

Abstract
Allogeneic (allo-) hematopoietic stem cell transplant (HSCT) is curative for many nonmalignant pediatric disorders, including hemoglobinopathies, bone marrow failure syndromes, and immunodeficiencies. There is great success using HLA-matched related donors for these patients; however, the use of alternative donors has been associated with increased graft failure, graft versus host disease (GVHD), and transplant-related mortality (TRM). HSCT using alternative donors with post-transplantation cyclophosphamide (PT/Cy) for GVHD prophylaxis has been performed for hematologic malignancies with engraftment, GVHD, and TRM comparable to that seen with HLA-matched related donors. There are limited reports of HSCT in nonmalignant pediatric disorders other than hemoglobinopathies using alternative donors and PT/Cy. We transplanted eleven pediatric patients with life-threatening nonmalignant conditions using reduced intensity conditioning (RIC), alternative donors, and PT/Cy alone or in combination with tacrolimus and mycophenolate mofetil. We observed limited GVHD, no TRM, and successful engraftment sufficient to eliminate manifestations of disease in all patients. Allo-HSCT using alternative donors and PT/Cy shows promise for curing nonmalignant disorders; development of prospective clinical trials to confirm these observations is warranted.
Keywords: Reduced-intensity conditioning, HLA-haploidentical, cyclophosphamide, immunodeficiency, bone marrow failure syndromes
Introduction
Allogeneic (Allo-) hematopoietic stem cell transplant (HSCT) is curative for many nonmalignant disorders, including hemoglobinopathies, bone marrow failure syndromes, and primary immunodeficiencies (PID). Advances in supportive care have greatly improved the success of allo-HSCT for this patient population [1, 2]. However, the use of HSCT is limited by the lack of human leukocyte antigen (HLA)-matched donors; only 50% of children in need of an allo-HSCT have an HLA-matched donor, and this number is less than 20% in some minority populations [3]. For patients with certain nonmalignant disorders, an HLA-matched family member may also be affected, further limiting related donor options. Use of alternative donor sources has historically led to increased risk of acute and chronic graft-versus-host disease (GVHD) and graft rejection. Moreover, transplant-related mortality (TRM) remains high particularly when conventional myeloablative conditioning is used [4]. Many children with nonmalignant disorders have significant comorbidities at the time of HSCT, including infection and active inflammation, which also contribute to poor outcomes. Reduced-intensity conditioning (RIC) strategies have demonstrated improved survival with decreased TRM [5-7]. Accordingly, allo-HSCT for nonmalignant disorders holds promise as a curative therapy if delivered using a platform that minimizes both short-term and long-term toxicities, maximizes engraftment and cure, and expands the donor pool.
In this context, post-transplantation cyclophosphamide (PT/Cy), when administered within a specific time frame after HSCT, selectively depletes the alloreactive donor T cells responsible for GVHD and graft rejection, while preserving non-alloreactive resting memory T cells responsible for adaptive immunity, as well as blood stem cells necessary for successful engraftment [8, 9]. Use of PT/Cy as GVHD prophylaxis, with or without additional immunosuppression, has been used with alternative donor sources in hematologic malignancies, leading to successful engraftment, as well as rates of GVHD, TRM, and graft failure comparable to HLA-matched related donors [10-12], without increased risk of posttransplant lymphoproliferative disorder [13]. Importantly, HSCT using alternative donors and PT/Cy has been performed for hemoglobinopathies, with GVHD and TRM comparable to that seen with HLA-matched related donors [14, 15], but reports using this strategy for other nonmalignant pediatric disorders, such as immunodeficiencies and bone marrow failure syndromes, are limited [16, 17]. Herein, we describe our institutional experience treating eleven pediatric patients with life-threatening, nonmalignant conditions using alternative donor sources, RIC, and PT/Cy for GVHD prophylaxis.
Materials and Methods
Patients
This study was approved by the institutional review board of The Johns Hopkins Hospital. All pediatric patients (ages 1 month to 21 years) who underwent allo-HSCT at The Johns Hopkins Hospital Bloomberg Children’s Center from January 1, 2009 until December 31, 2014 for a nonmalignant condition, and for whom there was no available matched sibling donor, were included. Nonmalignant conditions included primary immunodeficiencies (PID), hemophagocytic lymphohistiocytosis (HLH), bone marrow failure syndromes (BMFS), and disorders of erythrocytes or platelets. Alternative donors included matched unrelated donors (MUD), mismatched unrelated donors (MMUD), or haploidentical related donors (haplo). Data from the medical record including patient demographics, transplant and clinical data, complications, laboratory and radiologic diagnostic studies, therapy received, overall outcomes, and transplant-related complications were abstracted and reviewed.
Preparative Regimens
Preparative regimens used are shown in Figure 1. Patients received RIC with alemtuzumab, fludarabine, and melphalan, with or without the addition of low-dose total body irradiation (TBI) of 200 cGy (Figures 1A and 1B). The exceptions to this conditioning regimen were patients with dyskeratosis congenita (DKC), due to their underlying sensitivity to alkylator chemotherapy, who instead received alemtuzumab, fludarabine, and low-dose TBI (Figure 1C). Alemtuzumab dosing was based on weight, with children >10 kg receiving a total of 48 mg over three days, with a test dose of 3 mg followed by a dose escalation schedule of 10 mg/15 mg/20 mg. Patients <10 kg received a total of 33 mg of alemtuzumab over three days, with a test dose of 3 mg followed by a dosing schedule of 10 mg/10 mg/10 mg. The patients transplanted prior to 2013 received alemtuzumab beginning on day -21. From 2013 and onwards, alemtuzumab was given beginning on day -14, at the same doses, based on improved chimerism data with intermediate dosing of alemtuzumab [18]. All patients received fludarabine 150 mg/m2 divided over five days (or 1 mg/kg/day for five days for patients <10 kg), either starting on day -8 or on day -6. The dosing for melphalan was 140 mg/m2 (or 3.4 mg/kg for patients <10 kg), either given as a single dose on day -2 or divided over two days on days -3 and -2. Low dose total body irradiation (TBI) of 200 cGy was given on day -1.
Figure 1.

Preparatory regimens, including (A) alemtuzumab, fludarabine, melphalan, total body irradiation (TBI); (B) alemtuzumab, fludarabine, and melphalan; (C) alemtuzumab, fludarabine, TBI. Cy = cyclophosphamide; G-CSG = granulocyte colony stimulating factor; MMF = mycophenolate mofetil; TBI = total body irradiation.
There were two exceptions to the above preparative regimens. A patient with immune dysregulation polyendocrinopathy (IPEX) syndrome received alemtuzumab starting on day -21, followed by fludarabine, melphalan, and low-dose TBI dosed as described above, along with cyclophosphamide 14.5 mg/kg/day on days -6 and -5. One of the patients with chronic granulomatous disease (CGD) received alemtuzumab starting on day -21, fludarabine at the same dosing as above, and busulfan 0.8 mg/kg IV q6h on days -5,-4,-3, and the dose was adjusted to achieve an area under the curve (AUC) between 913-1100 micro mol/L*min, based on previously published data for patients with CGD [19].
GVHD prophylaxis
All patients received cyclophosphamide 50 mg/kg/day on days +3 and +4. The patients who received haplo related or MMUDs also received mycophenolate mofetil (MMF) 15mg/kg/dose PO TID, with maximum daily dose 3 gm/d, and tacrolimus 0.015mg/kg/dose IV every 12 hours, both starting on day +5. The MMF continued through day +35. The tacrolimus was transitioned to oral as tolerated by the patient, the dose was adjusted to maintain a trough level between 5 and 15 ng/mL, and was continued through day +180. The two patients with DKC, both of whom had MUD HSCT, only received one dose of cyclophosphamide 50 mg/kg on day +3, due to concern for alkylator sensitivity, combined with MMF and tacrolimus at the dosing described above.
Definitions of Clinical Outcomes
Neutrophil recovery time, or engraftment, was defined as the number of days from HSCT to the first of 3 consecutive days with an absolute neutrophil count above 0.5 × 109/L. Lymphocyte recovery time was defined as the number of days from HSCT to the first of 3 consecutive days with an absolute lymphocyte count above 0.5 × 109/L. Platelet recovery time was defined as platelet count greater than 20 × 109/L without platelet transfusion in the preceding 7 days. Routine donor chimerism analysis was performed on days +30 and +60 after HSCT on peripheral blood. Mixed chimerism was defined as >5% and < 95% donor chimerism, and full chimerism as ≥ 95% donor chimerism, both in whole blood. Primary graft failure was defined as ≤5% donor chimerism in peripheral blood by day +60. Secondary graft failure was defined as loss of donor engraftment (<5% donor chimerism) after achieving neutrophil recovery with detection of donor chimerism in whole blood. Acute GVHD was graded per standard criteria [20], and chronic GVHD was graded per the 2005 National Institutes of Health Working Group Report [21]. Overall survival (OS) was defined as the time from HCST to death from any cause. Event-free survival (EFS) was defined as the time from HSCT to death or re-transplantation. Transplant-related mortality (TRM) was defined as death related to the transplant.
Statistical analysis
Descriptive statistics were used to summarize baseline patient and transplant characteristics. The probability of OS was estimated using the Kaplan-Meier method with 95% confidence intervals (CIs) [22]. Cumulative incidences of relapse, TRM, and GVHD were estimated by competing-risk analysis using Gray’s method [23]. Relapse and TRM were competing risks for each other. Death, relapse, and graft failure were competing risks for GVHD. Data were analyzed with the R program, version 2.12 (R Core Development Team, Vienna, Austria) and Prism version 5.01 (Graphpad software, La Jolla CA).
Results
Patient Characteristics
From Jan 1, 2009 to Dec 31, 2014, eleven patients with nonmalignant conditions underwent allo-HSCT following RIC using alternative donors and PT/Cy for GVHD prophylaxis. Patient characteristics are summarized in Table 1 and 2. The median age at transplant was 8 years, ranging from 4 months to 21 years. Eight patients (73%) were male, and 3 patients (27%) were female. Indications for HSCT included primary immunodeficiencies with personal or family history of life-threatening infections, hemophagocytic lymphohistiocytosis (HLH) relapsing after standard therapy or inherited, bone marrow failure syndrome not responsive to other therapies or associated with life-threatening bleeding, and lymphoproliferative disorder. Four patients (36%) were termed CMV at risk, defined as either the patient and/or the donor being CMV seropositive; of these, one patient was CMV IgG positive and one patient’s CMV status was indeterminate because he was receiving replacement Ig at the time of BMT. No patients had any known active viral infections at the time of transplant.
Table 1.
Patient Characteristics.
| Patient Number | Diagnosis | Gender | Indication for Transplant | Infections pre-HSCT | Age at Transplant | Donor | Conditioning Regimen |
|---|---|---|---|---|---|---|---|
| 1 | IPEX | Male | PID with severe eczema/allergies, enteropathy with TPN dependence, and FTT | 10 months | Haplo related | Alem, Flu, Cy, Mel, TBI | |
| 2 | Birc4/XIAP deficiency | Male | HLH | 11 months | Haplo related | Alem, Flu, Mel, TBI | |
| 3 | DKC | Female | BMF | 19 years | 10/10 MUD | Alem, Flu, TBI | |
| 4 | CGD | Male | PID; brother with life-threatening infections | 5 months | 10/10 MUD | Alem, Flu, Mel | |
| 5 | CGD | Male | PID with life-threatening infections, enteritis, FTT | Klebsiella pneumonia, Paecilomyces pneumonia | 5 years | Haplo related | Alem, Flu, Mel, TBI |
| 6 | DKC | Male | BMF | 14 years | 10/10 MUD | Alem, Flu, TBI | |
| 7 | CGD | Male | PID with life-threatening infections; enteritis, FTT | MRSA bacteremia, fungal pneumonia | 8 years | 10/10 MUD | Alem, Flu, Bu |
| 8 | Hyper IgM | Male | LPD | 12 years | 10/10 MUD | Alem, Flu, Mel, TBI | |
| 9 | DBA | Male | Steroid refractory | 3 years | 10/10 MUD | Alem, Flu, Mel | |
| 10 | HLH | Female | Chemotherapy refractory | 21 years | 5/8 MMUD | Alem, Flu, Mel, TBI | |
| 11 | GT | Female | Platelet-refractory bleeding | 8 years | Haplo related | Alem, Flu, Mel |
Alem, alemtuzumab, BMF, bone marrow failure; Bu, busulfan; CGD, chronic granulomatous disease; Cy, cyclophosphamide, DBA Diamond-Blackfan anemia; DKC, dyskeratosis congenita; Flu, fludarabine; FTT, failure to thrive; GT, Glanzmann’s thrombasthenia; HLH, hemophagocytic lymphohistiocytosis; HSCT, hematopoietic stem cell transplant; IPEX, immune dysregulation polyendocrinopathy X-linked; LPD, lymphoproliferative disease; Mel, melphalan; MMUD mismatched unrelated donor; MRSA, methicillin-resistant staphylococcus aureus; MUD, matched unrelated donor; PID, primary immunodeficiency; TBI, total body irradiation; TPN, total parenteral nutrition.
Table 2.
Patient, Donor, and Allograft Characteristics.
| Characteristic | Number | Percent |
|---|---|---|
|
| ||
| Age at HSCT, years | ||
| Median | 8 | |
| Range | 0.4-21 | |
|
| ||
| Male Recipients | 8 | 73 |
|
| ||
| Graft Source | ||
| Bone Marrow | 11 | 100 |
|
| ||
| Donor Source: | ||
| 10/10 HLA-matched unrelated | 6 | 55 |
| 5/8 Mismatched unrelated | 1 | 9 |
| HLA haploidentical related | 4 | 36 |
|
| ||
| Relationship of haplo donors | ||
| Parent | 3 | 75 |
| Sibling | 1 | 25 |
|
| ||
| GVHD Prophylaxis: | ||
| PT/Cy | 4 | 36 |
| PT/Cy + CNI + MMF | 7 | 64 |
|
| ||
| Donor age, years | ||
| Median | 30 | |
| Range | 19-43 | |
|
| ||
| Male donors | 6 | 55 |
|
| ||
| Female-into-male allografting | 3 | 27 |
|
| ||
| CMV at risk | 4 | 36 |
|
| ||
| Total nucleated cells/kilogram IBW infused | ||
| Median | 5.38 × 108 | |
| Range | 2.06 × 108 − 22.8 × 108 | |
|
| ||
| CD34+ cells/kilogram | ||
| Median | 5.98 × 106 | |
| Range | 2.09 × 106 − 11.0 × 106 | |
|
| ||
| ABO mismatch | ||
| Compatible | 8 | 73 |
| Major mismatch | 3 | 27 |
ABO, blood group type; CMV, cytomegalovirus; CNI, calcineurin inhibitor; GVHD, graft versus host disease; HSCT, hematopoietic stem cell transplant; HLA, human leukocyte antigen; IBW, ideal body weight; MMF, mycophenolate mofetil; PT/Cy, post-transplant cyclophosphamide.
Donor and Graft Characteristics
Donor and graft characteristics are shown in Table 2. The median donor age was 30 years, ranging from 19 to 43 years. The median total nucleated cells per kilogram of recipient ideal body weight was 5.38 ×108, with a range of 2.06 × 108 to 22.8 × 108 total nucleated cells, and the median CD34+ cells per kilogram of recipient weight was 5.98 × 106, ranging from 2.09 ×106 to 11.0 × 106 CD34+ cells. Six patients (55%) received 10/10 HLA-matched unrelated donors, 1 patient (9%) received a 5/8 mismatched unrelated donor, and 4 patients (36%) received haploidentical related donors. The donor source for all patients was unmanipulated bone marrow.
Outcomes
Engraftment/Chimerism
Neutrophil engraftment occurred at a median of 19 days, with a range of 14 to 33 days. Lymphocyte engraftment occurred at a median of 28 days, with a range of 14 to 41 days. Platelet engraftment occurred at a median of 27 days, with a range of 15 to 55 days.
The median follow up time is 25 months, with a range of 12 to 72 months. All patients had evidence of donor chimerism by day 30 post-transplant. All patients have had sustained donor engraftment sufficient to eliminate the manifestations of their underlying diseases. Nine patients (82%) achieved 100% donor chimerism by day 30 measured in peripheral blood. Nine (90%) out of the ten evaluable patients at 1-year post-transplant are full donor chimeras (100% donor) in whole blood off immunosuppression. One of these nine patients, with DKC (patient 3), had stable mixed donor chimerism in the CD3+ cell compartment off immunosuppression; this patient is more than three years post HSCT and has 100% donor chimerism in whole blood and 89% donor chimerism in the CD3+ cell compartment. The one evaluable patient with mixed chimerism is a patient with chronic granulomatous disease (CGD) (patient 4). This patient had stable mixed donor chimerism and was maintained on a calcineurin inhibitor to prevent graft rejection for two years post-HSCT; the patient is now 33 months post HSCT and has 8% and 11% donor chimerism in whole blood and the CD3+ cell compartment respectively. Importantly, the patient has not had any serious infections, indicating ongoing remission from his underlying disease. The patient with immune dysregulation polyendocrinopathy X-linked (IPEX) syndrome (patient 1) had slowly decreasing donor chimerism, ultimately with <5% detectable donor cells in both the whole blood and CD3+ cell compartments. He had no evidence of disease for more than two years, but then developed recrudescence of his enteropathy, asthma, and eczema. No HLA antibodies against his donor were detectable. Therefore, at 37 months post-HSCT, he was retransplanted using a myeloablative preparative regimen (busulfan 32 mg/m2 IV Q6 hours on days -7, -6, -5, and -4 and cyclophosphamide 50mg/kg daily on days -3 and -2), the same haploidentical related donor, and GVHD prophylaxis of PT/Cy, MMF, and tacrolimus at the dosing described above; the patient is now 20 months post the myeloablative HSCT and has full donor chimerism in both whole blood and CD3+ cell compartments, with the desired result of no signs or symptoms of IPEX.
Graft-versus-host disease
One patient (patient 10) developed grade 2 (skin-only) acute GVHD on day +21, which resolved with systemic steroids. Two patients (patients 4 and 7) developed grade 1 (skin-only) acute GVHD on days +31 and +105. Both patients were treated successfully with either cyclosporine and steroids (patient 4) and/or systemic and topical steroids (patient 7). Both of these patients had received PT/Cy only for GVHD prophylaxis, so GVHD treatment was initiated for grade 1 skin-only in order to prevent progression to more severe GVHD. Patient 7 later developed chronic GVHD on day 179, initially treated with cyclosporine, and then with psoralen and ultraviolet A light therapy (PUVA). The patient is currently off chronic GVHD treatment without any evidence of GVHD. The patients who developed GVHD received either MUD transplants with PT/Cy alone (n=2) or MMUD transplants with PT/Cy, MMF, and tacrolimus (n=1) for GVHD prophylaxis.
Survival
With a median follow-up of 25 months, ranging 12 to 72 months, overall survival is 100%, and event-free survival is 91%.
Transplant-related complications
Two patients developed veno-occlusive disease (VOD) (patients 2 and 4), as defined by the Baltimore criteria [24]. Both of these patients’ preparative regimens included melphalan. Prior to transplant, patient 2 had a history of elevated ferritin, peaking at 15,308 ng/mL, and alanine transaminase (ALT) and aspartate transaminase (AST) elevated at 3-5x the upper limits of normal (ULN), in the context of an HLH flair. He developed VOD consisting of respiratory failure requiring intubation, creatinine peaking at three times his baseline, bilirubin peaking at seven times his baseline, and AST and ALT elevations at 7-11x ULN. Patient 4, an infant with CGD, had no known pre-existing liver disease; after development of VOD he required supplemental oxygen, had a doubling of his baseline creatinine, and developed bilirubin, AST, and ALT elevations. Both patients were treated with defibrotide, and both had complete resolution of VOD.
No patients developed hemorrhagic cystitis. There were no transplant-related mortalities.
Hospital Admissions
The median time of hospitalization, including the preparative regimen, through day +100, was 37 days (range 33 to 105 days). Nine patients had fewer than 40 total hospital days. In the first 100 days following HSCT, there were a total of four readmissions in three patients. All four admissions were for fever; in three no source was identified, and one was secondary to primary EBV infection (see below).
Infections
Three patients had bacteremia with coagulase negative staphylococcus on days -4, 0 and +14 (patients 1, 6, and 3). There were no other bacterial infections. One patient (patient 9) developed primary EBV infection consisting of viremia, pharyngitis, hepatosplenomegaly, and lymphadenopathy on day +83, and was successfully treated with rituximab and ganciclovir. There were no occurrences of CMV reactivation or CMV disease, and there were no episodes of fungemia or invasive fungal infections.
Organ Toxicity
There has been no evidence of long-term liver or renal toxicity in any of the patients. We did observe a rise in the median values of creatinine, total bilirubin, aspartate transaminase (AST), and alanine transaminase (ALT) at +20 and +100 days post-transplant, which normalized by 6 months and 1 year post-transplant (Figure 2A-D). Day +20 was the median time to neutrophil engraftment, and was chosen as a time point to capture early toxicities from the conditioning regimen and from PT/Cy.
Figure 2.
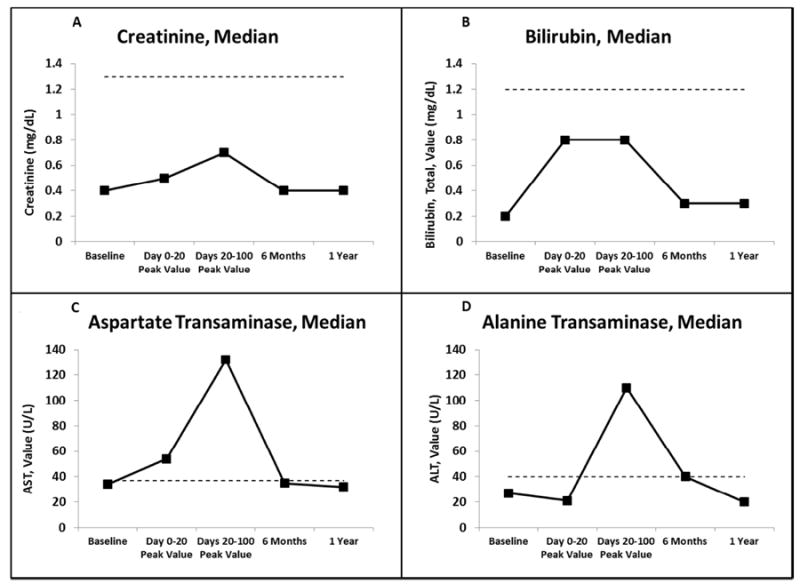
Median laboratory values at specified time points. Dotted line represents upper limit of normal. (A) creatinine (mg/dL); (B) bilirubin, total (mg/dL); (C) aspartate transaminase (AST) (U/L); (D) alanine transaminase (ALT) (U/L).
Discussion
This is the first report of pediatric patients with a variety of nonmalignant disorders treated with HSCT using RIC, alternative donors, and PT/Cy. With a median of 25 months of follow-up, overall survival is 100%, and all patients have had sustained donor engraftment sufficient to eliminate manifestations of their underlying diseases. With small numbers, we had low rates of acute and chronic GVHD, and all were successfully treated without any associated complications. There were no TRMs. The infectious complication rate was quite low, with just three cases of coagulase negative staphylococcus bacteremia, one case of primary EBV, no cases of CMV reactivation or disease, and no invasive fungal infections. Two patients developed severe VOD, both of whom were successfully treated with defibrotide. No patients developed hemorrhagic cystitis.
Engraftment after alternative donor HSCT, particularly with umbilical cord blood transplant (UCBT), has been a challenge, even with myeloablative regimens, due to factors such as alloimmunization from frequent transfusions and the presence of a chemotherapy-naive host [25-29]. Early experience with the use of umbilical cord blood (UCB) transplant following the fludarabine/melphalan/alemtuzumab conditioning in patients with PIDs led to a high number of rejections. This experience is borne out by CIBMTR data. Between 2008 and 2010, of the 56 patients who underwent UCBT for HLH (2/3 myeloablative, 1/3 RIC), 13 had primary and 7 had secondary graft failure, for an overall graft failure rate of 36%. One-year survival was sub-optimal, at 67%. Patients with PID undergoing UCB procedures during these three years rejected 35% and 34% of the time with RIC (n= 31) and myeloablative (n=68) approaches, respectively (personal communication with Mary Eapen, CIBMTR). Recently, a RIC UCBT regimen including hydroxyurea, alemtuzumab, fludarabine, melphalan, and thiotepa, showed better results with 77% OS, 3 out of 22 patients having graft failure, and a high incidence of viral infections [30]. Relative delay in immune reconstitution with cord grafts and difficulty generating cytotoxic T cell therapy from donor cells pose significant challenges in this population with frequent infectious complications. UCB products contain far fewer T cells than are present in collections of adult stem cells. Despite appropriate CD34+ cell numbers per kilogram of body weight after transplantation with double UCB products, immune recovery remains delayed, and infections remain a leading cause of mortality in this group of patients [31-33]. This is further evidence that expanding the donor pool with alternative bone marrow donors, including both mismatched unrelated and related HLA-haploidentical donors, is greatly needed.
In the largest report to date of reduced-intensity conditioning HSCT for patients with nonmalignant diseases, 46% of patients developed mixed whole blood chimerism [4]. The incidence of mixed chimerism in this population was highest in patients who received proximal alemtuzumab (starting day - 12 or earlier) or a distal dose (starting day -23, -22, or -21) of 2.5 mg/kg or greater. In comparison, in our cohort, no patients received proximal alemtuzumab. Of the patients who did receive distal alemtuzumab, dosing was age-based, and two of the patients did receive >2.5 mg/kg. One of these patients is a full donor chimera off immunosuppression, and the other has mixed donor chimerism off immunosuppression, and remains free of his underlying disease. Additionally, Balashov et al. report a cohort of 37 patients with PID transplanted using alternative donors and TCRαβ and CD19 depletion. In this study, there was a 27% graft failure rate [34], compared to our graft failure rate of 9%. Additionally, TCRαβ and CD19 depletion is expensive and requires specialized equipment and personnel, whereas PT/Cy is inexpensive and easily transportable to hospitals with limited access to resources.
We acknowledge the heterogeneity in our patient population as well as in our conditioning regimens. Although heterogeneous in their defects, individuals with nonmalignant disorders share some of the common problems of chronic infections, inflammation, and shortened life expectancy, and therefore the goals of reducing the intensity of the conditioning regimen, reducing rates of GVHD and TRM, and maximizing sustained donor engraftment and cure are the same. Though the patients in this cohort received subtly different conditioning regimens, all were reduced intensity alemtuzumab, fludarabine, and melphalan based, with the exception of one patient who received busulfan instead of melphalan, and the DKC patients, for whom there was an effort to minimize alkylator therapy. Going forward, we have simplified our conditioning regimen to include alemtuzumab, fludarabine, and melphalan, without TBI, given equally high rates of engraftment with or without TBI in our pilot data. It is also possible that we can continue to diminish the intensity of the preparative regimen, for example, by decreasing the dose of melphalan, to further limit unwanted toxicities of HSCT like VOD.
In the future, it will also be important to study the pharmacokinetics and pharmacogenomics of alemtuzumab when given with PT/Cy, with a focus on how this therapeutic combination effects immune reconstitution, as it is possible that the doses of these medications can be better optimized in order to increase cure and decrease unacceptable toxicities.
Several important benefits of PT/Cy are noteworthy. Firstly, PT/Cy is inexpensive, easy to administer, and readily available, making this regimen feasible in locations with less resources. PT/Cy also allows utilization of haploidentical related donors, a novel donor pool for patients with nonmalignant donors in need of HSCT. Given that HLA-matched donors can only be found for 50% of patients (and for less than 20% of African American patients), there is tremendous need to develop novel strategies for all patients, especially racial minorities [3]. Historically, GVHD, particularly in the setting of HLA-disparity, has constrained the applicability and availability of allo-HSCT for malignant and especially nonmalignant diseases. Success with PT/Cy and HLA haploidentical donors in patients with hematologic malignancies has already opened up allo-HSCT to a broader group of patients who may benefit. Finally, limiting posttransplant immunosuppression, specifically by eliminating prolonged calcineurin inhibition (CNI) in the setting of HLA-matched donors, permits reconstitution of the immune system in an environment free of ongoing pharmacologic regulation. As previously described [35], recovery of absolute lymphocyte numbers following PT/Cy was earlier and relatively rapid compared to CNI-based prophylaxis. Rapid immune reconstitution would be predicted to reduce the risk of transplant-related infections, as we observed in our small cohort. There were no CMV reactivations in patients who received PT/Cy, and no TRM. Given two of the three patients that developed low grade GVHD received PT/Cy only, our regimen going forward includes PT/Cy, MMF, and tacrolimus for all patients, regardless of their donor, with earlier cessation of tacrolimus at Day 90, as currently being studied in hematologic malignancy patients (Yvette Kasamon, personal communication) for those with HLA-matched donors. This is significantly shorter than most post-transplant immunosuppressive regimens for nonmalignant disorders [5, 30]. Our continued goal is to successfully cure these patients with no GVHD and minimal short- and long-term morbidity.
In conclusion, in a small patient cohort, we have shown that allo-HSCT using RIC with alternative donors and PT/Cy can be successfully administered to patients with nonmalignant conditions. This is a population with tremendous need for a safe, minimally-toxic, and efficacious alternative donor transplant regimen. We have demonstrated excellent overall survival and engraftment, with low rates of GVHD and other complications. These results are promising, and a prospective trial with larger patient numbers to optimize this regimen is warranted.
Highlights.
Reduced intensity alternative donor BMT can be used to treat nonmalignant disorders.
High-dose cyclophosphamide is a safe and effective GVHD prophylaxis regimen.
Outcomes include no TRM, no serious infections, and successful engraftment.
Acknowledgments
The authors would like to thank the patients and their families, as well as the physicians, advanced practice providers, nurses, care managers, transplantation coordinators, and other providers and staff members in the Pediatric Blood and Marrow Transplantation Program at The Johns Hopkins Hospital Sidney Kimmel Comprehensive Cancer Center who participated in the care of these patients. We also would like to thank Dr. Kenneth R. Cooke for his careful review of the manuscript.
Disclosures
Financial disclosure statement: This work was supported by the Giant Food Children’s Cancer Research Fund.
Footnotes
Conflict of interest statement: There are no conflicts of interest to report.
Data from this study were presented as an oral abstract at the 2015 ASBMT/CIBMTR Tandem meeting.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mateos MK, et al. Transplant-related mortality following allogeneic hematopoeitic stem cell transplantation for pediatric acute lymphoblastic leukemia: 25-year retrospective review. Pediatric Blood & Cancer. 2013;60(9):1520–1527. doi: 10.1002/pbc.24559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brissot E, et al. Improvement of overall survival after allogeneic hematopoietic stem cell transplantation for children and adolescents: a three-decade experience of a single institution. Bone Marrow Transplant. 2015 doi: 10.1038/bmt.2015.250. [DOI] [PubMed] [Google Scholar]
- 3.Gragert L, et al. HLA Match Likelihoods for Hematopoietic Stem-Cell Grafts in the U.S. Registry. New England Journal of Medicine. 2014;371(4):339–348. doi: 10.1056/NEJMsa1311707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marsh RA, et al. Experience with Alemtuzumab, Fludarabine, and Melphalan Reduced- Intensity Conditioning Hematopoietic Cell Transplantation in Patients with Nonmalignant Diseases Reveals Good Outcomes and That the Risk of Mixed Chimerism Depends on Underlying Disease, Stem Cell Source, and Alemtuzumab Regimen. Biology of Blood and Marrow Transplantation. 2015;21(8):1460–1470. doi: 10.1016/j.bbmt.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marsh JC, et al. Alemtuzumab with fludarabine and cyclophosphamide reduces chronic graft- versus-host disease after allogeneic stem cell transplantation for acquired aplastic anemia. Blood. 2011;118(8):2351–2357. doi: 10.1182/blood-2010-12-327536. [DOI] [PubMed] [Google Scholar]
- 6.Marsh RA, et al. Reduced-intensity conditioning significantly improves survival of patients with hemophagocytic lymphohistiocytosis undergoing allogeneic hematopoietic cell transplantation. Blood. 2010;116(26):5824–5831. doi: 10.1182/blood-2010-04-282392. [DOI] [PubMed] [Google Scholar]
- 7.Cooper N, et al. Stem cell transplantation with reduced-intensity conditioning for hemophagocytic lymphohistiocytosis. Blood. 2006;107(3):1233–1236. doi: 10.1182/blood-2005-05-1819. [DOI] [PubMed] [Google Scholar]
- 8.Kanakry CG, et al. Aldehyde Dehydrogenase Expression Drives Human Regulatory T Cell Resistance to Posttransplantation Cyclophosphamide. Science. Translational Medicine. 2013;5(211):211ra157. doi: 10.1126/scitranslmed.3006960. 211ra157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luznik L, Fuchs E. High-dose, post-transplantation cyclophosphamide to promote graft-host tolerance after allogeneic hematopoietic stem cell transplantation. Immunologic Research. 2010;47(1-3):65–77. doi: 10.1007/s12026-009-8139-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luznik L, et al. HLA-Haploidentical Bone Marrow Transplantation for Hematologic Malignancies Using Nonmyeloablative Conditioning and High-Dose, Posttransplantation Cyclophosphamide. Biology of Blood and Marrow Transplantation. 2008;14(6):641–650. doi: 10.1016/j.bbmt.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kanakry CG, et al. Multi-Institutional Study of Post-Transplantation Cyclophosphamide As Single-Agent Graft-Versus-Host Disease Prophylaxis After Allogeneic Bone Marrow Transplantation Using Myeloablative Busulfan and Fludarabine Conditioning. Journal of Clinical Oncology. 2014;32(31):3497–3505. doi: 10.1200/JCO.2013.54.0625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kasamon YL, et al. Nonmyeloablative HLA-Haploidentical Bone Marrow Transplantation with High-Dose Posttransplantation Cyclophosphamide: Effect of HLA Disparity on Outcome. Biology of Blood and Marrow Transplantation. 2010;16(4):482–489. doi: 10.1016/j.bbmt.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanakry JA, et al. Absence of Post-Transplantation Lymphoproliferative Disorder after Allogeneic Blood or Marrow Transplantation Using Post-Transplantation Cyclophosphamide as Graft-versus-Host Disease Prophylaxis. Biology of Blood and Marrow Transplantation. 2013;19(10):1514–1517. doi: 10.1016/j.bbmt.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brodsky RA, et al. Reduced intensity HLA-haploidentical BMT with post transplantation cyclophosphamide in nonmalignant hematologic diseases. Bone Marrow Transplant. 2008;42(8):523–527. doi: 10.1038/bmt.2008.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bolanos-Meade J, et al. HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood. 2012;120(22):4285–4291. doi: 10.1182/blood-2012-07-438408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parta M, et al. Haploidentical Hematopoietic Cell Transplantation with Post-Transplant Cyclophosphamide in a Patient with Chronic Granulomatous Disease and Active Infection: A First Report. Journal of Clinical Immunology. 2015;35(7):675–680. doi: 10.1007/s10875-015-0204-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thakar MS, et al. Cyclophosphamide-Based In Vivo T-Cell Depletion for HLA-Haploidentical Transplantation in Fanconi Anemia. Pediatric Hematology and Oncology. 2012;29(6):568–578. doi: 10.3109/08880018.2012.708708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marsh RA, et al. An Intermediate Alemtuzumab Schedule Reduces the Incidence of Mixed Chimerism Following Reduced-Intensity Conditioning Hematopoietic Cell Transplantation for Hemophagocytic Lymphohistiocytosis. Biology of Blood and Marrow Transplantation. 2013;19(11):1625–1631. doi: 10.1016/j.bbmt.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gungor T, et al. Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. The Lancet. 2014;383(9915):436–448. doi: 10.1016/S0140-6736(13)62069-3. [DOI] [PubMed] [Google Scholar]
- 20.Przepiorka D, et al. 1994 Consensus Conference on Acute GVHD Grading. Bone Marrow Transplant. 1995;6(15):825–828. [PubMed] [Google Scholar]
- 21.Filipovich AH, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. Diagnosis and Staging Working Group Report. Biology of Blood and Marrow Transplantation. 2005;11(12):945–956. doi: 10.1016/j.bbmt.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 22.Kaplan E, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–480. [Google Scholar]
- 23.Gooley TA, et al. Estimation of failure probabilities in the presence of competing risks: new representations of old estimators. Statistics in Medicine. 1999;18(6):695–706. doi: 10.1002/(sici)1097-0258(19990330)18:6<695::aid-sim60>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 24.Jones R, et al. Venoocclusive disease of the liver following bone marrow transplantation. Transplantation. 1987;6(44):778–83. doi: 10.1097/00007890-198712000-00011. [DOI] [PubMed] [Google Scholar]
- 25.Prasad VK, Kurtzberg J. Umbilical cord blood transplantation for non-malignant diseases. Bone Marrow Transplant. 2009;44(10):643–651. doi: 10.1038/bmt.2009.290. [DOI] [PubMed] [Google Scholar]
- 26.Eapen M, et al. Long-Term Survival and Late Deaths after Hematopoietic Cell Transplantation for Primary Immunodeficiency Diseases and Inborn Errors of Metabolism. Biology of Blood and Marrow Transplantation. 2012;18(9):1438–1445. doi: 10.1016/j.bbmt.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gennery AR, Cant AJ. Cord blood stem cell transplantation in primary immune deficiencies. Current Opinion in Allergy & Clinical Immunology. 2007;7(6):528–534. doi: 10.1097/ACI.0b013e3282f1d6b6. [DOI] [PubMed] [Google Scholar]
- 28.Bhattacharya A, et al. Single centre experience of umbilical cord stem cell transplantation for primary immunodeficiency. Bone Marrow Transplant. 2005;36(4):295–299. doi: 10.1038/sj.bmt.1705054. [DOI] [PubMed] [Google Scholar]
- 29.Fernandes JF, et al. Transplantation in patients with SCID: mismatched related stem cells or unrelated cord blood? Blood. 2012;119(12):2949–2955. doi: 10.1182/blood-2011-06-363572. [DOI] [PubMed] [Google Scholar]
- 30.Parikh SH, et al. A Novel Reduced-Intensity Conditioning Regimen for Unrelated Umbilical Cord Blood Transplantation in Children with Nonmalignant Diseases. Biology of Blood and Marrow Transplantation. 2014;20(3):326–336. doi: 10.1016/j.bbmt.2013.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ballen KK, et al. Double Unrelated Reduced-Intensity Umbilical Cord Blood Transplantation in Adults. Biology of Blood and Marrow Transplantation. 2007;13(1):82–89. doi: 10.1016/j.bbmt.2006.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brunstein CG, et al. Umbilical cord blood transplantation after nonmyeloablative conditioning: impact on transplantation outcomes in 110 adults with hematologic disease. Blood. 2007;110(8):3064–3070. doi: 10.1182/blood-2007-04-067215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Komanduri KV, et al. Delayed immune reconstitution after cord blood transplantation is characterized by impaired thymopoiesis and late memory T-cell skewing. Blood. 2007;110(13):4543–4551. doi: 10.1182/blood-2007-05-092130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Balashov D, et al. Single-Center Experience of Unrelated and Haploidentical Stem Cell Transplantation with TCRαβ and CD19 Depletion in Children with Primary Immunodeficiency Syndromes. Biology of Blood and Marrow Transplantation. 2015;21(11):1955–1962. doi: 10.1016/j.bbmt.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 35.Holtick U, et al. OCTET-CY: a phase II study to investigate the efficacy of post-transplant cyclophosphamide as sole graft-versus-host prophylaxis after allogeneic peripheral blood stem cell transplantation. European Journal of Haematology. 2015:n/a–n/a. doi: 10.1111/ejh.12541. [DOI] [PMC free article] [PubMed] [Google Scholar]