

Abstract
Inorganic colloidal nanoparticles (NPs) stabilized by a layer of hydrophobic surfactant on their surfaces have poor solubility in the aqueous phase, thus limiting their application as biosensors under physiological conditions. Here we report a simple model to ionize various types of hydrophobic colloidal NPs, including FePt, cubic Fe3O4, Pd, CdSe, and NaYF4 (Yb 30%, Er 2%, Nd 1%) NPs, to multicharged (positive and negative) NPs via ligand exchange. Surfaces of neutral hydrophobic NPs were converted to multicharged ions, thus making them soluble in water. Furthermore, peroxidase-like activity was observed for ionic FePt, Fe3O4, Pd, and CdSe NPs, of which FePt and CdSe catalyzed the oxidation of the colorless substrate 3,3′,5,5′-tetramethylbenzidine (TMB) to the blue-colored product in the absence of H2O2, while Pd and Fe3O4 catalyzed the oxidization of TMB in the presence of H2O2. With the benefit of the ionic functionalization protocols described herein, colloidal NPs should gain wider use as biomarkers, nanozymes, and biosensors.
Introduction
Colloidal NPs with excellent physical and chemical properties have been employed in applications ranging from solar cells1 to light-emitting diodes2 and photocatalysts.3 As biosensors, colloidal nanoparticles, including quantum dots,4 gold nanoparticles,5 upconversion nanoparticles,6 and alloyed plasmonic nanoparticles,7 have attracted intense interest because of their unique optical properties. However, despite success in the synthesis of hydrophobic colloidal NPs,8−11 the development of reproducible high-quality nanocrystal biosensors remains challenging.12 To stabilize the colloidal NPs in organic media at high temperature, a layer of hydrophobic surfactant is often needed,8 thus resulting in poor solubility in aqueous phases.
To improve the application of hydrophobic colloidal NPs in biochemical and biomedical research, most current methods use ligand exchange13 or amphiphilic ligand encapsulation14 to transfer the hydrophobic colloidal NPs to an aqueous phase. Typically, ligand exchange has been conducted in a two-phase system with hydrophobic colloidal NPs in a nonpolar phase and the reacting ligands in a polar phase, but this leads to serious aggregation and poor solubility. Amphiphilic ligand encapsulation has been performed at high temperature and requires extra washing steps, thus leading to low transfer efficiency and poor long-term stability. Although molecular metal chalcogenide complexes provide stable hydrophilic NPs,15 their biocompatibility and toxicity are concerns in biosensor applications.
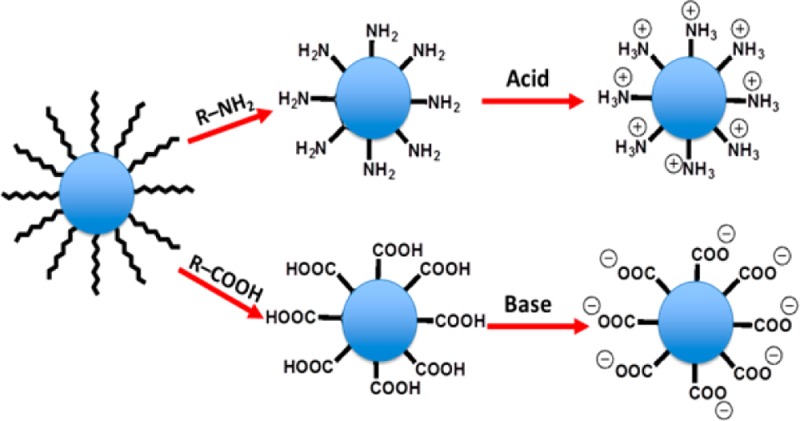
In previous work, we described a facile method to transfer hydrophobic magnetic nanoparticles to hydrophilic phase.16 However, that method could not be generalized to other hydrophobic nanocrystals, such as quantum dots. To solve these solubility and cytotoxicity problems, we have designed a simple model system to transfer hydrophobic colloidal NPs to an aqueous phase via ligand exchange and ionization. The process is conducted in a single phase (THF solvent) with gentle heating. Four different compounds, including 4-aminothiophenol (4-ATP), 4-mercaptobenzoic acid (4-MCBA), dopamine, and 3,4-dihydroxyhydrocinnamic acid (3,4-DHCA), were all tested in separate experiments to replace the long hydrocarbon chain ligands on colloidal NP surfaces, as shown in Figure 1a,b. After ligand exchange, aqueous HCl was added to the ligand exchange solutions containing either 4-ATP or dopamine to protonate the −NH2 groups. Likewise, aqueous NaOH was added to ligand exchange solutions containing either 4-MCBA or 3,4-DHCA to deprotonate the −COOH groups. Thus, the neutral hydrophobic NP surfaces were converted to multicharged ions, called ionic NPs (INPs), which were collected by centrifugation and redispersed in water.
Figure 1.

Ionization of hydrophobic colloidal nanoparticles. (a) Schematic representation of ionization of alloy metal (FePt) and metal oxide (Fe3O4) NPs with dopamine and 3,4-DHCA. (b) Schematic representation of ionization of noble metal (Pd) NPs and quantum dots (CdSe) with 4-ATP and 4-MCBA. TEM images of (c) ionic FePt, (g) cubic Fe3O4, (k) Pd, and (o) CdSe nanoparticles with 3,4-DHCA and 4-MCBA in water, respectively. TEM images of (d) FePt, (h) cubic Fe3O4, (l) Pd, and (p) CdSe nanoparticles in hexane. TEM images of (e) ionic FePt, (i) cubic Fe3O4, (m) Pd, and (q) CdSe nanoparticles with dopamine and 4-ATP in water, respectively. Corresponding photographic images of (f) FePt, (j) cubic Fe3O4, (n) Pd, and (r) CdSe nanoparticles in hexane and water after ligand exchange and ionization. Scale bar: FePt, Pd, and CdSe are 50 nm. Cubic Fe3O4 is 100 nm.
Compared with previously reported phase transfer methods of hydrophobic nanoparticles, the method we developed here has the following advantages: (1) Unlike traditional ligand exchange, in which nanoparticles are dissolved in nonpolar solvents and replacement ligands are dissolved in polar solvents to form a two-phase ligand exchange environment, tetrahydrofuran as solvent can dissolve both hydrophobic nanoparticles and the replacement ligands to provide a single-phase environment to facilitate ligand exchange. (2) Ionization of nanoparticles by adding a small amount of NaOH or HCl can precipitate the nanoparticles immediately in tetrahydrofuran, and the precipitated nanoparticles can be easily dispersed in water without any further surface modification, such as adding PEG. (3) For each kind of NP, two replacement ligands with the same binding groups but different terminal groups were selected for ligand exchange. After ligand exchange, each kind of NP can be ionized using either NaOH or HCl to form negatively or positively charged hydrophilic NPs, respectively. The ionized NPs, whether positively or negatively charged, can be dispersed in water very well because of their mutual electronic repulsion.
Experimental Section
Synthesis of Hydrophobic Colloidal Nanoparticles
FePt Alloy Nanoparticles
These NPs were synthesized in a manner similar to previously reported procedures.9 In a typical synthesis of FePt, platinum acetylacetonate (0.5 mmol), 1,2-hexadecanediol (1.5 mmol), and dioctyl ether (20 mL) were mixed and degassed three times before heating to 100 °C. Oleic acid (0.16 mL), oleylamine (0.17 mL), and Fe(CO)5 (0.13 mL) were added. Then the mixture was heated to 300 °C and refluxed for 30 min under argon flow. After reflux, the reaction system was rapidly cooled to room temperature. FePt nanoparticles were washed with ethanol and hexane and finally redispersed in tetrahydrofuran (THF).
Fe3O4 Nanocubes
Iron oleate precursor was first synthesized using previously reported procedures.10 Typically, 10.8 g of iron chloride (FeCl3·6H20, 40 mmol) and 36.5 g of sodium oleate (120 mmol) were dissolved in a solvent mixture composed of 80 mL of ethanol, 60 mL of distilled water, and 140 mL of hexane. The resulting solution was heated to 60 °C and refluxed for 4 h. When the reaction was finished and cooled to room temperature, the upper organic layer containing the iron oleate complex was washed three times with distilled water using a separatory funnel. After removal of hexane, the resulting iron oleate complex was in a waxy solid form. To synthesize Fe3O4 nanocubes,17 iron oleate (0.9 g, 1 mmol) and sodium oleate (0.32 g, 1.05 mmol) were added to a three-necked flask (25 mL) with a solvent of 1-octadecene (5 g). The reaction mixture was heated to 200 °C and maintained for 1 h, followed by heating the mixture to 320 °C. After 40 min, the reaction solution was quickly cooled to room temperature by blowing air across the reaction flask. The resulting Fe3O4 nanocubes were washed with hexane and acetone/ethanol. After purification the product was dispersed in THF.
Pd Nanoparticles
These NPs were synthesized on the basis of reported procedures.18 In a typical synthesis, Pd(acac)2 (0.1 g) was added to 1 mL of trioctylphosphine to form an orange solution. Then 10 mL of oleylamine was introduced and the mixture was degassed for 10 min. The resulting solution was slowly heated to 250 °C (5 °C/min). After 30 min, the reaction system was cooled quickly, washed with ethanol and hexane, and finally redispersed in THF.
CdSe Semiconductor Nanoparticles
These NPs were synthesized using a procedure similar to the previously published method.19 Precursor cadmium myristate was first prepared using the following procedure: To a solution of sodium myristate in methanol (0.025 M, 240 mL) was added cadmium nitrate in methanol (0.05 M, 40 mL) dropwise to form a white precipitate, which was washed twice with methanol and dried under vacuum overnight. To synthesize CdSe nanocrystals, selenium powder (0.05 mmol) and cadmium myristate (0.1 mmol) were added to a flask with 1-octadecene (5.0 g). The mixture was degassed for 30 min under vacuum at room temperature. Then the solution was heated to 240 °C and maintained for 20 min. The CdSe nanoparticles were washed with ethanol and toluene and redispersed in tetrahydrofuran (THF).
Upconversion Nanoparticles
NaYF4 (Yb 30%, Er 2%, Nd 1%) NPs were synthesized in a manner similar to previously reported procedures.20 Typically, Y(CH3CO2)3 (0.67 mmol), Yb(CH3CO2)3 (0.3 mmol), Er(CH3CO2)3 (0.02 mmol), and Nd(CH3CO2)3 (0.01 mmol) were added to a 50 mL flask containing oleic acid (7.5 mL) and 1-octadecene (17.5 mL). The resulting mixture was heated to 150 °C, maintained there for 0.5 h, and then cooled to room temperature. Subsequently, a methanol solution (6 mL) containing NH4F (4 mmol) and NaOH (2.5 mmol) was added and stirred at 50 °C for 0.5 h. The reaction mixture was then heated to 100 °C to remove the methanol. Finally, the reaction solution was heated to 290 °C and maintained for 2 h under argon flow. The resulting nanoparticles were washed with hexane and ethanol and redispersed in THF.
Ionization of Colloidal Nanoparticles
Ionization of FePt with Dopamine
Dopamine (50 mg) was first dissolved in 300 μL of deionized water, followed by adding 4.7 mL of THF. The mixture was transferred to a 25 mL three-necked flask and heated to 50 °C under argon flow. To the 25 mL three-necked flask were added FePt (15 mg) nanoparticles in THF (2 mL) and the mixture was incubated for 5 h at 50 °C. After incubation, 100 μL of HCl (1 M) was added to the mixture to form a precipitate, which was collected by centrifugation and redispersed in ultrapure Millipore water (18.2 Ω).
Ionization of FePt with 3,4-Dihydroxyhydrocinnamic Acid (3,4-DHCA)
To a 25 mL three-necked flask was added 3,4-dihydroxyhydrocinnamic acid (50 mg) dissolved in 5 mL of THF. The mixture was heated to 50 °C. Then 15 mg of FePt nanoparticles dissolved in 2 mL of THF was added to the flask and the mixture was incubated for 5 h. After incubation, 200 μL of NaOH (0.5 M) was added to the mixture to form a precipitate, which was collected by centrifugation and redispersed in ultrapure Millipore water (18.2 Ω).
Ionization of Pd with 4-Aminothiophenol (4-ATP)
Replacement ligand 4-aminothiophenol (40 mg) was dissolved in 5 mL of THF in a 25 mL three-necked flask and heated to 50 °C. Then 10 mg of Pd nanoparticles dissolved in 2 mL of THF was added to the flask and the mixture was incubated for 5 h. After incubation, 100 μL of HCl (1 M) was added to the mixture to form a precipitate, which was collected by centrifugation and redispersed in ultrapure Millipore water (18.2 Ω).
Ionization of Pd with 4-Mercaptobenzoic Acid (4-MCBA)
Replacement ligand 4-mercaptobenzoic acid (40 mg) was dissolved in 5 mL of THF in a 25 mL three-necked flask and heated to 50 °C. Then, 10 mg of Pd nanoparticles dissolved in 2 mL of THF was added to the flask and the mixture was incubated for 5 h. After incubation, 200 μL of NaOH (0.5 M) was added to the mixture to form a precipitate, which was collected by centrifugation and redispersed in ultrapure Millipore water (18.2 Ω).
Ionization of Fe3O4 Nanocubes with Dopamine
Dopamine (50 mg) was first dissolved in 300 μL of deionized water, followed by adding 4.7 mL of THF. The above mixture was transferred to a 25 mL three-necked flask and heated to 50 °C under argon flow. To the 25 mL three-necked flask were added Fe3O4 nanocubes (15 mg) dissolved in THF (2 mL), and the mixture was incubated for 5 h at 50 °C. After incubation, 100 μL of HCl (1 M) was added to the mixture to form a precipitate, which was collected by centrifugation and redispersed in ultrapure Millipore water (18.2 Ω).
Ionization of Fe3O4 Nanocubes with 3,4-Dihydroxyhydrocinnamic Acid
To a 25 mL three-necked flask was added 3,4-dihydroxyhydrocinnamic acid (50 mg) dissolved in 5 mL of THF. The mixture was heated to 50 °C. Then, 15 mg of Fe3O4 nanocubes dissolved in 2 mL of THF was added to the flask and the mixture was incubated for 5 h. After incubation, 200 μL of NaOH (0.5 M) was added to the mixture to form a precipitate, which was collected by centrifugation and redispersed in ultrapure Millipore water (18.2 Ω).
Ionization of CdSe with 4-Aminothiophenol
Replacement ligand 4-aminothiophenol (40 mg) was dissolved in 5 mL of THF in a 25 mL three-necked flask and heated to 50 °C. Then, 10 mg of CdSe nanoparticles dissolved in 2 mL of THF was added to the flask and the mixture was incubated for 5 h. After incubation, 100 μL of HCl (1 M) was added to the mixture to form a precipitate, which was collected by centrifugation and redispersed in ultrapure Millipore water (18.2 Ω).
Ionization of CdSe with 4-Mercaptobenzoic acid
Replacement ligand 4-mercaptobenzoic acid (40 mg) was dissolved in 5 mL of THF in a 25 mL three-necked flask and heated to 50 °C. Then, 10 mg of CdSe nanoparticles dissolved in 2 mL of THF was added to the flask and the mixture was incubated for 5 h. After incubation, 200 μL of NaOH (0.5 M) was added to the mixture to form a precipitate, which was collected by centrifugation and redispersed in ultrapure Millipore water (18.2 Ω).
Ionization of UCNPs [NaYF4 (Yb 30%, Er 2%, Nd 1%)] with Dopamine
Dopamine (50 mg) was first dissolved in 300 μL of deionized water, followed by adding 4.7 mL of THF. The above mixture was transferred to a 25 mL three-necked flask and heated to 50 °C under argon flow. To the 25 mL three-necked flask were added UCNPs (15 mg) dissolved in THF (2 mL), and the mixture was incubated for 5 h at 50 °C. After incubation, 100 μL of HCl (1 M) was added to the mixture to form a precipitate, which was collected by centrifugation and redispersed in ultrapure Millipore water (18.2 Ω).
Ionization of UCNPs [NaYF4 (Yb 30%, Er 2%, Nd 1%)] with 3,4-Dihydroxyhydrocinnamic Acid
To a 25 mL three-necked flask was added 3,4-dihydroxyhydrocinnamic acid (50 mg) dissolved in 5 mL of THF. The mixture was heated to 50 °C. Then 15 mg of UCNPs dissolved in 2 mL of THF was added to the flask and the mixture was incubated for 5 h. After incubation, 200 μL of NaOH (0.5 M) was added to the mixture to form a precipitate, which was collected by centrifugation and redispersed in ultrapure Millipore water (18.2 Ω).
Transmission Electron Microscopy (TEM)
Imaging was carried out using a Hitachi H-7000 transmission electron microscope at 100 kV. Five microliter samples of colloidal nanocrystals in hexane or water were dropped onto a carbon-coated copper grid (Ted Pella) and then dried for TEM.
ζ-Potential
ζ-Potentials were determined at room temperature using a Zetasizer Nano-ZS (Malvern).
FT-IR Spectra
Spectra were recorded with a near- and mid-IR spectrometer (a Nicolet Nexus 670) in KBr pellets.
Optical Absorption Spectroscopy
UV–vis absorption spectra were recorded using a Shimadzu UV-1800. Nanocrystals were dissolved in hexane or water for measurement.
Photoluminescence Measurement
A Millenia eV laser (second harmonic of Nd:YAG, 532 nm) was used to pump a Spectra-Physics Tsunami femtosecond Ti:sapphire laser with a repetition rate of 80 MHz. The output of the Ti:sapphire laser was tuned to 980 nm, had pulse widths of <100 fs, and had a power of 375 mW. Steady-state emission spectra were collected on a Fluoromax-3 spectrophotometer.
Peroxidase-like Activity Mimic
In a typical test, FePt–3,4-DHCA or FePt–dopamine (5 μL, 3.5 mg/mL) was added to 200 μL of TMB solution (1.5 mM, pH 3) in a quartz cuvette (200 μL). The kinetics of the peroxidase-like activity of FePt was monitored by monitoring the absorbance of oxidized TMB (652 nm) on a Shimadzu UV-1800. The initial velocities were measured by adding 17.5 μg of ionic FePt to 200 μL of standard TMB solution (pH 3) with different concentrations at room temperature.
Results and Discussion
Various colloidal NPs, including alloyed metal, metal oxide, noble metal, and semiconductor nanoparticles, were transferred to the aqueous phase by this ionization process. In our model system, dopamine and 3,4-DHCA were used for metal alloys and metal oxides, respectively, while 4-ATP and 4-MCBA were used for noble metals and quantum dots, respectively. In particular, we selected FePt, cubic Fe3O4, Pd, and CdSe as representative samples of a metal alloy, metal oxide, noble metal, and semiconductor NPs, respectively.
The four replacement ligands fulfill several important requirements. (i) They are soluble in tetrahydrofuran with hydrophobic colloidal NPs forming a single-phase ligand-exchange environment. (ii) The nucleophilic thiol groups in 4-ATP and 4-MCBA form stable complexes with the surfaces of Pd and CdSe NPs to facilitate ligand exchange.16 Stable metallocyclic chelates are formed between the ligands and the undercoordinated metal atoms on the colloidal NP surface (FePt and Fe3O4); meanwhile, the two phenolic hydroxyl groups of dopamine and 3,4-DHCA also promote ligand exchange.21 (iii) They have either acidic (COOH) or basic (NH2) groups, which can be neutralized by NaOH or HCl to form ionic NPs.
Transmission electron microscopy (TEM) revealed that the monodisperse INPs retain their shape and size in the aqueous phase (Figure 1c–e,g–i,k–m,o–q). Infrared (IR) spectra [Figures S1–S4, Supporting Information (SI)] show characteristic C–H stretching peaks from oleic acid (OA)/oleylamine (OAm) or myristic acid (MA) stabilizers before ligand exchange. However, these peaks disappeared after replacement with dopamine and 3,4-DHCA, or 4-ATP and 4-MCBA, indicating successful exchange of the original ligands. To further determine the degree of ligand exchange, we used NMR to characterize the ligands before and after ligand exchange. Fe3O4 NPs were used as an example, because they can be dissolved by HCl easily to release the surface ligands. From the NMR results (Figure S7, SI), a near quantitative ligand exchange was achieved, demonstrating a high degree of ligand exchange. As support, the photographs in Figure 1 (panels f, j, n, and r) show that FePt, cubic Fe3O4, Pd, and CdSe NPs were transferred to the aqueous phase from the organic phase after ligand exchange and ionization. Regarding the stability of INPs, carboxyl terminal INPs can be stable in buffers over a period of 3 months without obvious aggregation. Amino-terminal INPs aggregated immediately when buffers were introduced. Under high temperature, both INPs aggregated when the temperature exceeded 80 °C.
After ligand exchange, ζ-potentials were measured for surface characterization of INPs in the aqueous phase. The ionic colloidal NPs, whether using carboxylic acid- or amine-functionalized ligands, were redispersed in neutral (pH 7) ultrapure Millipore water (18.2 Ω). Table 1 shows that the INPs modified by amine-functionalized ligands and neutralized by HCl gave positive ζ-potentials, while the INPs modified by carboxylic acid-functionalized ligands and neutralized by NaOH gave negative ζ-potentials. When aqueous HCl or NaOH was introduced after ligand exchange, both amine and carboxyl groups were neutralized to form salts, either NP-NH3Cl or NP-COONa, which aggregated in tetrahydrofuran. In water, however, the positive or negative surface charge prevented aggregation, thus maintaining the colloidal state of INPs.
Table 1. ζ-Potentials of Ionic FePt (dopamine and 3,4-DHCA), Fe3O4 (dopamine and 3,4-DHCA), Pd (4-ATP and 4-MCBA), and CdSe (4-ATP and 4-MCBA).
| ζ
(mV) |
||||
|---|---|---|---|---|
| NP’s ligand | FePt | Fe3O4 | Pd | CdSe |
| dopamine | +31.3 | +31.6 | ||
| 3,4-DHCA | –41.9 | –52.1 | ||
| 4-ATP | +35.5 | +25.3 | ||
| 4-MCBA | –38.1 | –48.7 | ||
The availability of our INPs led us to explore their catalytic properties, especially because their ionic properties would commend their use in aqueous solutions and because their thermal stability may exceed that of most biological catalysts. Indeed, some nanozymes, which are nanomaterial-based enzyme mimics, offer high stability, low cost, and good catalytic efficiency,22,23 making them useful in immunoassays, biosensing, bioremediation, and cancer diagnostics.24,25 For greatest effectiveness, it is important to fabricate high-quality nanozymes of uniform size and defined structure to best fulfill specific tasks.26 Given that horseradish peroxidase is a heme–iron protein that is widely used in bioanalytical chemistry, we examined our FePt, Fe3O4, Pd, and CdSe nanoparticles for their ability to catalyze oxidation reactions. Of these, ionic FePt and CdSe NPs were found to catalyze the oxidation of the colorless substrate 3,3′,5,5′-tetramethylbenzidine (TMB), which is blue in the absence of hydrogen peroxide, while Pd and Fe3O4 NPs could catalyze the oxidation of TMB in the presence of H2O2. (Spectra of the colorless substrate and blue product are shown in Figure S8, SI.)
Mindful that most nanoparticles lack the cardinal features of enzymes (e.g., homogeneous composition, structurally defined active sites, substrate specificity, and high catalytic rate enhancements), we were interested in determining whether INP catalysis could be modeled phenomenologically by the Michaelis–Menten equation (v = kcat[Etot}/{1 + (Km/[S]}), where v is the initial velocity, kcat is the turnover number, [Etot] is total catalyst concentration, Km is the Michaelis constant, and [S] is the substrate concentration. In the absence of detailed information about likely binding sites, we assumed that there was one catalytic center per nanoparticle, but multiple independent centers on each particle would only affect the true value of kcat. (Future work using the Langmuir equation with unreactive substrates should clarify the actual number of sites per particle.) Reaction conditions were optimized to ensure that initial velocity data were obtained, insofar as product formation was linear with time and v was found to be directly proportional to INP concentration. The resulting rate data are presented in Figure 2, and the derived rate parameters are presented in Table 2.
Figure 2.

Michaelis–Menten kinetics {ν (initial velocity) versus [S]} for the oxidation of TMB catalyzed by ionic FePt, cubic Fe3O4, and Pd nanoparticles. The initial velocities of parts a and b were measured by adding 17.5 μg of ionic FePt to 200 μL of standard TMB solution (pH 3) with different concentrations at room temperature. The initial velocities of parts c–f were measured by adding 25 μg of ionic Fe3O4 or 20 μg of ionic Pd to 200 μL of standard TMB solution (pH 3) with 400 mM of H2O2. Insets: Lineweaver–Burk plots.
Table 2. Comparison of Michaelis–Menten Parameters for Ionic FePt, Cubic Fe3O4, and Pd with Different Ligandsa.
| INPs | Km (mM) | Vmax (M s–1) | [E0] (M) | kcat (s-1) | substrate (mM) |
|---|---|---|---|---|---|
| FePt–dopamine | 0.079 | 59.88 × 10–8 | 5.9 × 10–7 | 1.02 | H2O2 (0) and TMB |
| FePt–3,4-DHCA | 0.121 | 21.10 × 10–8 | 5.9 × 10–7 | 0.36 | H2O2 (0) and TMB |
| Fe3O4–dopamine | 0.599 | 162.1 × 10–8 | 1.2 × 10–8 | 135 | H2O2 (400) and TMB |
| Fe3O4–3,4-DHCA | 0.951 | 163.1 × 10–8 | 1.2 × 10–8 | 136 | H2O2 (400) and TMB |
| Pd–4-ATP | 0.113 | 19.53 × 10–8 | 3.4 × 10–7 | 0.58 | H2O2 (400) and TMB |
| Pd–4-MCBA | 0.260 | 200.0 × 10–8 | 3.4 × 10–7 | 5.88 | H2O2 (400) and TMB |
Km is the Michaelis constant, Vmax is the maximal reaction velocity, [E] is the ionic nanoparticle concentration, and kcat is the catalytic constant, where kcat = Vmax/[E].
We found that this peroxidase-like activities of the above NPs show different catalytic properties and pH dependencies. In the absence of H2O2, for example, FePt displays better catalytic activity than CdSe, which requires up to several hours (Figures S13–S15, SI) to realize an obvious color change with TMB. In the presence of H2O2, Fe3O4 shows better catalytic activity than Pd nanoparticles. To study the effect of pH on peroxidase-like activity, we used pH values ranging from 3 to 12 in TMB solutions. All INPs showed the best catalytic activity at pH 3 (Figure S9, SI). Thus, pH 3 and room temperature were adopted as the standard conditions for the steady-state kinetics assay. Typical Michaelis–Menten curves were observed for FePt, Fe3O4, and Pd with several replacement ligands (Figure 2), and the parameters (Table 2 and SI) were determined according to the fitted Michaelis–Menten model and Lineweaver–Burk plots (Figure 2).27 The Km values of amino-terminal INPs were lower than those of the carboxyl-terminal INPs, suggesting that INPs with terminal NH3+ have higher affinity for substrate than the INPs with terminal COO–. This can be attributed to the surface charges of the INPs and the aromatic amine structure of TMB. Amino-terminal INPs with strong positive charge for R–NH3+ can easily attract and template the nucleophilic aromatic TMB.
In the kinetic studies presented here, we treated the catalytic process as a one-substrate reaction, where molecular oxygen was kept at oxygen-saturated conditions or where hydrogen peroxide H2O2 is present in sufficient excess. Further investigation will be required to determine how metal ions on the nanoparticle surface facilitate catalysis by acting as Lewis acids and/or substrate templates.
A control experiment was conducted using ionic CdSe in the absence of H2O2, both in the dark and in visible light, and no oxidized TMB was observed from ionic CdSe in the dark. However, the amino-terminal ionic CdSe catalyzed TMB slowly under illumination (medium bipin base bulb, 32 W). The absorption peak of ionic CdSe was 626 nm. The UV–vis spectra showed no obvious wavelength shift, either before or after ligand exchange (Figure S12, SI). Under visible light irradiation, the excited electrons in the valence band were transferred to the conduction band with holes induced in the valence band. Valence band holes are powerful oxidants and could directly react with the electron donor TMB to form TMB radical cations and finally yield the oxidized product. This process indicates that the peroxidase-like activity of ionic CdSe could be attributed to its photocatalytic property.3
Ionic Fe3O4 nanocubes and Pd NPs do not catalyze TMB oxidation in the absence of H2O2. Fe3O4 synthesized in aqueous phase displayed peroxidase-like activity by the presence of rich Fe2+ on the surface.22 It has been reported that porous Pd NP assemblies can be used as horseradish peroxidase substitutes.28 Ionic Fe3O4 nanocubes and Pd NPs, which were synthesized in the organic phase, also exhibited excellent peroxidase-like activity, indicating that the peroxidase-like activity of Fe3O4 and Pd NPs originates from the NPs themselves. Ligand exchange and ionization transfer hydrophobic colloidal NPs to the aqueous phase, but without affecting their peroxidase-like activity.
Ionic functionalization of colloidal NPs via ligand exchange in a single phase provides a facile method to transfer the highly crystalline NPs synthesized through pyrolysis to an aqueous phase. Notably, ligand exchange and ionization can be generalized to other nanoparticle systems, such as upconversion nanoparticles (UCNPs), which are used extensively in bioimaging and photodynamic therapy. As a representative, NaYF4 (Yb 30%, Er 2%, Nd 1%) was selected and synthesized20 in order to study optical properties before and after ligand exchange and ionization. TEM indicated that both shape and size were maintained (Figure 3a–c), and no obvious change was observed in the photoluminescence spectra (Figure 3d). ζ-Potential measurements (Figure 3e) showed that dopamine-modified ionic NaYF4 has a positive surface charge and that the 3,4-DHCA-modified ionic NaYF4 has a negative surface charge.
Figure 3.
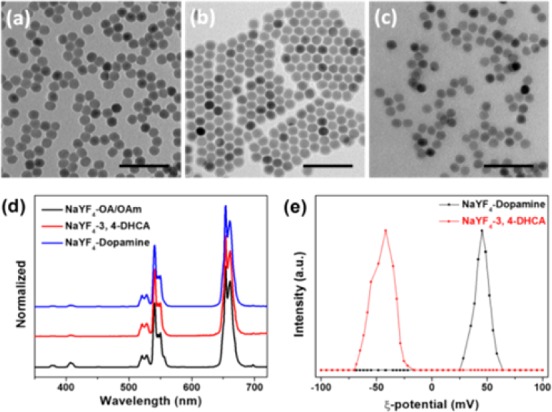
TEM and photoluminescence spectra of NaYF4 (Yb 30%, Er 2%, Nd 1%) before and after ligand exchange and ionization. TEM of (a) ionic NaYF4 with 3,4-DHCA in water, (b) NaYF4 before ligand exchange in hexane, and (c) ionic NaYF4 with dopamine in water. (d) Photoluminescence spectra of NaYF4 before and after ligand exchange and ionization. (e) ζ-Potential measurement for NaYF4–dopamine shows positive surface charge, and NaYF4–3,4-DHCA shows negative surface charge in water.
In conclusion, ionic functionalization of hydrophobic colloidal nanoparticles to form hydrophilic ionic NPs via ligand exchange offers the opportunity to develop reproducible high-quality nanoparticle biosensors with maximal performance under physiological conditions and to achieve large-scale application and impact. Advanced nanoparticle biosensors can be engineered for visualization and detection on the basis of the ionic NPs. Future work can also benefit from the rational design of nanozymes, allowing natural horseradish peroxidase to be replaced by nanozymes, which are cheaper and more stable.
Acknowledgments
The authors are grateful to Dr. K. S. Schanze, R. Winkel, Y. Huang, J. Zhang, and K. Kelly for technical support in characterizing the samples and Dr. K. R. Williams for useful discussion. This work is supported by grants awarded by the National Institutes of Health (GM079359, GM111386, and CA133086). It is also supported by the National Key Scientific Program of China (2011CB911000), NSFC grants (NSFC 21221003 and NSFC 21327009) and China National Instrumentation Program 2011 YQ03012412.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b08533.
Detailed FT-IR, ζ-potential, NMR, optical absorption spectra, and characterization of the peroxidase-like activity mimic (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Huynh W. U.; Dittmer J. J.; Alivisatos A. P. Science 2002, 295, 2425–2427. 10.1126/science.1069156. [DOI] [PubMed] [Google Scholar]
- Dai X.; Zhang Z.; Jin Y.; Niu Y.; Cao H.; Liang X.; Chen L.; Wang J.; Peng X. Nature 2014, 515, 96–99. 10.1038/nature13829. [DOI] [PubMed] [Google Scholar]
- Han Z.; Qiu F.; Eisenberg R.; Holland P. L.; Krauss T. D. Science 2012, 338, 1321–1324. 10.1126/science.1227775. [DOI] [PubMed] [Google Scholar]
- Chan W. C. W.; Nie S. Science 1998, 281, 2016–2018. 10.1126/science.281.5385.2016. [DOI] [PubMed] [Google Scholar]
- Elghanian R.; Storhoff J. J.; Mucic R. C.; Letsinger R. L.; Mirkin C. A. Science 1997, 277, 1078–1080. 10.1126/science.277.5329.1078. [DOI] [PubMed] [Google Scholar]
- Liu X.; Deng R.; Zhang Y.; Wang Y.; Chang H.; Huang L.; Liu X. Chem. Soc. Rev. 2015, 44, 1479–1508. 10.1039/C4CS00356J. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Lorenzo L.; de la Rica R.; Alvarez-Puebla R. A.; Liz-Marzan L. M.; Stevens M. M. Nat. Mater. 2012, 11, 604–607. 10.1038/nmat3337. [DOI] [PubMed] [Google Scholar]
- Yin Y.; Alivisatos A. P. Nature 2005, 437, 664–670. 10.1038/nature04165. [DOI] [PubMed] [Google Scholar]
- Sun S.; Murray C. B.; Weller D.; Folks L.; Moser A. Science 2000, 287, 1989–1992. 10.1126/science.287.5460.1989. [DOI] [PubMed] [Google Scholar]
- Park J.; An K.; Hwang Y.; Park J.; Noh H.; Kim J.; Park J.; Hwang N.; Hyeon T. Nat. Mater. 2004, 3, 891–895. 10.1038/nmat1251. [DOI] [PubMed] [Google Scholar]
- Wang F.; Han Y.; Lim C.; Lu Y.; Wang J.; Xu J.; Chen H.; Zhang C.; Hong M.; Liu X. Nature 2010, 463, 1061–1065. 10.1038/nature08777. [DOI] [PubMed] [Google Scholar]
- Howes P. D.; Chandrawati R.; Stevens M. M. Science 2014, 346, 1247390. 10.1126/science.1247390. [DOI] [PubMed] [Google Scholar]
- Michalet X.; Pinaud F. F.; Bentolila L. A.; Tsay J. M.; Doose S.; Li J. J.; Sundaresan G.; Wu A. M.; Gambhir S. S.; Weiss S. Science 2005, 307, 538–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T.; Ge J.; Hu Y.; Yin Y. Nano Lett. 2007, 7, 3203–3207. 10.1021/nl071928t. [DOI] [PubMed] [Google Scholar]
- Kovalenko M. V.; Scheele M.; Talapin D. V. Science 2009, 324, 1417–1420. 10.1126/science.1170524. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Chen T.; Wu C.; Qiu L.; Hu R.; Li J.; Cansiz S.; Zhang L.; Cui C.; Zhu G.; You M.; Zhang T.; Tan W. J. Am. Chem. Soc. 2014, 136, 12552–12555. 10.1021/ja5060324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalenko M.; Bodnarchuk M.; Lechner M.; Hesser G.; Schaffler F.; Heiss W. J. Am. Chem. Soc. 2007, 129, 6352–6353. 10.1021/ja0692478. [DOI] [PubMed] [Google Scholar]
- Kim S.; Park J.; Jang Y.; Chung Y.; Hwang S.; Hyeon T.; Kim Y. W. Nano Lett. 2003, 3, 1289–1291. 10.1021/nl0343405. [DOI] [Google Scholar]
- Chen O.; Chen X.; Yang Y.; Lynch J.; Wu H.; Zhuang J.; Cao C. Angew. Chem., Int. Ed. 2008, 47, 8638–8641. 10.1002/anie.200804266. [DOI] [PubMed] [Google Scholar]
- Xie X.; Gao N.; Deng R.; Sun Q.; Xu Q.; Liu X. J. Am. Chem. Soc. 2013, 135, 12608–12611. 10.1021/ja4075002. [DOI] [PubMed] [Google Scholar]
- Shultz M. D.; Reveles J. U.; Khanna S. N.; Carpenter E. E. J. Am. Chem. Soc. 2007, 129, 2482–2487. 10.1021/ja0651963. [DOI] [PubMed] [Google Scholar]
- Gao L.; Zhuang J.; Nie L.; Zhang J.; Zhang Y.; Gu N.; Wang T.; Feng J.; Yang D.; Perrett S.; Yan X. Nat. Nanotechnol. 2007, 2, 577–583. 10.1038/nnano.2007.260. [DOI] [PubMed] [Google Scholar]
- Wei H.; Wang E. Chem. Soc. Rev. 2013, 42, 6060–6093. 10.1039/c3cs35486e. [DOI] [PubMed] [Google Scholar]
- Lin Y.; Ren J.; Qu X. Acc. Chem. Res. 2014, 47, 1097–1195. 10.1021/ar400250z. [DOI] [PubMed] [Google Scholar]
- Gao Z.; Hou L.; Xu M.; Tang D. Sci. Rep. 2014, 4, 3966. 10.1038/srep03966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandari R.; Coppage R.; Knecht M. R. Catal. Sci. Technol. 2012, 2, 256–266. 10.1039/C1CY00350J. [DOI] [Google Scholar]
- Purich D. L.Enzyme Kinetics Catalysis & Control: A Reference of Theory and Best-Practice Methods; Academic Press: London, 2010. [Google Scholar]
- Han M.; Liu S.; Bao J.; Dai Z. Biosens. Bioelectron. 2012, 31, 151–156. 10.1016/j.bios.2011.10.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.