ki67 proliferation index is reduced in the anterior lobes of CPKV mice 1 week post-castration, by 2 weeks post-castration, the proliferation index returned to levels similar to those found in intact (non-castrated) CPKV mice (Figure 7b). Such fast regrowth and early lethality are in sharp contrast to our previous study of castration in the Pten null model, in which tumors returned to their original sizes 4-8 weeks post-castration and very few animals succumbed to CRPC-related mortality.35, 36
Figure 7. HDACi treatment can effectively inhibit the development of CRPC by targeting castration-resistant mesenchymal-like tumor cells.
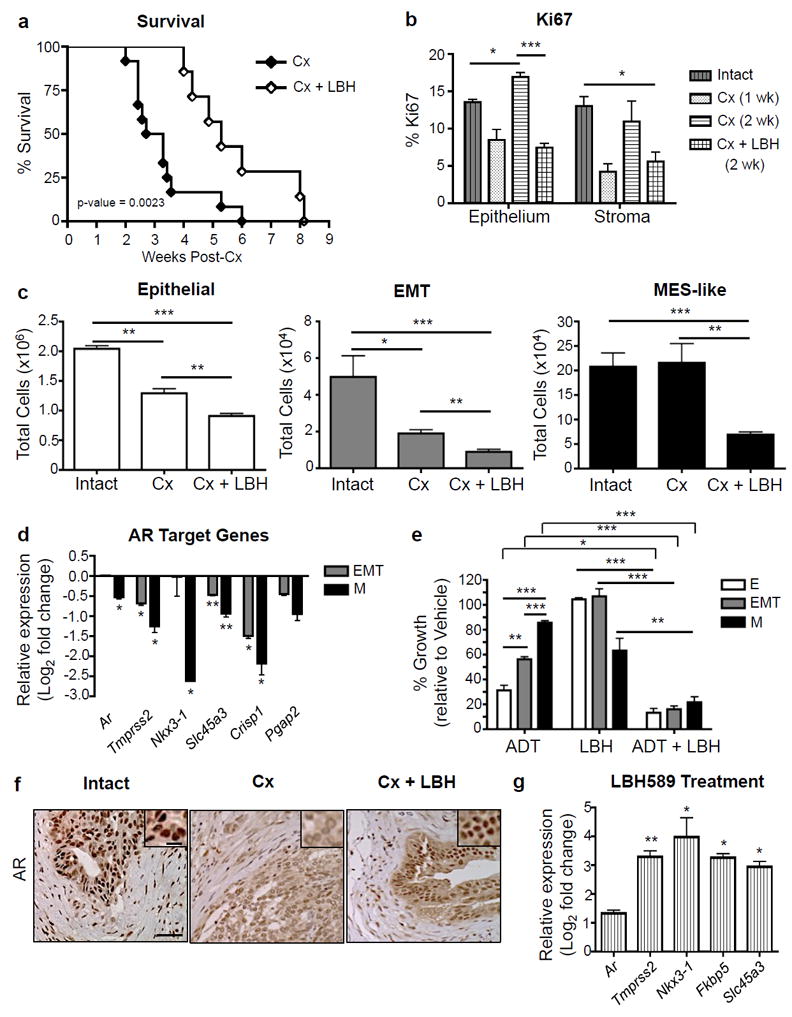
(a) While castration (Cx) of CPKV mice (n=12) at 6 weeks of age led to early lethality, LBH589 treatment in combination with Cx (n=7) significantly increased overall survival. Data were combined from 3 independent experiments. (b) LBH589 treatment in combination with Cx significantly lowered the Ki67 proliferation index in both the epithelium and stroma of CPKV prostates. n=3 for Intact and Cx samples; n=4 for Cx + LBH samples. (c) Castrated (Cx) mice (n=4) have no change in MES-like tumor cell numbers compared to intact, vehicle-treated mice (n=3). LBH589 treatment in combination with castration (n=6) successfully targets castration-resistant MES-like tumor cells and further reduces epithelial and EMT tumor cell numbers in vivo. Data were combined from 2 independent experiments. (d) AR target gene expression is significantly downregulated in MES-like (M) tumor cells compared to epithelial tumor cells isolated from the prostates of 10-12 week old CPKV mice as assessed by RNA-seq analysis. Expression is relative to gene expression values found in epithelial tumor cells. (e) 7 day treatment of PKV cells with media lacking androgens (CDT-FBS) significantly impeded the growth of epithelial (E) and EMT but not MES-like (M) tumor cells. Low doses of LBH589 (1nM) in combination with ADT sensitize MES-like (M) tumor cells to androgen withdrawal-induced growth inhibition. (f) LBH589 treatment reestablishes nuclear AR expression after Cx in the anterior lobes of CPKV prostates. Scale bar, low magnification, 50 μm; high magnification, 10 μm. (g) LBH589 treatment enhances AR target gene expression in CPKV prostates compared to vehicle alone. Expression is relative to gene expression values found in vehicle-treated CPKV mice. Data in b, c, d, e, and g are represented as mean ± SEM. *, Data in e and g were collected from 2-3 independent experiments done in triplicate. p<0.05; **, p<0.01; ***, p<0.001.
To investigate the causes for such an early lethality, we compared epithelial, EMT, and MES-like tumor cell numbers before and 2 weeks after castration, and found that while the epithelial and EMT tumor cell populations were significantly decreased in response to castration, the MES-like tumor cell population remained relatively stable (Figure 7c, right panel). RNA-seq analysis revealed that AR expression, along with a number of AR target genes, is significantly downregulated in the MES-like tumor cell population compared to the epithelial tumor cell population (Figure 7d), which may explain the poor responsiveness of MES-like cells to castration. Indeed, when the PKV cell line was treated with media stripped of androgens (charcoal/dextran treated (CDT) FBS), we observed a similar phenotype: while epithelial and EMT tumor cell growth was inhibited, ADT had a minimal effect on MES-like tumor cell growth (Figure 7e). These data suggest that MES-like tumor cells, through downregulation of the AR signaling axis, are inherently castration-resistant, and likely contribute to the early lethality observed in castrated CPKV mice.
HDACi treatment can effectively inhibit the development of CRPC by targeting castration-resistant mesenchymal-like tumor cells
Given the substantial effect that HDACi treatment has on MES-like tumor cell growth and survival both in vitro and in vivo (Figures 5 and 6), we reasoned that HDACi therapy in combination with castration might inhibit castration-resistant disease and in turn improve the overall survival of CPKV mice post-castration. At 6 weeks of age, CPKV mice were castrated and administered either LBH589 or vehicle alone. CPKV mice receiving LBH589 had significantly reduced tumor burden 2 weeks post-castration as determined by decreased anterior lobe size (Supplementary Figure 5a) and significantly reduced prostate weight (Supplementary Figure 5b). Remarkably, LBH589 treatment was able to significantly improve the overall survival of castrated CPKV mice, nearly doubling the median survival from ~21 days to ~37 days post-castration (Figure 7a). Moreover, castrated mice receiving LBH589 treatment had a significantly diminished Ki67 proliferation index in both the epithelial and stromal compartments compared to mice receiving vehicle alone (Figure 7b). In sharp contrast to castration alone, LBH589 treatment in combination with castration was able to significantly reduce the number of MES-like tumor cells, as well as further diminish the epithelial and EMT tumor cell populations (Figure 7c, right panel). Therefore, LBH589 treatment can effectively improve the overall survival of castrated CPKV mice by targeting castration-resistant MES-like tumor cells.
HDACi treatment induces reactivation of AR signaling and sensitizes mesenchymal-like tumor cells to ADT-induced apoptosis
Finally, we wanted to determine the impact of HDACi treatment on AR signaling in vivo. While castration alone led to weaker and more cytoplasmic AR protein expression in the anterior lobes, LBH589 treatment in combination with castration led to restoration of strong nuclear AR staining, similar to that found in intact CPKV prostates (Figure 7f, see high powered inserts). As further confirmation of HDACi-induced reactivation of AR signaling, CPKV mice treated with LBH589 had significant induction of AR downstream target gene expression, including increased expression of Tmprss2, Nkx3-1, Fkbp5, and Slc45a3 (Figure 7g). Previous studies have suggested that acetylation of AR in its flexible hinge region is required for maximal AR activation and transcriptional activity through its regulation of the DNA binding, nuclear translocation, and transactivation of AR.37, 38 Moreover, HDAC1 has been shown to interact directly with AR and repress AR activity through its effects on AR acetylation.39 As a mechanism behind heightened AR nuclear localization and transcriptional activity following LBH589 treatment, we explored whether AR acetylation itself was enhanced upon LBH589 treatment. As early as 6 hours post-treatment, PKV cells treated with LBH589 had dramatically increased AR acetylation levels compared to cells treated with vehicle alone (Supplementary Figure 5c). Interestingly, the ratio of acetylated AR to total AR levels after LBH589 treatment is similar to the ratio found in androgen-dependent LNCaP human prostate cancer cells, showing that HDACi treatment with LBH589 leads to AR nuclear localization and increased AR transcriptional activity as would be found in an androgen-dependent context.
To investigate if LBH589 treatment could sensitize MES-like tumor cells to growth inhibition and apoptosis induced by ADT, PKV cells were treated with LBH589 in the absence of androgens (CDT FBS). A low concentration of LBH589 (1nM), which by itself only partially reduces MES-like tumor cell numbers, was indeed able to synergize with ADT to sensitize castration-resistant MES-like tumor cells to ADT-induced growth inhibition and significantly reduce MES-like tumor numbers compared to either treatment alone (Fig. 7E). Moreover, while androgen withdrawal or 1nM LBH589 treatment alone were unable to induce apoptosis, androgen withdrawal in combination with LBH589 treatment induced significant apoptosis in all three tumor cell populations, including castration-resistant MES-like tumor cells (Supplementary Figure 5d). These results suggest that HDACi therapy leads to the reactivation of AR signaling in AR-independent MES-like tumor cells, making them sensitive to ADT-induced apoptosis. Overall, while castration leads to expansion of the castration-resistant MES-like tumor cell population and early lethality in CPKV mice, LBH589 treatment in combination with castration significantly prolongs survival by successfully targeting the MES-like tumor cell population and thereby impeding the onset of CRPC.
Discussion
Visualizing epithelial-mesenchymal plasticity has been difficult due to its transient nature and the lack of defined biomarkers of epithelial-mesenchymal plasticity. Here, we created an in vitro model system of epithelial-mesenchymal plasticity from the previously described CPKV murine prostate cancer model17 that contains epithelial, EMT, and mesenchymal-like tumor cell populations harboring Pten deletion and conditional Kras activation. Using this system, we uncovered a novel mechanism of regulation of epithelial-mesenchymal plasticity mediated by epigenetic rather than genetic mechanisms through the chromatin remodeling protein HMGA2, and determined an effective therapeutic strategy for inhibiting HMGA2 activity, targeting treatment-resistant mesenchymal-like tumor cells, and preventing mCRPC with the HDACi LBH589. These findings provide some of the first in vivo evidence that direct targeting of epithelial-mesenchymal plasticity through epigenetic inhibitors can have therapeutic efficacy at blocking the onset of CRPC and preventing distant metastasis. Importantly, we are also able to identify HMGA2 and a number of EMT signature genes, including SNAIL, FN1, and FOXC2, as potential biomarkers of epithelial-mesenchymal plasticity and metastatic CRPC in the human setting.
The unexpected finding that MES-like tumor cells, unlike epithelial and EMT tumor cells, are resistant to PI3K and MAPK pathway inhibitors despite uniform Pten deletion and Kras activation in all cell types has important implications in the clinic, as it suggests that clinicians will need to consider the tumor initiation event (genetic event) as well as the tumor cell state/lineage (epigenetic event) when determining optimal treatment for a given patient. One possible resistance mechanism could be that activation of alternative survival or developmental/stem cell pathways, such as those regulated by HMGA2, allows for MES-like tumor cell growth and survival that is independent of PI3K and MAPK signaling.40, 41 Indeed, MES-like tumor cells have a dramatic change in the expression of various developmental, growth-signaling, survival, and stem cell pathways compared to epithelial and EMT tumor cells (Figure 2), and these alterations may contribute to therapeutic resistance.
The large number of epigenetic regulators altered in MES-like tumor cells suggests that epigenetic alterations may play a defining role in the differential transcriptional profile of MES-like tumor cells and their lack of sensitivity to PI3K and MAPK pathway inhibitors. Indeed, we discovered that epigenetic changes mediated by the chromatin remodeling protein HMGA2 are necessary for 1) epithelial-mesenchymal plasticity, 2) preservation of the mesenchymal state, and 3) maintenance of stemness activities. Hence, changes in epigenetic regulation via HMGA2 lead to a more stem-like state where MES-like tumor cells are less susceptible to targeted therapy. As an “architectural transcription factor” with the ability to affect the expression of thousands of genes by altering the structure of chromatin, HMGA2 may have many downstream targets,20 including novel regulators of epithelial-mesenchymal plasticity and stemness, which require further elucidation in our model. Our transcriptional profiling analysis of the EMT and MES-like tumor cell populations, combined with the ability to visualize and manipulate epithelial-mesenchymal plasticity in vitro with the PKV cell line, provides an important platform for uncovering novel regulators of epithelial-mesenchymal plasticity that could be targeted therapeutically. Moreover, as early EMT (E-EMT transition) and late EMT phases (EMT-M transition) can be separated, this system provides a unique model for dissecting and differentiating those alterations that are important for the initiation of EMT versus those that are essential for the maintenance of the mesenchymal state. In addition, it will also be important to unravel the global epigenetic changes in DNA methylation and histone acetylation/methylation patterns that take place in MES-like tumor cells in order to identify novel regulators of these redefined epigenetic states.
Importantly, we were able to therapeutically inhibit HMGA2 expression and eradicate treatment-resistant MES-like tumor cells with the Pan-HDACi LBH589. Interestingly, we found that LBH589 mediated many of its effects through enhanced acetylation of the nuclear transcription factors p53 and AR, which in turn promotes a more differentiated, androgen-dependent cell state that is re-sensitized to apoptosis. As dysregulated p53 signaling in MES-like tumor cells (Figure 2e) likely contributes to the general resistance of this cell state to therapy-induced apoptosis, a drug such as LBH589 that induces p53 activation is likely to have lasting therapeutic benefit by inducing apoptosis rather than cytostasis. Increased AR activity may also lead to the sensitization of MES-like tumor cells to cell death through its ability to regulate p53, as it has been previously demonstrated that NKX3.1, which is a downstream target of AR, can bind to HDAC1 and subsequently lead to increased p53 acetylation through an MDM2-dependent mechanism34. However, we cannot rule out the possibility that LBH589 treatment could also sensitize tumor cells to androgen withdrawal-induced cell death in a p53-independent manner, such as through the alteration of other AR transcriptional programs. In this regard, ChIP-seq analysis of AR-occupied DNA binding sites in the presence or absence of LBH589 will be needed to fully elucidate the altered transcriptional program regulated by the AR in response to HDAC inhibition. In addition to these mechanisms, the effect of LBH589 treatment on histone acetylation, chromatin remodeling, and other epigenetic changes is still worthy of further exploration.
As one major issue with the clinical use of HDACi therapy has been the cytotoxicity and adverse effects associated with treatment,42-44 it will be important to determine which specific HDAC isoforms are responsible for regulating HMGA2, p53, and AR expression and activity so that therapeutics that target specific HDAC isoforms can be designed in order to reduce off-target effects. Moreover, as different doses of HDACi treatment may be required to affect changes in acetylation of histone vs. non-histone protein targets, it will be important to fully validate the desired molecular target of LBH589 treatment before determining the optimal treatment regimen.
The efficacy of LBH589 (Panobinostat) as a single agent in the treatment of CRPC has not been very promising.42, 45 However, the design of the Phase II study of Panobinostat in CRPC patients specified a protocol-defined response of 50% PSA decline, which 0/34 patients met.45 As our study, in contrast to other preclinical studies, demonstrates that HDACi treatment enhances rather than represses AR activation and signaling in prostate tumor cells,46, 47 PSA levels are likely to either remain constant or even potentially rise as a consequence of LBH589 treatment. This suggests that new biomarkers are needed to identify patients with prostate cancer who are responsive to HDAC inhibitors. Our results suggest that HDACi treatment may be effective against a subset of CRPC patients with increased HMGA2 and EMT marker expression. As a previous report has shown that HMGA2 mRNA could be detected in peripheral blood samples of breast cancer patients by RT-PCR and that patients with HMGA2 expression had a worse prognosis than those without detectable levels, it will be interesting to see if HMGA2 mRNA levels in the blood are also predictive of mCRPC development and sensitivity to HDACi therapy.48 Since LBH589 promotes the reactivation of AR signaling and thus facilitates the transition of stem-like, MES-like tumor cells to a more differentiated, AR-dependent state, the combination of ADT or AR-targeted therapies with LBH589 may likely lead to apoptosis and eradication of prostate tumor cell types that are intrinsically castration-resistant and in turn improve the survival outcomes of patients with mCRPC, for which there is still no cure.
Materials and methods
Mouse strains
The Cre-;PtenL/L;KrasG/+;Vim-GFP (V), Cre+/-;PtenL/L;Kras+/+;Vim-GFP (CPV), and Cre+/-;PtenL/L;KrasG/+;Vim-GFP (CPKV) mouse models were generated as previously described.17 These strains have been maintained on a mixed strain background. All studies with animals were performed under the regulation of the division of Laboratory Animal Medicine at the University of California at Los Angeles (UCLA).
Cell lines and reagents
The PKV cell line was generated by FACS sorting CD45-CD31-Ter119-EpCAM+GFP- epithelial cells from the prostates of 10 week old CPKV mice and culturing them in 0.2% gelatin-coated 10 cm dishes with Dulbecco’s modified eagle medium (DMEM) (Sigma-Aldrich, St. Louis, MO, USA) containing 1% Pen/Strep, 10% Fetal Bovine Serum (FBS) (Omega Scientific, Tarzana, CA, USA), 25 μg /mL bovine pituitary extract, 5 μg /mL insulin (Invitrogen, Grand Island, NY, USA), and 6 ng/mL recombinant human EGF (BD Biosciences, San Jose, CA, USA). LNCaP cells were grown in RPMI-1640 media (Sigma-Aldrich) with 1% Pen/Strep and 10% FBS. For studies carried out in the context of androgen deprivation, media containing 10% charcoal dextran-treated (CDT) FBS (BD Biosciences) was used.
Short hairpin RNA knockdown
To generate the PKV-shScramble or PKV-shHmga2 cell lines, PKV cells were transduced with lentivirus containing the pLKO.shHmga2 or pLKO.Scramble plasmids, respectively, for 24 hours in the presence of 8 μg/ml polybrene. Cells with stable integration of the desired construct were then selected for using puromycin at 4 μg/ml for 5 days. The PKV-shScramble or PKV-shHmga2 cell lines were subsequently passaged three times before being used for experiments. Stable knockdown of HMGA2 expression was confirmed by Western blot analysis. pLKO.shHmga2 was a gift from Tyler Jacks & Monte Winslow (Addgene plasmid # 32399),49 and Scramble shRNA was a gift from David Sabatini (Addgene plasmid # 1864).50
In vivo dissemination model
500 000 PKV cells were resuspended in 200 μl of PBS and transplanted by tail vein injection into 6-8 week old male NOD/SCID/IL2Rγ-null (NSG) mice. 8 weeks after transplantation, mice were sacrificed, and the presence of lung macrometastases was assessed by gross examination of formalin-fixed lung samples under a dissecting microscope, as well as by examining H&E stained lung sections. Micrometastases were defined as Ki67- metastases less than 300 μm in diameter.
Drug treatment
For the in vitro drug studies, PKV cells were treated with PD035901, PKI-587, LBH589 or a combination thereof at the indicated concentrations. Dimethyl Sulfoxide (DMSO) was used as a vehicle control. PD325901 (PF00192513) and PKI-587 (PF05212834) were a generous gift from the Pfizer Pharmaceutical Company (New York, USA), while LBH589 was purchased from LC Laboratories (Woburn, MA, USA). Live cell counts were assessed using trypan blue exclusion. Percent growth was determined by first assessing the percentage of each cell population by FACS analysis, and then dividing the total cell number of each drug treated cell population by the total cell number of the same populations in vehicle treated cells. Apoptosis was determined by positive staining for 7AAD by FACS analysis.
For the in vivo drug studies using intact (non-castrated) CPKV mice, 10-week old CPKV mice were treated for 2 weeks with either vehicle alone (5.2% Tween 80 and 5.2% PEG 400 in Phosphate buffered saline (PBS)) or LBH589 (10 mg/kg, 5 days/week) by intraperitoneal (i.p.) injection before sacrificing. For the castration studies, 6-week old intact or castrated CPKV mice were treated with vehicle or LBH589 for 2 weeks, or until protocol-determined endpoints for morbidity required euthanasia for the survival study. Prostate weight was assessed after removal of the seminal vesicles and bladder from the prostate lobes. For the in vivo dissemination assay, NSG mice were treated with either vehicle or LBH589 beginning 24 hours after transplantation of PKV cells for a total of 8 weeks. Mice were randomized into different treatment cohorts using a Random Number Calculator (GraphPad). A minimum of 4-5 mice were used in each experimental treatment cohort.
Accession numbers
Gene expression datasets used in this study are available at Gene Expression Omnibus (GEO) under accession numbers GSE67879.
Statistical analysis
Sample sizes were determined using statistical power analysis. Graphpad Prism software (La Jolla, CA, USA) was used to calculate mean and standard deviation. Student’s t-test (assuming Gaussian distribution and equal variance between samples) was used to calculate the statistical significance between the two groups of data. P < 0.05 is considered significant.
Supplementary Material
Acknowledgments
The authors thank members of the Hong Wu lab for their critical comments and suggestions. We thank Drs. Yang Zong and Owen Witte for generously supplying us with HMGA2 antibodies, and Dr. Shumin Wu for supplying protein lysates from mouse ES cells.
Grant Support:
MR was supported by NIH T32 CA009056. WG was supported by a General Financial Grant from the China Postdoctoral Science Foundation (2015M570010). DJM was supported by NIH F32 CA112988-01, CIRM TG2-01169 and a Prostate Cancer Foundation Young Investigator Award. This work has been supported in part by awards from the Prostate Cancer Foundation (to HW), and grants from the NIH (P50 CA097186 and P01 CA163227 to PSN, P50 CA092131 to YX and HW, and R01 CA107166, RO1 CA121110, and U01 CA164188 to HW).
Footnotes
Disclosure of Potential Conflicts of Interest: The authors do not have potential conflicts of interest.
Supplementary Information accompanies this paper on the Oncogene website.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA: a cancer journal for clinicians. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Attard G, de Bono JS. Translating scientific advancement into clinical benefit for castration-resistant prostate cancer patients. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17:3867–3875. doi: 10.1158/1078-0432.CCR-11-0943. [DOI] [PubMed] [Google Scholar]
- 3.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. The New England journal of medicine. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. The New England journal of medicine. 2012;367:1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 5.Rescigno P, Buonerba C, Bellmunt J, Sonpavde G, De Placido S, Di Lorenzo G. New perspectives in the therapy of castration resistant prostate cancer. Current drug targets. 2012;13:1676–1686. doi: 10.2174/138945012803529956. [DOI] [PubMed] [Google Scholar]
- 6.Shah RB, Mehra R, Chinnaiyan AM, Shen R, Ghosh D, Zhou M, et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer research. 2004;64:9209–9216. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- 7.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baca SC, Garraway LA. The genomic landscape of prostate cancer. Frontiers in endocrinology. 2012;3:69. doi: 10.3389/fendo.2012.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brannon AR, Sawyers CL. “N of 1” case reports in the era of whole-genome sequencing. The Journal of clinical investigation. 2013;123:4568–4570. doi: 10.1172/JCI70935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haffner MC, Mosbruger T, Esopi DM, Fedor H, Heaphy CM, Walker DA, et al. Tracking the clonal origin of lethal prostate cancer. The Journal of clinical investigation. 2013;123:4918–4922. doi: 10.1172/JCI70354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanaka H, Kono E, Tran CP, Miyazaki H, Yamashiro J, Shimomura T, et al. Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nature medicine. 2010;16:1414–1420. doi: 10.1038/nm.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun Y, Wang BE, Leong KG, Yue P, Li L, Jhunjhunwala S, et al. Androgen deprivation causes epithelial-mesenchymal transition in the prostate: implications for androgen-deprivation therapy. Cancer research. 2012;72:527–536. doi: 10.1158/0008-5472.CAN-11-3004. [DOI] [PubMed] [Google Scholar]
- 13.Armstrong AJ, Marengo MS, Oltean S, Kemeny G, Bitting RL, Turnbull JD, et al. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Molecular cancer research : MCR. 2011;9:997–1007. doi: 10.1158/1541-7786.MCR-10-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bitting RL, Schaeffer D, Somarelli JA, Garcia-Blanco MA, Armstrong AJ. The role of epithelial plasticity in prostate cancer dissemination and treatment resistance. Cancer metastasis reviews. 2014;33:441–468. doi: 10.1007/s10555-013-9483-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marin-Aguilera M, Codony-Servat J, Reig O, Lozano JJ, Fernandez PL, Pereira MV, et al. Epithelial-to-mesenchymal transition mediates docetaxel resistance and high risk of relapse in prostate cancer. Molecular cancer therapeutics. 2014;13:1270–1284. doi: 10.1158/1535-7163.MCT-13-0775. [DOI] [PubMed] [Google Scholar]
- 16.Mulholland DJ, Kobayashi N, Ruscetti M, Zhi A, Tran LM, Huang J, et al. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer research. 2012;72:1878–1889. doi: 10.1158/0008-5472.CAN-11-3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruscetti M, Quach B, Dadashian EL, Mulholland DJ, Wu H. Tracking and Functional Characterization of Epithelial-Mesenchymal Transition and Mesenchymal Tumor Cells During Prostate Cancer Metastasis. Cancer research. 2015 doi: 10.1158/0008-5472.CAN-14-3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nature medicine. 2013;19:1438–1449. doi: 10.1038/nm.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fusco A, Fedele M. Roles of HMGA proteins in cancer. Nature reviews Cancer. 2007;7:899–910. doi: 10.1038/nrc2271. [DOI] [PubMed] [Google Scholar]
- 21.Nishino J, Kim I, Chada K, Morrison SJ. Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf Expression. Cell. 2008;135:227–239. doi: 10.1016/j.cell.2008.09.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li O, Vasudevan D, Davey CA, Droge P. High-level expression of DNA architectural factor HMGA2 and its association with nucleosomes in human embryonic stem cells. Genesis. 2006;44:523–529. doi: 10.1002/dvg.20242. [DOI] [PubMed] [Google Scholar]
- 23.Rommel B, Rogalla P, Jox A, Kalle CV, Kazmierczak B, Wolf J, et al. HMGI-C, a member of the high mobility group family of proteins, is expressed in hematopoietic stem cells and in leukemic cells. Leukemia & lymphoma. 1997;26:603–607. doi: 10.3109/10428199709050896. [DOI] [PubMed] [Google Scholar]
- 24.Zong Y, Huang J, Sankarasharma D, Morikawa T, Fukayama M, Epstein JI, et al. Stromal epigenetic dysregulation is sufficient to initiate mouse prostate cancer via paracrine Wnt signaling. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E3395–3404. doi: 10.1073/pnas.1217982109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watanabe S, Ueda Y, Akaboshi S, Hino Y, Sekita Y, Nakao M. HMGA2 maintains oncogenic RAS-induced epithelial-mesenchymal transition in human pancreatic cancer cells. The American journal of pathology. 2009;174:854–868. doi: 10.2353/ajpath.2009.080523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo Y, Li W, Liao H. HMGA2 induces epithelial-to-mesenchymal transition in human hepatocellular carcinoma cells. Oncology letters. 2013;5:1353–1356. doi: 10.3892/ol.2013.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun M, Song CX, Huang H, Frankenberger CA, Sankarasharma D, Gomes S, et al. HMGA2/TET1/HOXA9 signaling pathway regulates breast cancer growth and metastasis. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:9920–9925. doi: 10.1073/pnas.1305172110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zha L, Wang Z, Tang W, Zhang N, Liao G, Huang Z. Genome-wide analysis of HMGA2 transcription factor binding sites by ChIP on chip in gastric carcinoma cells. Molecular and cellular biochemistry. 2012;364:243–251. doi: 10.1007/s11010-012-1224-z. [DOI] [PubMed] [Google Scholar]
- 29.Gu L, Frommel SC, Oakes CC, Simon R, Grupp K, Gerig CY, et al. BAZ2A (TIP5) is involved in epigenetic alterations in prostate cancer and its overexpression predicts disease recurrence. Nature genetics. 2015;47:22–30. doi: 10.1038/ng.3165. [DOI] [PubMed] [Google Scholar]
- 30.Ferguson M, Henry PA, Currie RA. Histone deacetylase inhibition is associated with transcriptional repression of the Hmga2 gene. Nucleic acids research. 2003;31:3123–3133. doi: 10.1093/nar/gkg403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee S, Jung JW, Park SB, Roh K, Lee SY, Kim JH, et al. Histone deacetylase regulates high mobility group A2-targeting microRNAs in human cord blood-derived multipotent stem cell aging. Cellular and molecular life sciences : CMLS. 2011;68:325–336. doi: 10.1007/s00018-010-0457-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo J, Li M, Tang Y, Laszkowska M, Roeder RG, Gu W. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:2259–2264. doi: 10.1073/pnas.0308762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 34.Lei Q, Jiao J, Xin L, Chang CJ, Wang S, Gao J, et al. NKX3.1 stabilizes p53, inhibits AKT activation, and blocks prostate cancer initiation caused by PTEN loss. Cancer cell. 2006;9:367–378. doi: 10.1016/j.ccr.2006.03.031. [DOI] [PubMed] [Google Scholar]
- 35.Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer cell. 2003;4:209–221. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 36.Mulholland DJ, Xin L, Morim A, Lawson D, Witte O, Wu H. Lin-Sca-1+CD49fhigh stem/progenitors are tumor-initiating cells in the Pten-null prostate cancer model. Cancer research. 2009;69:8555–8562. doi: 10.1158/0008-5472.CAN-08-4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fu M, Rao M, Wang C, Sakamaki T, Wang J, Di Vizio D, et al. Acetylation of androgen receptor enhances coactivator binding and promotes prostate cancer cell growth. Molecular and cellular biology. 2003;23:8563–8575. doi: 10.1128/MCB.23.23.8563-8575.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haelens A, Tanner T, Denayer S, Callewaert L, Claessens F. The hinge region regulates DNA binding, nuclear translocation, and transactivation of the androgen receptor. Cancer research. 2007;67:4514–4523. doi: 10.1158/0008-5472.CAN-06-1701. [DOI] [PubMed] [Google Scholar]
- 39.Gaughan L, Logan IR, Cook S, Neal DE, Robson CN. Tip60 and histone deacetylase 1 regulate androgen receptor activity through changes to the acetylation status of the receptor. The Journal of biological chemistry. 2002;277:25904–25913. doi: 10.1074/jbc.M203423200. [DOI] [PubMed] [Google Scholar]
- 40.Muranen T, Selfors LM, Worster DT, Iwanicki MP, Song L, Morales FC, et al. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer cell. 2012;21:227–239. doi: 10.1016/j.ccr.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, Piris A, et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer discovery. 2014;4:816–827. doi: 10.1158/2159-8290.CD-13-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rathkopf D, Wong BY, Ross RW, Anand A, Tanaka E, Woo MM, et al. A phase I study of oral panobinostat alone and in combination with docetaxel in patients with castration-resistant prostate cancer. Cancer chemotherapy and pharmacology. 2010;66:181–189. doi: 10.1007/s00280-010-1289-x. [DOI] [PubMed] [Google Scholar]
- 43.Molife LR, Attard G, Fong PC, Karavasilis V, Reid AH, Patterson S, et al. Phase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC) Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2010;21:109–113. doi: 10.1093/annonc/mdp270. [DOI] [PubMed] [Google Scholar]
- 44.Bradley D, Rathkopf D, Dunn R, Stadler WM, Liu G, Smith DC, et al. Vorinostat in advanced prostate cancer patients progressing on prior chemotherapy (National Cancer Institute Trial 6862: trial results and interleukin-6 analysis: a study by the Department of Defense Prostate Cancer Clinical Trial Consortium and University of Chicago Phase 2 Consortium. Cancer. 2009;115:5541–5549. doi: 10.1002/cncr.24597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rathkopf DE, Picus J, Hussain A, Ellard S, Chi KN, Nydam T, et al. A phase 2 study of intravenous panobinostat in patients with castration-resistant prostate cancer. Cancer chemotherapy and pharmacology. 2013;72:537–544. doi: 10.1007/s00280-013-2224-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Welsbie DS, Xu J, Chen Y, Borsu L, Scher HI, Rosen N, et al. Histone deacetylases are required for androgen receptor function in hormone-sensitive and castrate-resistant prostate cancer. Cancer research. 2009;69:958–966. doi: 10.1158/0008-5472.CAN-08-2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu X, Gomez-Pinillos A, Liu X, Johnson EM, Ferrari AC. Induction of bicalutamide sensitivity in prostate cancer cells by an epigenetic Puralpha-mediated decrease in androgen receptor levels. The Prostate. 2010;70:179–189. doi: 10.1002/pros.21051. [DOI] [PubMed] [Google Scholar]
- 48.Langelotz C, Schmid P, Jakob C, Heider U, Wernecke KD, Possinger K, et al. Expression of high-mobility-group-protein HMGI-C mRNA in the peripheral blood is an independent poor prognostic indicator for survival in metastatic breast cancer. British journal of cancer. 2003;88:1406–1410. doi: 10.1038/sj.bjc.6600935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Winslow MM, Dayton TL, Verhaak RG, Kim-Kiselak C, Snyder EL, Feldser DM, et al. Suppression of lung adenocarcinoma progression by Nkx2-1. Nature. 2011;473:101–104. doi: 10.1038/nature09881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.