

Abstract
We report on a child with the brachytelephalangic type of chondrodysplasia punctata, a very rare form of the disease. At birth, the patient was originally diagnosed with the Conradi-Hunermann type, a more common and severe type. A pediatric radiologist questioned the diagnosis and followed up with the patient, who is now three years old. Based on the clinical and radiographic findings, it was concluded that he had the brachytelephalangic type. This unique case demonstrates the necessity of communication among all health care personnel taking care of the patient. The diagnosis greatly affected the child’s future and education.
Abbreviations: CT, computed tomography; ASD, atrial septal defect
Case report
The patient, a male, was born at 38 weeks to a 33-year-old, G4P3 woman by cesarean section (the mother had previous cesarean sections) after an uncomplicated antenatal course. The mother’s screening tests were negative. The patient’s apgar scores were 8 and 9 at 1 and 5 minutes, respectively. He had midline facial hypoplasia with mild respiratory distress and was admitted to a special care nursery and placed on oxygen, since his saturation levels were in the 80s. A sepsis evaluation was conducted, and IV fluids (along with ampicillin and gentamicin) were administered. Cultures were found to be negative. The patient went on to undergo a direct laryngoscopy and bronchoscopy (DL&B) that revealed narrowed nasal passages bilaterally. A stent was placed in the right naris. He did not pass the newborn hearing screen.
Multiple radiological studies were performed (Figure 1-1, Figure 1-2, Figure 1-3, Figure 1-4, Figure 1-5). An echocardiogram showed a patent foramen versus small ASD. A renal ultrasound was normal. A CT scan of the head showed a normal brain and a questionable small subdural hematoma along the right side of the tentorium. A 3D CT scan of the face revealed midface hypoplasia with relative frontal bossing and a normal mandible, a mega cisterna magna, and stippled calcifications along the cervical spine and tracheal cartilage. A skeletal survey showed hypoplastic distal phalanges of the hands and punctuate calcifications in the tracheal cartilage, in the periarticular soft tissues, and in the ossification centers of most long bones.
Figure 1-1.
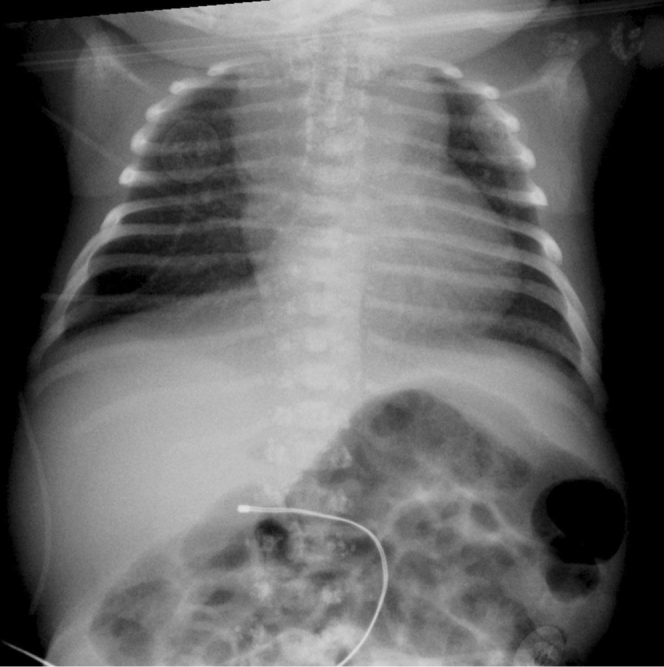
Infant male with the brachytelephalangic type of chondrodysplasia punctata. A chest radiograph at two days of age shows tracheal ring cartilage calcifications and punctuate calcifications of the lateral elements of thoracic vertebrae.
Figure 1-2.
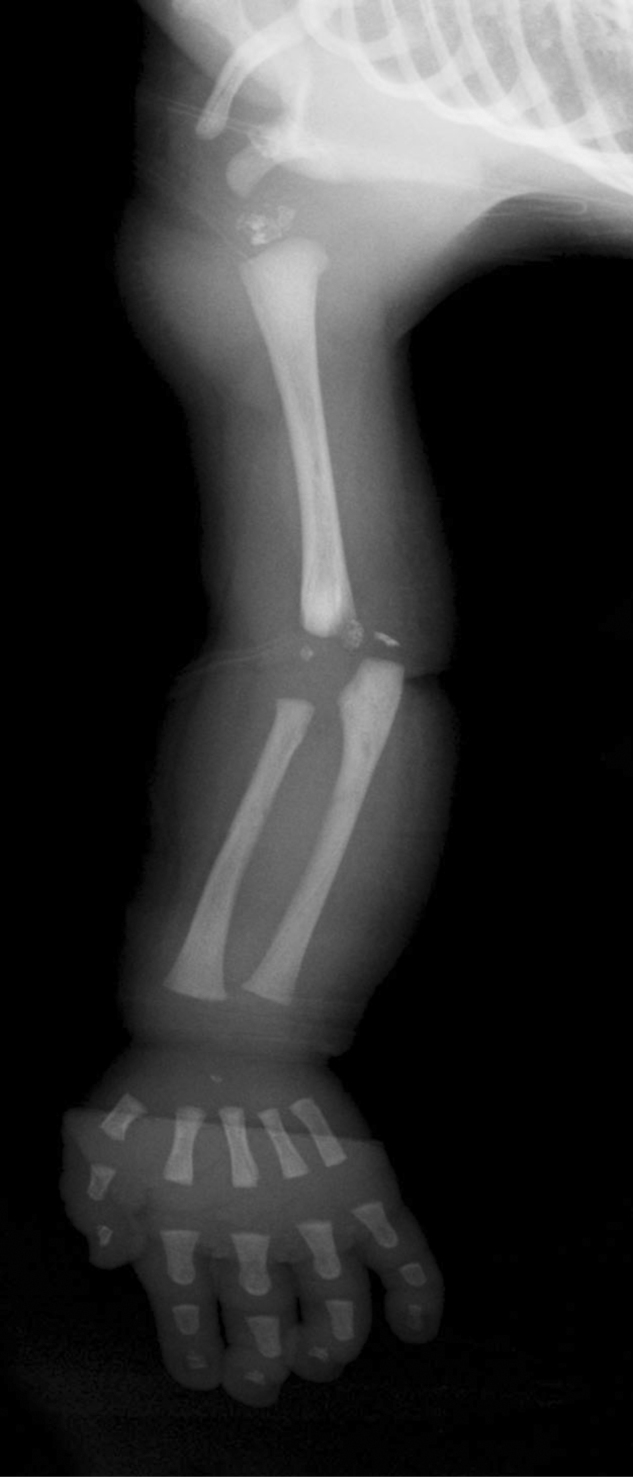
Infant male with the brachytelephalangic type of chondrodysplasia punctata. Examination of the right upper extremity at two days of age shows punctuate calcifications in the proximal ulna and humerus, and short distal phalanges.
Figure 1-3.
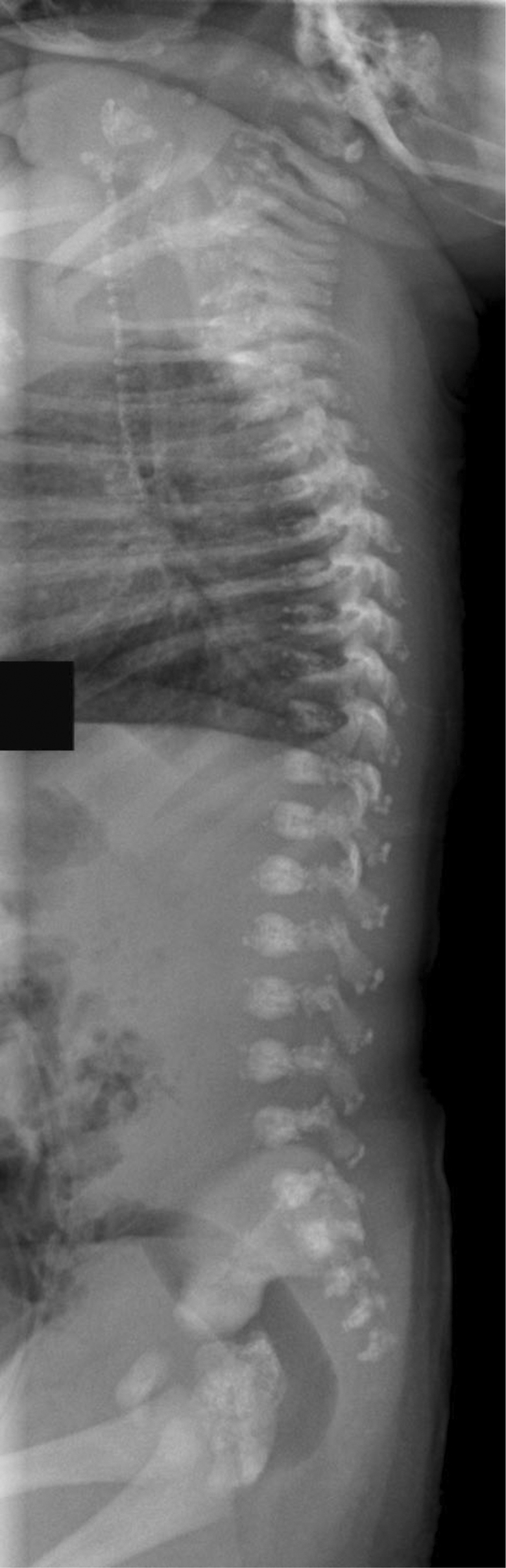
Infant male with the brachytelephalangic type of chondrodysplasia punctata. A lateral view of the spine taken two days after birth shows calcifications in the tracheal cartilage rings, and stippled calcifications in the lateral elements and spinous processes of the vertebrae.
Figure 1-4.
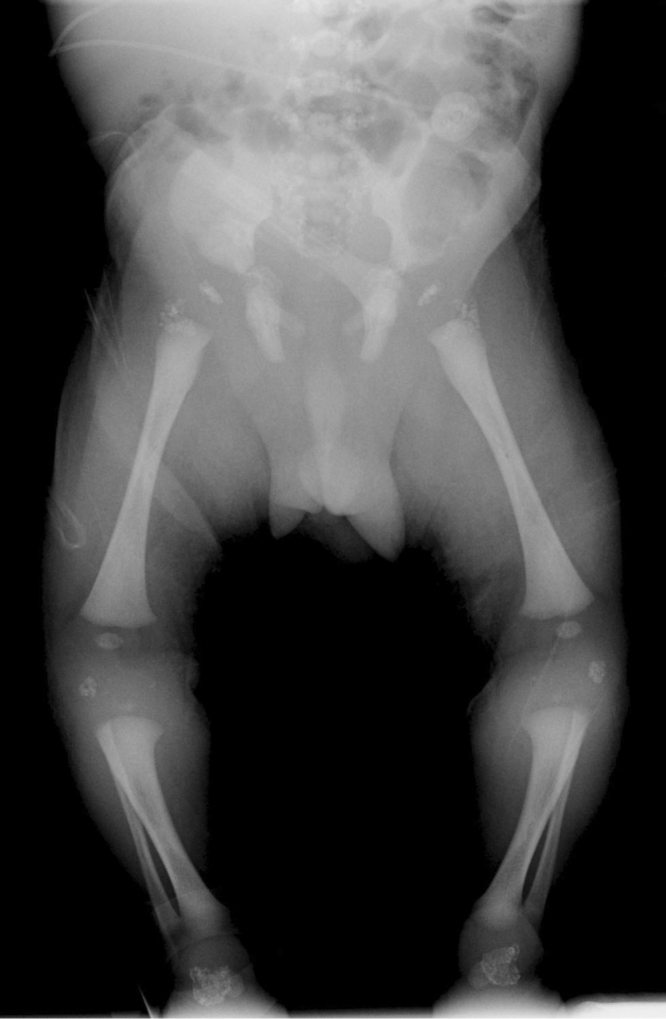
Infant male with the brachytelephalangic type of chondrodysplasia punctata. Examination of the lower extremities at two days of age reveals punctuate calcifications of the proximal femurs, patellae, and taluses.
Figure 1-5.
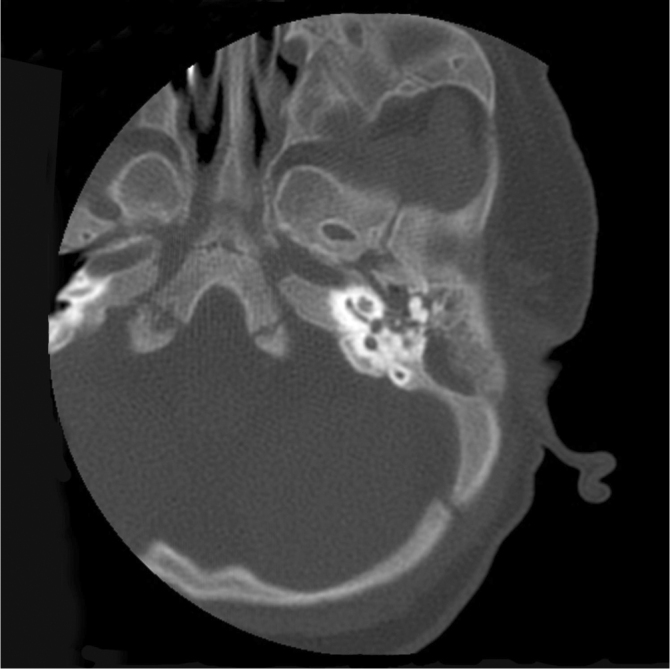
Infant male with the brachytelephalangic type of chondrodysplasia punctata. A CT scan of the temporal bone at four months of age shows, among other findings of midface hypoplasia, sclerosis of the bone surrounding the inner ear but no abnormality of the inner ear structures.
The patient was discharged home one week later, and followup was scheduled with multiple medical specialists. He was diagnosed with Conradi-Hunermann type of chondrodysplasia punctata based on physical and diagnostic exams. Shortly before he left the hospital, the pediatric radiologist who read the skeletal survey went to see the patient and was concerned that the child did not show the skin changes, such as ichthyosis, usually seen with the Conradi-Hunermann type. At this point, the radiologist worried that this might not be the correct diagnosis.
After discharge, the patient did well and was followed by multiple medical subspecialists. The pediatric radiologist who questioned the diagnosis later went on to reassess the case. Some of the punctuate calcifications present shortly after birth were seen at 6 months of age (Fig. 6), but showed near-total resolution by the age of one.
Figure 1-6.
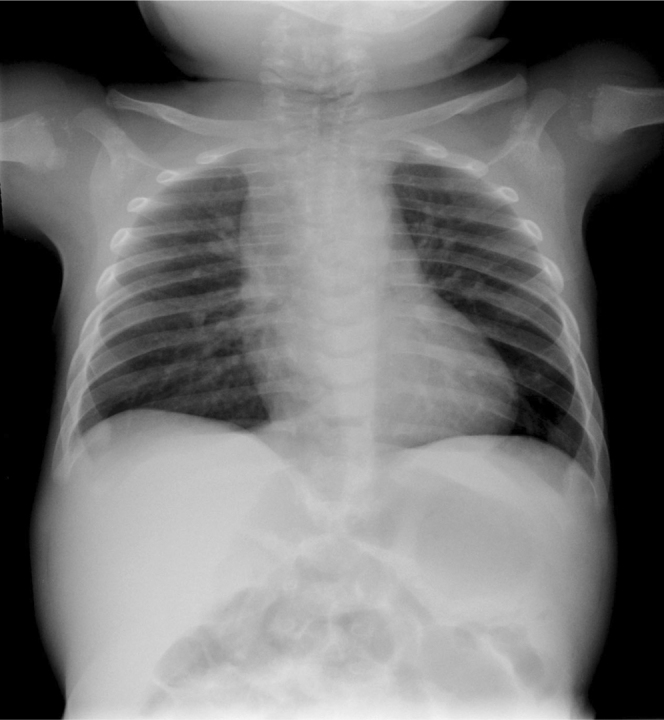
Infant male with the brachytelephalangic type of chondrodysplasia punctata. A chest radiograph at six months of age shows persistent calcifications of the tracheal cartilages and punctuate calcifications of the humeral epiphyses.
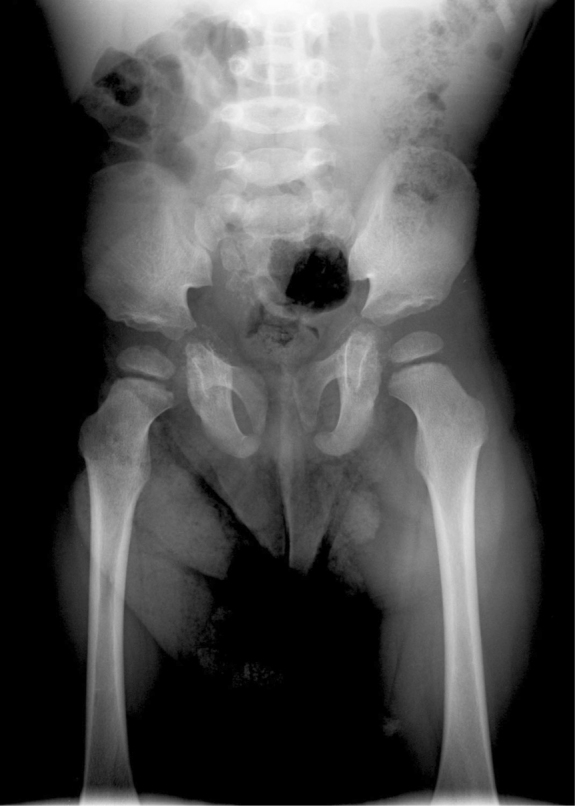
At the age of two, growth was normal, and the only abnormalities were facial dysmorphism, shortness of the distal phalanges of the fingers, decreased acetabular angle, some persistant abnormal calcifications in the innominate bones (Fig. 7), and hearing loss (Fig. 8).
Figure 1-7.
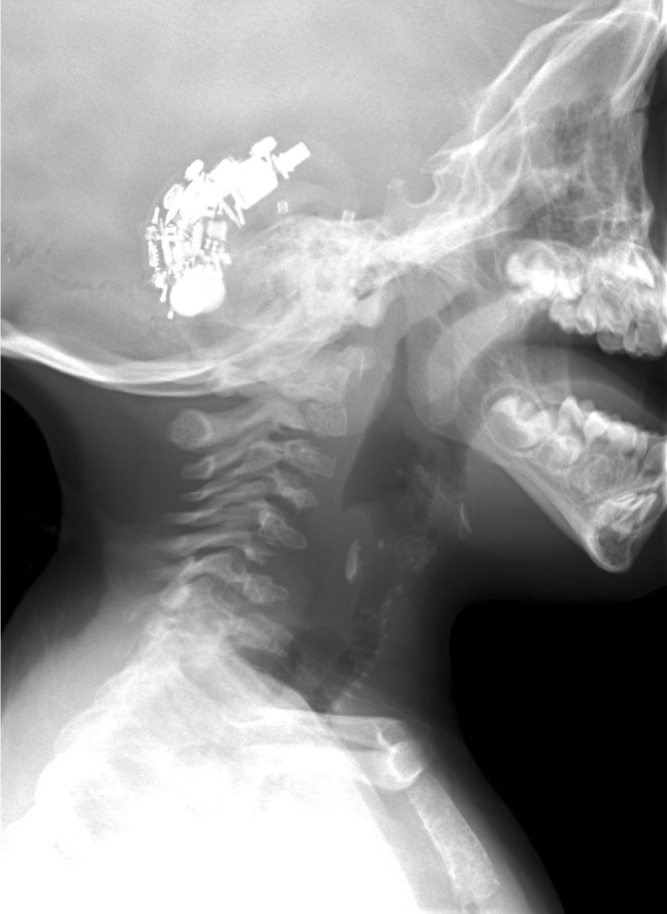
Infant male with the brachytelephalangic type of chondrodysplasia punctata. A view of the lateral nasopharynx at three years of age shows calcifications of the tracheal cartilages and the thyroid cartilage. Hearing aids also appear.
Figure 1-8.
Infant male with the brachytelephalangic type of chondrodysplasia punctata. Examination of the pelvis at two years of age shows a decreased acetabular angle and some persistent abnormal calcifications at the level of the innominate bones.
At present, the patient is three years old. He has met all the appropriate milestones and now displays normal development for his age. The mother states that the child is a very bright and pleasant child. These findings, and the fact that he has had a very mild course of disease, goes along with the characteristics of the brachytelephalangic type.
Discussion
Chondrodysplasia punctata is a heterogeneous group of disorders that includes a number of conditions displaying punctuate calcifications in the cartilage of growing bones. The radiographically detectable abnormality among this group is a congenital defect in calcium deposition of endochondral bones. Subsequently, there is calcific stippling of cartilage and periarticular soft tissues during infancy, leading to skeletal anomalies. Many other nonskeletal problems are also seen. The overall incidence is 1:10,000 live births. Several types have been described, and the more common forms include rhizomelic, autosomal dominant, X-linked dominant, and X-linked recessive (1).
In 1989, Maroteaux described a new form of chondrodysplasia punctata named the brachytelephalangic type. The key to this diagnosis is hypoplasia of the distal phalanges of the fingers. The newborn’s distal phalanges have a triangular appearance with a proximal apex. The child has short phalanges with slightly irregular metaphyses and relatively larger epiphyses. Other manifestations include facial dysmorphism similar to that found in Binder’s maxillofacial dyostosis phenotype, no asymmetry of the limbs, and hypotonia. Patients can have a small tracheal opening with an anterior larynx that may lead to airway problems. The punctuate calcifications (stippling) are seen in newborns, with disappearance in infancy and childhood (2).
One hypothesis is that this form, which is observed in males, is due to an isolated mutation of the Xp localized gene. In Maroteaux’s paper, he discusses four clinical cases of this type. The similarities among the cases included no asymmetry of the limbs, moderate growth disturbance, facial dysmporphism, and phalangeal hypoplasia. There were localized calcifications at birth, and there was a good prognosis for these cases.
Although most patients have been reported to have a benign course, this may not always be the case, mainly due to severe cervical spine abnormalities that can occur. Herman et al. report on two patients with phenotypic features of brachytelephalangic chondrodysplasia punctata who had severe cervical spine stenosis secondary to dysplastic cervical vertebrae (3).
Other authors have reported on patients with the brachytelephalangic type. Curry et al. and Wulfsberg et al. reported similar cases, but their patients also had ichthyosis and mental retardation. The genetics involved deletion of terminal Xp in the Curry et al. patients, and an XY translocation in one case of Wulfsberg et al. Petit et al. described two male patients who had no skin abnormalities but who had optic nerve hypoplasia and delayed psychomotor development. Their genetics involved interstitial deletion in Xp22.3. Ballabio et al. demonstrated a case in which the patient also had optic nerve hypoplasia and delayed psychomotor development. The genetics involved an XY translocation. Toriello et al. presented a unique case involving a brother and sister. In addition to the brachytelephalangic characteristics, they had ocular colobomata, developmental delay, and normal chromosomes. It was thought that the inheritance was autosomal recessive, making it different from the other reported cases. Another case of a female with brachytelephalangic chondrodysplasia punctata was reported by Peter et al. and brought into question the genetic heterogeneity of this syndrome (4, 5).
The differential diagnosis also includes the other types of chrondrodysplasia punctata, the top of the list being the Sheffield type. Sheffield et al. presented 23 cases, some presenting with the phalangeal abnormality and similar facial dysmorphisms. The majority of the cases were males. Other manifestations include stippling replacing ossification of the calcaneus in infancy, sacral and coccygeal stippling, laryngeal calcification, and sagittal clefts of the vertebrae (6).
Warfarin embryopathy is another condition that manifests in a similar manner as the brachytelephalangic type of chondrodysplasia punctata. The two most common anomalies in this disorder are nasal hypoplasia and chondroplasia punctata. A depressed nasal bridge and diffuse bone stippling appear on X-ray, with calcifications in the axial skeleton, vertebrae, and epiphyses of long bones (7).
This case demonstrates how various forms of chondrodysplasia punctata can be difficult to diagnose due to the extensive heterogeneity of both phenotypical and genetic features. The case also emphasizes the value of combining clinical and radiographic features, and the importance of communication among different specialties. This gives the patient and family a better evaluation of potential growth physically and mentally. Fortunately, in this case, the rare diagnosis for the patient turned out to have a much more promising prognosis.
Footnotes
Published: March 13, 2010
References
- 1.Bennett C, Berry A, Maxwell D. Chondrodysplasia punctata: Another possible X-linked recessive case. Am J Med Genet. 1992;44(6):795–799. doi: 10.1002/ajmg.1320440615. [PubMed] [DOI] [PubMed] [Google Scholar]
- 2.Maroteaux P. Brachytelephalangic chondrodysplasia punctata: Possible X-linked recessive form. Human Genetics. 1989;82(2):167–170. doi: 10.1007/BF00284052. [PubMed] [DOI] [PubMed] [Google Scholar]
- 3.Herman T, Lee B, McAlister W. Brachytelephalangic chondrodysplasia punctata with marked cervical stenosis and cord compression: Report of two cases. Pediatric Radiology. 2002;32(6):452–456. doi: 10.1007/s00247-001-0638-7. [PubMed] [DOI] [PubMed] [Google Scholar]
- 4.Zizka J, Charvat A, Balicek P. Brachytelephalangic chondrodysplasia punctata with distinctive phenotype and normal karytope. Am J Med Genet. 1998;76(3):213–216. doi: 10.1002/(sici)1096-8628(19980319)76:3<213::aid-ajmg3>3.0.co;2-r. [PubMed] [DOI] [PubMed] [Google Scholar]
- 5.Petit C, Melki J, Levilliers J. An interstitial deletion in Xp22.3 in a family with X-linked recessive chondrodysplasia punctata and short stature. Human Genetics. 1990;85(2):247–250. doi: 10.1007/BF00193206. [PubMed] [DOI] [PubMed] [Google Scholar]
- 6.Sheffield L, Danks D, Mayne V. Chondrodysplasia punctata—23 cases of a mild and relatively common variety. J Pediat. 1976;89(6):916–923. doi: 10.1016/s0022-3476(76)80596-3. [PubMed] [DOI] [PubMed] [Google Scholar]
- 7.Finkelstein Y, Chiayat D, Schechter T. Warfarin embryopathy following low-dose maternal exposure. J Obst Gyne Can. 2005;27(7):702–706. doi: 10.1016/s1701-2163(16)30550-3. [PubMed] [DOI] [PubMed] [Google Scholar]