

Abstract
Background
Recessive mutations in PLA2G6 have been associated with different neurodegenerative disorders, including infantile neuroaxonal dystrophy, neurodegeneration with brain iron accumulation and more recently, early-onset dystonia parkinsonism.
Method
Targeted-next generation sequencing using a custom Neurology panel, containing 758 OMIM-listed genes implicated in neurological disorders, was carried out in two index cases from two different Saudi families displaying early-onset levodopa-responsive Parkinsonism with pyramidal signs and additional clinical features. The detected mutations were verified in the index cases and available family members by direct sequencing.
Results and conclusion
We identified a previously described PLA2G6 homozygous p.R741Q mutation in three affected and two asymptomatic individuals from two Saudi families. Our finding reinforces the notion of the broadness of the clinical spectrum of PLA2G6-related neurodegeneration.
Electronic supplementary material
The online version of this article (doi:10.1186/s13104-016-2102-7) contains supplementary material, which is available to authorized users.
Keywords: PLA2G6, Parkinsonism, Saudi patients
Background
Oxidative stress is considered a key pathophysiological mechanism underlying dopaminergic neuronal loss in Parkinson’s disease (PD). Excess iron is one main contributor to oxidative stress that promotes the formation of harmful free radicals via the Fenton reaction. High iron content, in addition to other factors, including the presence of reactive oxygen species (ROS)-generating enzymes and dopamine oxidation, renders dopaminergic neurons of the substantia nigra vulnerable to oxidative stress [1]. Another source of ROS in the brain is the metabolism of arachidonic acid, which is released from membrane phospholipids by the hydrolytic activity of calcium-independent group VI phospholipase A2 (iPLA2-VI) [2].
Recessive mutations in PLA2G6 [MIM 603604], the gene encoding (iPLA2β/iPLA2-VI), have been associated with different neurodegenerative disorders, including infantile neuroaxonal dystrophy (INAD), neurodegeneration with brain iron accumulation (NBIA) and more recently, early-onset dystonia parkinsonism [3]. The involvement of iPLA2-VI in oxidative stress [2], the identification of PLA2G6 mutations in patients with parkinsonian features [3], and the presence of α-synuclein Lewy body pathology in five dystonia-parkinsonism cases with PLA2G6 genetic abnormalities [4] suggest a possible role for this gene in PD pathogenesis.
Utilizing targeted-next generation sequencing (NGS) of 758 OMIM-listed neurological disorders-associated genes, we identified a homozygous p.R741Q mutation previously described in PLA2G6 in three cases with early-onset Parkinsonism [MIM 612953] displaying additional clinical features. Our finding reinforces the notion of the broadness of the clinical spectrum of PLA2G6-related neurodegeneration.
Methods
Subjects
A total of 17 individuals (3 affected and 14 unaffected) from two families originating from the Eastern province of Saudi Arabia were recruited after obtaining written informed consent from all study subjects for participation in the study and for the publication of their genetic and clinical data. This study was approved by the King Faisal Specialist Hospital and Research Center (KFSHRC) IRB, Research Ethics Approval Committee (RAC# 2110035).
Clinical assessment
Standardized clinical investigation was undertaken in all patients by senior neurologists. Neuroimaging examinations were performed using Axial brain fluid attenuated inversion recovery (FLAIR) MRI and F-18-fluorodeoxyglucose positron emitting tomography (18-FDG-PET). Mini Mental State examination (MMSE) and Hoehn & Yahr (H&Y) stage were used to evaluate cognitive function and disease progression, respectively. Clinical features of the patients are summarized in Table 1. For detailed clinical history, see Additional file 1.
Table 1.
Summary of the demographic, clinical and neuroimaging information of the study subjects
| Patient ref no. | Family 66 | Family 97 | ||
|---|---|---|---|---|
| 66-E | 66-K | 66-L | 97-E | |
| Gender/AAE (y) | Female/35 | Male/28 | Male/31 | Female/30 |
| AAO (y) | 26 | 22 | 23 | 25 |
| Initial symptoms | Depression/bradykinesia | Depression/tremor | Bradykinesia | Neuropsychiatric symptoms |
| Bradykinesia | +++ | ++ | +++ | ++ |
| Tremors | Yes | Yes | No | No |
| Postural instability | +++ | ++ | +++ | +++ |
| Dystonia | No | No | No | No |
| Loss of ambulation | 3 y after onset | ambulatory | 3 y after onset | 2 y after onset |
| Pyramidal signs | ++ | + | ++ | + |
| Cerebellar signs | No | No | No | No |
| Levodopa response | Yes -minimal | Yes -minimal | Yes -minimal | Yes -moderate |
| Levodopa-induced dyskinesia | ++ | – | ++ | – |
| Frontotemporal atrophy on MRI | ++ | + | ++ | ++ |
| Iron accumulation | No | No | No | No |
| PET scan | Frontotemporal lobe | Frontotemporal lobe | Not done | Frontotemporal lobe |
| Hoehn & Yahr stage | 4.5 | 4 | 5 | 4 |
| Neuropsychiatric symptoms | ++ | ++ | + | +++ |
| Emotional Liability | +++ | + | + | +++ |
| Cognitive decline | + | ++ | + | + |
| Semantic disorder | + | + | + | – |
| Autonomic symptoms | Yes | Yes | Yes | No |
| Bladder disturbances | Urgency | Urge incontinence | Urgency | – |
| Sweating | Yes | Yes | No | – |
| Flushing | No | Yes | No | – |
| Sleep disorders | Yes | Yes | Yes | Yes |
| REM-sleep behavior disorder | Yes | No | No | – |
| Vivid dreaming | No | Yes | No | Yes |
| Sleep fragmentation | Yes | Yes | Yes | – |
| Death | Alive | Alive | Died after 8 y | Alive |
AAE age at examination; y years; AAO age at onset; REM rapid eye movement; + mild; ++ moderate; +++ severe
Targeted-NGS
Pathogenic mutations in parkinsonism-related genes, such as PARKIN [MIM 602544], PINK1 [MIM 608309], DJ-1 [MIM 602533], SNCA [MIM 163890], LRRK2 [MIM 609007], UCHL1 [MIM 191342], and FBXO7 [MIM 605648], had previously been excluded in all index cases by means of Sanger sequencing before undergoing targeted-NGS [5]. Custom Neurology panel, developed by the Saudi Mendeliome Group as part of the Saudi Human Genome Program [6], was utilized to evaluate peripheral blood DNA from the probands of both families. The panel includes 758 OMIM-listed genes implicated in neurological disorders. Library building, NGS, and bioinformatics analysis was carried out as previously described [6]. Short-listed variants (those that passed the filtering criteria) were validated by Sanger sequencing and subsequently screened in available affected and unaffected family members.
Genotyping
Parents and the index case from each family were genotyped using the Affymetrix Axiom Arrays according to the manufacturer’s recommendations (Affymetrix, Santa Clara, CA 95051, USA), genotypes were called using Genotyping Console™ (GTC version 4.2), and generated files were further integrated for markers flanking the PLA2G6 locus; 72 markers were selected in the haplotype analysis. In addition to the Axiom analysis, conventional genotyping was performed using five previously reported microsatellite markers within chromosome 22 flanking PLA2G6 (D22S426, D22S1045, D22S445, D22S1156, D22S423) [7]. The PCR amplicons were electrophoretically separated on ABI Prism 3100 Genetic Analyzer and the data was analyzed using GeneMapper 5 (Applied Biosystems, Foster City, CA, USA).
Results
Mutation detection and haplotype analysis
Targeted-NGS analysis revealed a previously described PLA2G6 homozygous mutation [c.2222G > A (p.R741Q)] [8] (Fig. 1a) shared by both probands (66-E and 97-E) as well as an affected sibling (66-K) (Fig. 1b, c). The mutation was absent in the 1000 Genomes Project database, in addition to our in-house Saudi human genome database (~1000 controls), and was confirmed by bidirectional Sanger sequencing. We next sought to assess the segregation of this mutation, and, to that end, DNA samples from affected and unaffected family members of both families (FM 66 and FM 97) were screened for the p.R741Q mutation. The mutation appears to segregate with the disease in FM 97 as clinically unaffected members were either heterozygotes or wild-type (Fig. 1c). As for FM 66, the presence of this homozygous mutation in two asymptomatic members (66-D and 66-G) is suggestive of incomplete penetrance (Fig. 1b).
Fig. 1.
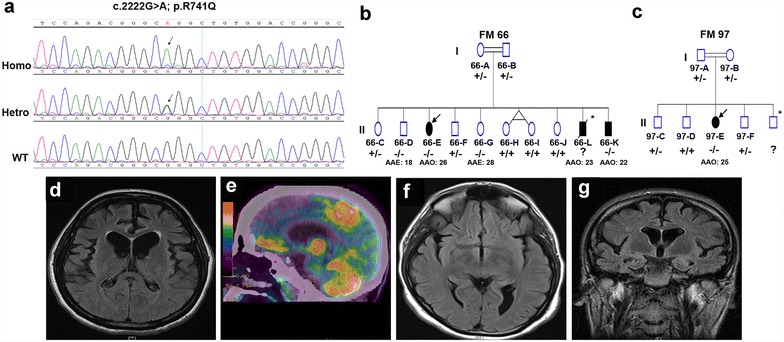
p.R741Q mutation of PLA2G6 detected in two families with early-onset parkinsonism. a Chromatogram of the c.2222G > A mutation. b, c Pedigrees of the two families showing the genotypes of the mutation. b Pedigree of family 66 (FM 66). c Pedigree of family 97 (FM 97). d–g Example radiological imaging of the affected individuals. d, f Axial brain fluid attenuated inversion recovery (FLAIR) MRI sequence of the frontotemporal region of 66-E (d) and 66-K (f) showing moderate (d) and mild (f) atrophy. e 18-FDG-PET scan of 66-E showing moderate decrease in glucose uptake in the frontoparietal regions. g Coronal FLAIR MRI sequence showing moderate frontotemporal atrophy of 97-E. * denotes no DNA sample available. AAE age at examination in years; AAO age at onset in years
Since both families originate from the same geographical area and share a specific mutation, we suspected them to have descended from a common ancestor. This notion was examined by genotyping 72 SNP markers along with five previously described microsatellite markers [7] spanning a 3.3 Mb segment on chromosome 22 harboring the PLA2G6 locus. The analysis revealed a shared haplotype between the father of 66-E and the parents of 97-E spanning a 0.49 Mb segment on chr22q13.1 containing part of PLA2G6. The probands, on the other hand, shared the same haplotype across a larger region (1.4 Mb) (Fig. 2). Overall, the genotyping results suggest that the p.R741Q mutation is likely to be inherited from a common ancestor.
Fig. 2.
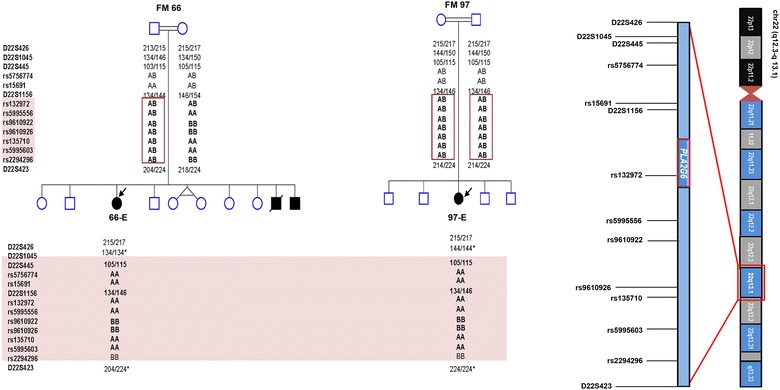
Haplotype analysis of the two families suggests a connection to a common ancestor. The Father of 66-E and the parents of 97-E carry a common haplotype extending 7 markers (top). The probands (66-E and 97-E) from both families share the same haplotype for 11 (SNP/STS) markers (shaded in pink) flanking PLA2G6 on Chr22 including one intragenic (rs132972) (bottom). Schematic representation of Chr22 adapted from UCSC genome browser showing the position of some of the markers included in the analysis (right). * The different genotypes could be due to recombination events
Clinical features of the patients homozygous for p.R741Q mutation
At the age of 26 years, the proband of FM 66 (patient 66-E) presented with bradykinesia, tremors, neuropsychiatric symptoms, and sleep disturbances. A year later, she became almost anarthric, using sign language, and was confined to a wheelchair due to poor balance. She showed marked generalized rigidity affecting axial more than appendicular muscles; however, no dystonia was observed. During her admission, she was started on Levodopa/Carbidopa therapy. This caused a stereotyped, predictable episode of agitation, crying and moaning with semi-rhythmic dyskinetic and dystonic movement-including craniofacial dystonia- with clenching of hands, scratching, and mutilating movements. These movements would start 30–40 min after a Levodopa dose and last for 1.5–2 h. This occurred even with the smallest possible dose of Levodopa, i.e. 50 mg/day. Alternatively, the patient was switched to dopamine agonist therapy in the form of Pramipexole. The dose was gradually increased to 3 mg/day with some improvements in the bradykinesia; her walking improved moderately, but she still required assistance with standing and walking. However, her agitation and emotional lability did not match the symptomatic motor improvement. MRI brain scan showed moderate atrophy in the frontotemporal region (Fig. 1d) with absence of iron deposition in the basal ganglia which was confirmed by T2* sequence. Dopamine transporter single-photon emission computed tomography (DaT-SPECT) scan revealed symmetrical reduced uptake bilaterally in the basal ganglia (image not available) and 18-FDG-PET showed moderate decrease in glucose uptake in the frontoparietal regions (Fig. 1e).
At the age of 22 years, the affected sibling of the proband (patient 66-K) started showing signs of motor dysfunction, including bradykinesia, poor balance, and symmetrical tremors, that led him to have three road traffic accidents in one year. Signs of autonomic dysfunction, sleep disorder, and psychiatric problems were reported. Brain MRI (Fig. 1f) and PET imaging revealed similar but milder findings compared to the proband (66-E). Moderate cognitive deficit was shown on neurological assessment. The patient manages limited daily activities on a combination therapy of dopamine agonist, amantadine, MAO-Inhibitors, and atypical antipsychotic. The proband also has another affected sibling (patient 66-L-deceased) that was a former patient at KFSHRC. See Additional file 1 for detailed clinical history.
The proband from the second family (97-E) was leading a normal life as a full time nurse until the age of 25 when she developed neuropsychiatric symptoms. A few months later, motor symptoms, such as rigidity, bradykinesia and poor balance, were evident. The initial diagnosis was drug-induced Parkinsonism, and, subsequently, she was treated with anticholinergic and electroconvulsive therapy. She became relentlessly dependent, needing two people to help her stand up and walk on her tip-toes. The examination revealed severe Parkinsonism (H&Y stage >4.5) with mild cognitive impairment (MMSE score of 18). Brain MRI showed moderate frontotemporal lobar atrophy (Fig. 1g) with no iron deposition in the basal ganglia and 18-FDG-PET scan showed bilateral frontal and parietal reduction of glucose metabolism. Like in the previous cases (66-E and 66-L), Levodopa therapy triggered adverse emotional–side effects in this patient. She had frequent episodes of loud crying, tearing, clenching of the mouth at times or bringing her head and neck backward, refusal to eat, self-scratching and stereotyped repetitive movements involving the arms and legs. She became more disturbed after Levodopa therapy and developed craniofacial dystonia and, consequently, was started on a combination of second line medication, including Clozapine, Amantadine, Clonazepam and Pramipexole.
Discussion
Here we report on the clinical and neuroimaging findings of patients from two different families presenting with early-onset Parkinsonism in whom the reported PLA2G6 mutation (c.2222G > A; p.R741Q) [8] was detected. The two families, with no reported relationship, originate from the Eastern province of Saudi Arabia; however, haplotype analysis suggests that the mutation may be inherited from a common ancestor (Fig. 2).
The clinical features of our patients overlap with what have been reported in the original cases harboring the p.R741Q mutation [8] with the exception that there was no dystonia reported. Additional features, including sleep and autonomic problems, were also noticed (Table 1). Of note, response to levodopa was limited by the adverse emotional side-effects.
All recruited patients, two of which are siblings (66-E and 66-K), were born to consanguineous parents (Fig. 1b, c). The autosomal recessive transmission of this mutation, as demonstrated by the genotypes and pedigrees, is consistent with the first report of this mutation in a Pakistani/Indian family and with other reports on different PLA2G6 mutations in PARK14-linked Parkinsonism patients from other populations [3, 8]. The parents of 97-E were heterozygous carriers and the siblings were either heterozygotes or wild-type; they all have passed the expected age of disease-onset at the time of examination without showing any symptoms (Fig. 1c) which suggests that the mutation co-segregates with the disease as previously reported [8]. Unlike FM 97, the mutation does not appear to segregate with the disease in FM 66 as not only the affected individuals were homozygous carriers, but also two asymptomatic individuals; one (66-G) above and the other (66-D) under the age of disease-onset reported in the proband (Fig. 1b). Defining the penetrance of PLA2G6 mutations in PARK14-linked Parkinsonism patients is complicated by the age of onset, that can vary widely among patients (8–37 years) and within families [3, 8, 9], which could lead to pseudo-incomplete penetrance. Pseudo-incomplete penetrance is a term used when an inaccurate assumption of non-penetrance has been made due to incomplete clinical examination or absence of symptoms at the time of examination [10]. Moreover, asymptomatic homozygous mutation carriers may exhibit preclinical signs; for instance, Shi and colleagues described a slightly reduced uptake in the right posterior putamen of a homozygous carrier of p.D331Y mutation in PLA2G6 who was clinically unaffected at the time of examination [9]. In this regard, including DAT imaging to the initial clinical assessment and following-up asymptomatic homozygous mutation carriers should warrant accurate penetrance determination.
PLA2G6 is ubiquitously expressed with widespread presence in all areas of mammalian brain [11]. Its product, iPLA2-VI, which catalyzes fatty acids hydrolysis from phospholipids and lysophospholipids, plays a key role in maintaining cell membrane homeostasis. The high lipid content of the brain renders the CNS especially sensitive to lipid metabolism dysregulation. For instance, arachidonic acid, a product of PLA2 activation, contributes to the generation of ROS which indirectly induce cellular lipid peroxidation compromising membrane integrity and fluidity and/or disrupting permeability and ion homeostasis [2]. These alterations may underlie axonal dystrophy and iron accumulation typical of INAD and NBIA, respectively [11]. Similarly, the connection between defective phospholipid metabolism and Parkinsonism was speculated on the basis of iPLA2-VI role in ROS generation and lipid peroxidation. This was supported pathologically by the widespread Lewy body presence that is most pronounced in the neocortex documented in postmortem analysis of patients with PLA2G6 mutations and clinically by the presence of Parkinsonism features [4].
Distinct PLA2G6 mutations give rise to various phenotypes and have different effects on iPLA2-VI catalytic activity. For instance, impaired enzyme catalytic activity was reported in iPLA2-VI proteins extracted from 293FT cells expressing different mutations associated with either INAD or NBIA. However, results on the effect of dystonia-parkinsonism associated mutations were controversial. Engel et al. [12] reported normal catalytic activity of recombinant iPLA2-VI proteins containing missense mutations identified in dystonia-parkinsonism patients, including the p.R741Q substitution reported herein, while another study reported a reduced catalytic activity of the recombinant enzyme containing each of the (novel) mutations identified in Chines Han PD patients [13]. Although p.R741Q and other dystonia-parkinsonism associated mutations were shown not to alter the enzyme catalytic activity, their interference with substrate recognition or other regulatory elements of iPLA2-VI remains possible. Moreover, the discrepant findings could be explained by the fact that different mutations, not only within the same gene but also at the same amino acid residue, could elicit different effects on the protein function. This notion is further illustrated by the differential effects of substituting Arg741 residue with either Trp or Gln on iPLA2-VI catalytic activity and phenotypic outcome [12].
Conclusion
In conclusion, we described a homozygous p.R741Q mutation in PLA2G6 in 3 affected individuals from two families with clinical features resembling dystonia-parkinsonism. Six different homozygous PLA2G6 mutations were previously identified in 11 patients from 6 unrelated consanguineous Saudi families with variable phenotypes. Out of the 11 individuals, 6 presented with INAD, 4 had a phenotype resembling Karak-syndrome, and one presented with atypical NAD [14]. Our patients presented a phenotype different from what was reported by Salih and colleagues [14], despite their matching ethnicity. This emphasizes the clinical heterogeneity of PLA2G6 mutations not only across populations but also within a single population.
Authors’ contributions
NAA conceived and designed the study. SAB and HA contributed with patients recruitment, diagnosis and clinical assessment. DM, SAH, MA, ME and TF carried out the targeted-NGS experiments and analysis. EAA preformed the mutation validation experiments. AEM and DSK preformed genotyping experiments. NAA, AIT, EAA and BRA interpreted the data. BRA and SAB drafted the manuscript. NAA critically reviewed the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We would like to thank all team members at King Abdulaziz City for Science and Technology (KACST) and the Saudi Human Genome project team for their help in conducting the study. In addition we would like to thank sequencing and genotyping core facilities at Genetics Department at the Research Centre, KFSHRC. This work was supported by KACST grant No. 11-BIO1440-20.
Availability of data and materials
The data supporting the results of this article are included within the article and its additional files.
Competing interests
The authors declare that they have no competing interests.
Consent to publish
Written informed consent was obtained from all study subjects for the publication of their genetic and clinical data including images.
Ethics and consent to participate
Written informed consent was obtained from all study subjects for participation in the study and for the publication of their genetic and clinical data. This study was approved by the King Faisal Specialist Hospital and Research Center (KFSHRC) IRB, Research Ethics Approval Committee (RAC# 2110035).
Funding
This work was supported by KACST Grant No. 11-BIO1440-20. The Funding body was not involved in any stage of the study and had no role in the design of the study or collection, analysis and interpretation of data or in writing the manuscript.
Abbreviations
- Arg
arginine
- CNS
central nervous system
- DaT-SPECT
dopamine transporter single-photon emission computed tomography
- 18-FDG-PET
F-18-fluorodeoxyglucose positron emitting tomography
- FLAIR
fluid attenuated inversion recovery
- Gln
glutamine
- H&Y
hoehn & yahr
- INAD
infantile neuroaxonal dystrophy
- iPLA2-VI
calcium-independent group VI phospholipase A2
- IRB
Institutional Review Board
- KFSHRC
King Faisal Specialist Hospital and Research Center
- MAO
monoamine oxidase
- MMSE
mini mental state examination
- MRI
magnetic resonance imaging
- NBIA
neurodegeneration with brain iron accumulation
- NGS
next generation sequencing
- OMIM
online Mendelian inheritance in man
- PD
Parkinson’s disease
- RAC
Research Advisory Council
- ROS
reactive oxygen species
- Trp
tryptophan
Additional file
10.1186/s13104-016-2102-7 Detailed clinical history of the patients.
Contributor Information
Saeed A. Bohlega, Email: boholega@kfshrc.edu.sa
Bashayer R. Al-Mubarak, Email: BAl-Mubarak@kfshrc.edu.sa
Eman A. Alyemni, Email: eman.alyemni@gmail.com
Mohamed Abouelhoda, Email: mabouelhoda@yahoo.com.
Dorota Monies, Email: DMonies@kfshrc.edu.sa.
Abeer E. Mustafa, Email: xxabr5xx@gmail.com
Dania S. Khalil, Email: khalil.dania@gmail.com
Sara Al Haibi, Email: salhaibey@kfshrc.edu.sa.
Hussam Abou Al-Shaar, Email: hal-shaar@alfaisal.edu.
Tariq Faquih, Email: tariqf549@gmail.com.
Mohamed El-Kalioby, Email: mkalioby@mkalioby.com.
Asma I. Tahir, Email: atahir@kfshrc.edu.sa
Nada A. Al Tassan, Email: naltassan@kfshrc.edu.sa
References
- 1.Hwang O. Role of oxidative stress in Parkinson’s disease. Experimental neurobiology. 2013;22(1):11–17. doi: 10.5607/en.2013.22.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adibhatla RM, Hatcher JF. Phospholipase A(2), reactive oxygen species, and lipid peroxidation in CNS pathologies. BMB reports. 2008;41(8):560–567. doi: 10.5483/BMBRep.2008.41.8.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu CS, Lai SC, Wu RM, Weng YH, Huang CL, Chen RS, Chang HC, Wu-Chou YH, Yeh TH. PLA2G6 mutations in PARK14-linked young-onset parkinsonism and sporadic Parkinson’s disease. Am J Med Genet B, Neuropsychiatr Genet. 2012;159B(2):183–191. doi: 10.1002/ajmg.b.32012. [DOI] [PubMed] [Google Scholar]
- 4.Paisan-Ruiz C, Li A, Schneider SA, Holton JL, Johnson R, Kidd D, Chataway J, Bhatia KP, Lees AJ, Hardy J, et al. Widespread Lewy body and tau accumulation in childhood and adult onset dystonia-parkinsonism cases with PLA2G6 mutations. Neurobiol Aging. 2012;33(4):814–823. doi: 10.1016/j.neurobiolaging.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al-Mubarak BR, Bohlega SA, Alkhairallah TS, Magrashi AI, AlTurki MI, Khalil DS, AlAbdulaziz BS, Abou Al-Shaar H, Mustafa AE, Alyemni EA, et al. Parkinson’s Disease in Saudi Patients: A Genetic Study. PLoS ONE. 2015;10(8):e0135950. doi: 10.1371/journal.pone.0135950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saudi Mendeliome Group Comprehensive gene panels provide advantages over clinical exome sequencing for Mendelian diseases. Genome Biol. 2015;16:134. doi: 10.1186/s13059-015-0693-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morgan NV, Westaway SK, Morton JE, Gregory A, Gissen P, Sonek S, Cangul H, Coryell J, Canham N, Nardocci N, et al. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet. 2006;38(7):752–754. doi: 10.1038/ng1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paisan-Ruiz C, Bhatia KP, Li A, Hernandez D, Davis M, Wood NW, Hardy J, Houlden H, Singleton A, Schneider SA. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol. 2009;65(1):19–23. doi: 10.1002/ana.21415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi CH, Tang BS, Wang L, Lv ZY, Wang J, Luo LZ, Shen L, Jiang H, Yan XX, Pan Q, et al. PLA2G6 gene mutation in autosomal recessive early-onset parkinsonism in a Chinese cohort. Neurology. 2011;77(1):75–81. doi: 10.1212/WNL.0b013e318221acd3. [DOI] [PubMed] [Google Scholar]
- 10.Zlotogora J. Penetrance and expressivity in the molecular age. Genet Med. 2003;5(5):347–352. doi: 10.1097/01.GIM.0000086478.87623.69. [DOI] [PubMed] [Google Scholar]
- 11.Levi S, Finazzi D. Neurodegeneration with brain iron accumulation: update on pathogenic mechanisms. Front Pharmacol. 2014;5:99. doi: 10.3389/fphar.2014.00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Engel LA, Jing Z, O’Brien DE, Sun M, Kotzbauer PT. Catalytic function of PLA2G6 is impaired by mutations associated with infantile neuroaxonal dystrophy but not dystonia-parkinsonism. PLoS One. 2010;5(9):e12897. doi: 10.1371/journal.pone.0012897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gui YX, Xu ZP, Wen L, Liu HM, Zhao JJ, Hu XY. Four novel rare mutations of PLA2G6 in Chinese population with Parkinson’s disease. Parkinsonism Relat Disord. 2013;19(1):21–26. doi: 10.1016/j.parkreldis.2012.07.016. [DOI] [PubMed] [Google Scholar]
- 14.Salih MA, Mundwiller E, Khan AO, AlDrees A, Elmalik SA, Hassan HH, Al-Owain M, Alkhalidi HM, Katona I, Kabiraj MM, et al. New findings in a global approach to dissect the whole phenotype of PLA2G6 gene mutations. PLoS One. 2013;8(10):e76831. doi: 10.1371/journal.pone.0076831. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data supporting the results of this article are included within the article and its additional files.