

Abstract
Here, we propose an efficient strategy for enzyme- and hairpin-free nucleic acid detection called an entropy beacon (abbreviated as Ebeacon). Different from previously reported DNA hybridization/displacement-based strategies, Ebeacon is driven forward by increases in the entropy of the system, instead of free energy released from new base-pair formation. Ebeacon shows high sensitivity, with a detection limit of 5 pM target DNA in buffer and 50 pM in cellular homogenate. Ebeacon also benefits from the hairpin-free amplification strategy and zero-background, excellent thermostability from 20 °C to 50 °C, as well as good resistance to complex environments. In particular, based on the huge difference between the breathing rate of a single base pair and two adjacent base pairs, Ebeacon also shows high selectivity toward base mutations, such as substitution, insertion, and deletion and, therefore, is an efficient nucleic acid detection method, comparable to most reported enzyme-free strategies.
The efficient detection of nucleic acids, especially the strong capacity to identify single nucleotide polymorphisms (SNPs), is highly significant in biochemical studies and genetic diagnostics inasmuch as DNA is, intrinsically, an essential biotarget.1−5 However, the environmental interference of the cytoplasm (e.g., relatively high temperature and ubiquitous biomolecules) has made traditional nuclease-based signal amplification strategies difficult to use in complex biosystems, despite their outstanding performance in buffer solution.6−10 This calls for the development of enzyme-free nucleic acid-detection strategies with high detection sensitivity, high selectivity toward base mutation,2,6 and high stability in complex biological milieu.11,12
Among all reported enzyme-free strategies,13−18 the dynamic DNA-assembly-based enzyme-free signal amplification strategy has been regarded as having unparalleled advantages, such as low cost, easy construction, high tolerance to environmental disturbance, and uncompromised biocompatibility.19 Apart from conventional hybridization of two complementary DNA sequences, toehold-mediated strand displacement,20−23 which is known as a programmable form of dynamic DNA hybridization, can also be used to design powerful amplification systems, and some of them can even achieve polynomial or exponential amplification of input signals.22 Most reported DNA hybridization/displacement-based amplification strategies are driven by the released free energy associated with base-pair formation, which has proven especially suitable for signal acquisition.21,24 During the past five years, quite a few remarkable designs based on this strategy have been proposed, including the hybridization chain reaction (HCR)15 and catalytic hairpin assembly (CHA).16 Relying on these typical transduction and amplification strategies, more and more biosensors have been created with diverse reporting signals and a variety of targets.25−33
However, these strategies also have some weaknesses, such as circuit leakage resulting from catalyst-independent side reactions and environmental sensitivity related to pH, temperature, biomolecules, or random DNA sequences, which may lead to relatively high background and false-positive signals.34 For instance, the two hairpin substrates in a CHA circuit can potentially react nonspecifically, even in the absence of a single-stranded catalyst, and this nonspecific background degrades the signal-to-noise ratio.35 Moreover, many typical DNA amplification systems are hairpin-based processes,21,36 in which the opening of the hairpin structure by strand displacement is, to some extent, reversible, because the displaced strand is tethered in close proximity to the newly formed helix.37 Also, by easily forming multimers, hairpin structures have led to control problems in biosensing systems, thus limiting their application in multiplex environments. Therefore, when designing a DNA amplification-based biosensing system, both reliability and performance must be considered.
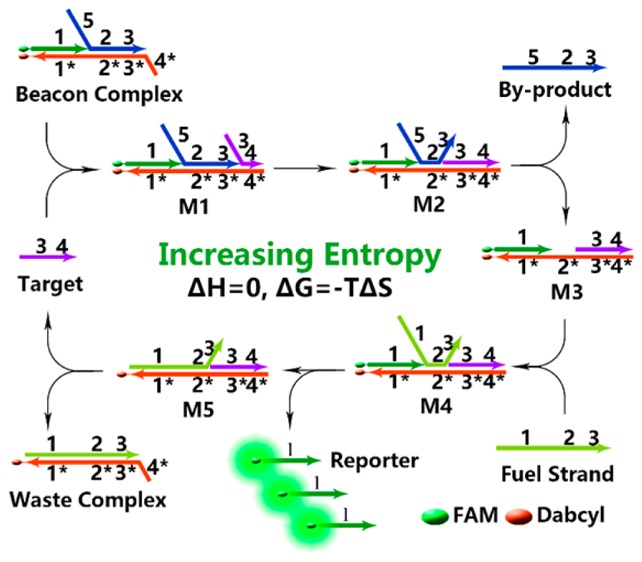
Inspired by the entropy-driven strategy,38 which was a milestone report in the field of DNA logic circuits proposed by Zhang and co-workers, we herein present an enzyme- and hairpin-free amplification system, termed as entropy beacon (Ebeacon, as shown in Figure 1), to overcome the drawbacks of other enzyme-free strategies. In this novel design, the displaced strand forms a double-stranded waste complex, which makes the reaction irreversible. Thus, use of double-stranded assembling substrates, instead of DNA hairpins, not only allows very flexible sequence design but also increases the stability of the assembled products. With this design, we acquired a nucleic acid detection limit of <5 pM with a zero-background,32 which is better than most of previously reported enzyme-free DNA amplification strategies (see Table S1 in the Supporting Information).24,26,29,37,39,40 Benefiting from the unique and exclusive entropy-driven force, Ebeacon is driven forward by increases in the entropy of the system, instead of free-energy release by new base-pair formation. The base pairs of Ebeacon remain unchanged during the amplification process, thus avoiding the interference of other nucleic acids and complex environments, while, at the same time, showing robust thermostability. Furthermore, this design shows outstanding recognition toward single-base mutation, including substitution, deletion, and insertion, when compared with other enzyme/nonenzyme strategies.9,14,41 This be attributed to the difference between the breathing rates of a single base pair and two adjacent base pairs. Thus, Ebeacon may find broad application as an efficient signal amplification element in the construction of various biosensing and biological systems.
Figure 1.
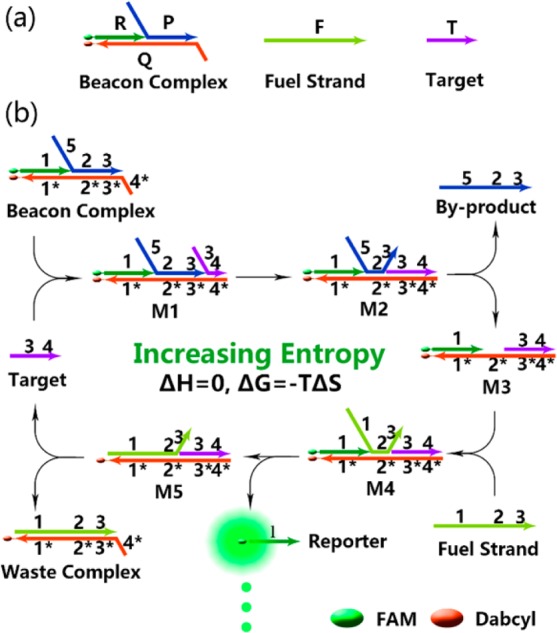
(a) Components of Ebeacon system: beacon complex, fuel strand (pale green) and target (purple). Beacon complex is a FAM/Dabcyl-labeled three-stranded double helix structure, consisting of a reporter strand (R, green), a quencher strand (Q, red) and a byproduct strand (P, blue). (b) Scheme of the entropy beacon (Ebeacon) amplification platform. The entire cycle amplification process is triggered by the addition of a target strand. A substoichiometric concentration of target DNA leads to the conversion of beacon complex to waste complex and the subsequent release of reporter strand. Arrows drawn on DNA strands represent 3′ termini. Domains are named by numbers, and complementarity is denoted by asterisks.
Experimental Section
Reagents
The sequences of oligonucleotides used in this paper are listed in Table S2 in the Supporting Information. DNA synthesis reagents were purchased from Glen Research (Sterling, VA). A solution of 0.1 M triethylamine acetate (pH 6.5) was used as HPLC buffer A, and HPLC-grade acetonitrile from Oceanpak (Sweden) was used as HPLC buffer B. A 1× TAE/Mg2+ buffer (40 mM Tris-acetic acid, 1 mM EDTA, and 12.5 mM magnesium acetate, balanced to pH 8.0) was used for all reactions. Stainsall was obtained from Sigma–Aldrich (Shanghai, China). All other chemicals were obtained from Shanghai Chemical Reagents (Shanghai, China) and used without further purification. Milli-Q water (resistance of >18 MΩ cm) was used to prepare all solutions.
Instruments
A PolyGen DNA-synthesizer was used for DNA synthesis. Probe purification was performed with an Agilent Model 1260 HPLC system that was equipped with a C18 column (Inertsil OSD-3, 5 μm, 250 × 4.6 mm) from GL Sciences, Inc. Ultraviolet–visible light (UV–vis) measurements were performed with a Biospec-nano spectrophotometer from Shimadzu for probe quantitation. Steady-state fluorescence measurements were performed on a Fluoromax-4 spectrofluorometer from Horiba with a temperature controller, using a quartz fluorescence cell with an optical path length of 1.0 cm. The excitation was made at 488 nm with recording emission range of 500–600 nm. All excitation and emission bandwidths were set at 5 nm. The pH measurements were carried out on a Mettler–Toledo Delta 320 pH meter.
Buffer Conditions
The buffer for all experiments was TAE (40 mM Tris-acetic acid, pH balanced to 8.0, 1 mM EDTA), with 12.5 mM MgCl2 added, unless otherwise stated.
Preparation of Cellular Homogenate
CCRF-CEM cells (1 × 107 cells) were first centrifuged for 5 min at 25 °C (1000 rpm), followed by the removal of supernantant. Precipitated cells were resuspended in 1 mL buffer solution. Then, the resuspended cells were strongly sonicated for 30 min in an ice–water bath. The resulting cellular homogenate was stored at 4 °C for further use.
DNA Synthesis and Purification
The DNA sequences were synthesized on a PolyGen DNA synthesizer. The synthesis protocol was set up according to the requirements specified by the reagents’ manufacturers. Following on-machine synthesis, the DNA products were deprotected and cleaved from CPG by incubating with 2 mL of AMA (ammonium hydroxide and 40% methylamine, 1:1) for 30 min at 65 °C in a water bath. The cleaved DNA product was transferred to a 15 mL centrifuge tube and mixed with 200 μL of 3.0 M NaCl and 5.0 mL of ethanol, after which the sample was placed in a freezer at −20 °C for ethanol precipitation. Afterward, the DNA product was spun at 4000 rpm at 4 °C for 30 min. The supernatant was removed, and the precipitated DNA product was dissolved in 400 μL of 0.1 M triethylammonium acetate (TEAA) for HPLC purification, which was performed with a cleaned C18 column on an Agilent 1260 HPLC system. The collected DNA product was dried and processed for detritylation by dissolving and incubating in 200 μL of 80% acetic acid for 20 min. The detritylated DNA product was mixed with 20 μL of 3.0 M NaCl and 500 μL of ethanol and placed in a freezer at −20 °C for 30 min. Afterward, the DNA product was spun at 14 000 rpm at 4 °C for 5 min. The DNA product was dried by a vacuum dryer, redissolved in ultrapure water, and desalted with desalting columns. The DNA products were quantified and stored in ultrapure water for subsequent experiments. Detailed sequences are presented in Table S2. The three-stranded beacon complexes were manually purified to ensure proper stoichiometry and improve purity. Sources of substrate impurity include synthesis errors and truncations, partially formed complexes from imperfect stochiometry, and dimerization, which may cause undesired system leakage and defects. Strands for beacon complex were prepared with nominally correct stochiometry at 10 μM and annealed in 1× TAE/Mg2+ buffer (40 mM Tris-Acetate, 1 mM EDTA, pH 8.0, with 12.5 mM Mg(Ac)2 added). The fuel strand was then added, triggering many poorly formed substrates to decay into products that can be removed by gel purification. DNA complex solution was collected by soaking gel pieces in 1× TAE/Mg2+ buffer for 24 h at room temperature. Finally, the DNA duplex sequences were quantified by UV spectrometry and kept in a buffer for future use.42−44
Native Gel Electrophoresis
Prior to each experiment, stock solutions of purified beacon complex and fuel strands were annealed and diluted to a concentration of 2 μM. For each lane, the final concentration of beacon complex and fuel strands was 400 nM with a total volume of 5 μL. Before loading the samples on a gel, reactions were run for 3 h at room temperature in TAE/Mg2+ buffer (40 mM Tris-acetic acid, 1 mM EDTA, and 12.5 mM magnesium acetate, pH 8.0), and reacted samples were supplemented with 1 μL of 6× BeyoRed DNA ladder from Beyotime. Samples were run in a 12% polyacrylamide gel with 1× TAE/Mg2+ buffer at 110 V for 2 h. The buffer temperature was controlled to maintain the samples at 4 °C throughout the run. The gel was stained with Stainsall stain solution (Sigma–Aldrich). Photos were taken above a white background after the purple in the gel faded. All annealing processes were performed with an Eppendorf Mastercycler Gradient thermocycler. The samples were cooled from 95 to 10 °C at a constant rate over the course of 85 min.
Fluorescence Measurements
All fluorescence measurements were performed on a Fluoromax-4 spectrofluorometer from Horiba with temperature controller, using a quartz fluorescence cell with an optical path length of 1.0 cm. For spectrofluorimetry studies, the excitation was recorded at 488 nm with a recording emission range of 500–600 nm. For fluorescence kinetics studies, the excitation was recorded at 488 nm, and the emission was recorded at 520 nm. After thoroughly mixing the components, the rate of fluorescence increase was monitored every 2 min. Unless otherwise specified, all excitation and emission bandwidths were set at 5 nm. Prior to each experiment, all cuvettes were washed with 70% ethanol and distilled water. In order to avoid the nonspecific sticking of DNAs to pipette tips and to acquire high-performance signals, a nonreactive 20 nt poly-T “carrier” strand at a concentration of 1 μM was introduced into all diluted stocks (1 μM and below).43
Results and Discussion
Construction and Thermodynamics Calculation of Ebeacon System
Ebeacon consists of three components: the beacon complex (B), the fuel strand (F), and the target (T) (Figure 1a). The beacon complex is, in turn, a three-stranded hybridization complex consisting of a reporter strand (R) with a FAM modification at the 5′-end, a quencher strand (Q) with a Dabcyl modification at the 3′-end, and a byproduct strand (P). The fuel strand is a single-stranded and full-length DNA that can hybridize with the quencher strand. The amplication process is shown in Figure 1b. The beacon complex is formed through hybridization of R, P, and Q, resulting in the quenching of FAM fluorescence. The quencher strand contains a single-stranded toehold, denoted as domain 4*. The target binds to domain 4* to form a four-stranded metastable complex M1, triggering a strand-displacement reaction between domain 3 on T and domain 3 on P. As a result, T completely binds to Q and forms a metastable complex M2, which may easily release strand P and convert to three-stranded complex M3. Then, strand F binds to domain 2* on strand Q and forms four-stranded complex M4. Finally, R is released after the strand-displacement reaction, leading to recovery of fluorescence. Meanwhile, T is regenerated and will trigger a new cycle. Until now, in the presence of target strand (T), the beacon complex is converted to a double-stranded waste complex (W) with the help of the fuel strand (F), releasing a byproduct strand (P). The entire system is driven forward by increasing entropy, because during each single catalysis round, the number of base pairs remains unchanged, i.e., domains 1/1*, 2/2*, 3/3* are double-stranded before and after the conversion.
The entire process is shown as a reaction equation in Figure 2, with the parameters labeled. The corresponding thermodynamic theory is also calculated. As shown in Figure 2, the Gibbs free energy change for this reaction in dilute solutions is
| 1 |
where ΔH is the change in system enthalpy, ΔS the change in entropy, and T the thermodynamic temperature. The total number of base pairs in the reactants and products is unchanged, giving ΔH ≈ 0. Hence, the reaction is driven forward thermodynamically by the entropic gain of the liberated molecules and the driving force, at any moment, is TΔS.
Figure 2.
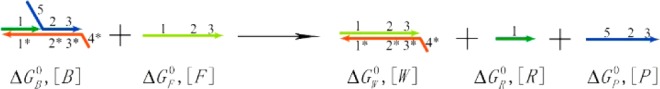
Reaction equation of the Ebeacon system with thermodynamic parameters.
The final concentration of all species in this entropy-driven system can now be estimated. The Gibbs free energy change is given by
| 2 |
where Q is the reaction quotient, relative to standard conditions (Q = [([R]/c0)([P]/c0)([W]/c0)]/[([B]/c0)([F]/c0)]), and ΔGX0 is the standard free energy of species X under standard conditions, which herein are represented by our TAE buffer condition with 12.5 mM Mg2+, 25 °C, and c0 = 1 M.
ΔGX0 (expressed in units of kcal/mol) can be calculated by using free software, such as Mfold and NUPACK,45 giving
| 3 |
When the reaction reaches equilibrium, which means ΔG = 0, according to eq 2 and 3, we can calculate Q = 9.61.
According to the former equation, we know that Q = {([R]/c0)([P]/c0)([W]/c0)}/{([B]/c0)([F]/c0)]} and c0 = 1 M. For a system with initial concentrations of B and F both 10 nM and the final concentration of R is x (expressed in units of nM), we can write the following equation:
| 4 |
Using the bisection method, x is estimated to be between 9.999 and 9.9999 nM, which means a potential systemic fractional conversion of more than 99.99%, without regard for the reaction time.
Target DNA-Triggered Zero-Background Signal Enhancement
Electrophoresis was first used to show that the conversion from beacon complex to waste complex was a result of the presence of target DNA (Figure 3a). The purified beacon complex shows a sharp and neat band (lane 1). When target DNA of the same concentration was added, a toehold-mediated strand migration occurred, and part of the beacon complex formed complex M1 with the release of byproduct strand (lane 2). Only negligible leakage, which accounted for the system background, was observed during the 3 h reaction time when both beacon complex and fuel strand were present (lane 3). In the presence of a substoichiometric concentration of target DNA (0.1×), almost all beacon complexes were converted to waste complex with obvious bands of released reporter sequence and byproduct sequence.
Figure 3.
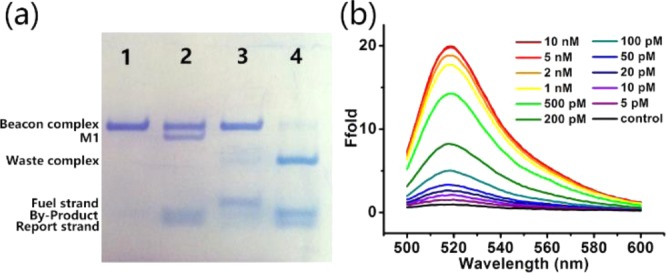
Ebeacon-based amplified detection of nucleic acids. (a) Native PAGE of the entropy beacon system. Lane 1: B: 2 μM × 5 μL; lane 2: B: 2 μM × 5 μL + T: 20 μM × 0.5 μL; lane 3: B: 2 μM × 5 μL + F: 20 μM × 0.5 μL; lane 4: B: 2 μM × 5 μL + F: 20 μM × 0.5 μL + T: 2 μM × 0.5 μL (0.1×). Reaction time = 3 h. (b) Response of the Ebeacon probes to different concentrations of target DNA after incubation for 3 h. Concentration of the target DNA (from top to bottom of curve at 485 nm): 10 nM, 5 nM, 2 nM, 1 nM, 500 pM, 200 pM, 100 pM, 50 pM, 20 pM, 10 pM, 5 pM, control.
Having observed the conversion from beacon complex to waste complex in the electrophoresis experiment, we next measured the fluorescence emission spectra of Ebeacon probes at an excitation wavelength of 488 nm in a buffer solution containing the target DNA at varying concentrations (see Figures 3b and 4a). The initial system contained 10 nM beacon complex and 12 nM fuel strand, and the reaction time was still 3 h. When no target DNA was present, a very weak emission peak at 520 nm was observed, corresponding to the negligible leakage observed in electrophoresis. At this point, the beacon complex remained a stable three-stranded structure, despite the existence of the fuel strand. However, upon the addition of target DNA to the solution, the system was quickly perturbed. A series of strand-displacement reactions occurred, culminating with the hybridization of the fuel strand with the quencher strand, as well as the release of the reporter strand. We found that an increasing number of target sequences correlated with an increasing number of released reporter strands, leading, in turn, to higher fluorescence signal over the same reaction time. The stoichiometric concentration (10 nM) of target DNA caused an ∼20-fold enhancement in fluorescence, which demonstrated the outstanding response of Ebeacon as a DNA amplification sensing platform.
Figure 4.
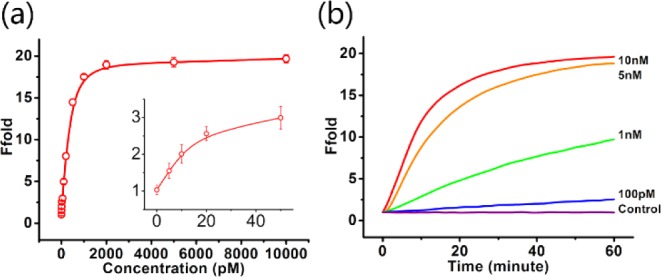
Sensitivity of Ebeacon system for target DNA detection. (a) Calibration curve of the sensing system for target DNA. The curve was plotted with the initial rate of fluorescence enhancement vs DNA concentration. From bottom to top: 0 pM, 5 pM, 10 pM, 20 pM, 50 pM, 100 pM, 200 pM, 500 pM, 1 nM, 2 nM, 5 nM, and 10 nM. The inset shows the responses at low DNA concentrations. Error bars show the standard deviations of measurements taken from three independent experiments. (b) Fluorescence kinetics monitoring conversion of entropy beacon system to the waste strand and release of the reporter strand. No release is discernible in the absence of the target strand (control). However, rapid release of the reporter sequence is observed upon introducing 10 nM target strand. Substoichiometric target strand concentrations demonstrate turnover, with the conversion rate decreasing monotonically with target concentration.
Time-dependent fluorescence enhancement arising from different concentrations of target DNA was then measured (Figure 4b). To balance the reaction time and the fractional conversion, we chose 1 h as the reaction time for this assay. Absent the target sequence, the dynamics results showed that system leakage remained insignificant and unchanged because a zero background was observed.32 In order to further show the sensitivity of Ebeacon for low concentrations of target DNA, by extending the reaction time to 3 h, we were able to reliably distinguish between <5 pM target DNA and the background (see Figure S1 in the Supporting Information). The calibration curve of the sensing system for target DNA also gave a similar limit of detection. The effects of toehold length and fuel strand concentration on reaction rate were also studied (see Figures S2 and S3 in the Supporting Information).
Thermal Stability of the Ebeacon System
Author: Despite the obvious advantages of DNA-based sensors, one important limitation is their thermal instability, resulting from the close relationship between hydrogen bond strength and ambient temperature.35 This is also a major source of false-positive signals associated with such sensors. This fact prompted us to further investigate the effect of temperature on the proposed entropy-driven biosensing platform. With increasing reaction temperature from 20 °C to 50 °C, the background remained relatively stable for 1 h (see Figure 5a, as well as Figure S4 in the Supporting Information). Even at 50 °C, the enhancement in background signal was less than 1-fold, compared with the initial signal. This exciting result demonstrated the outstanding thermostability of the Ebeacon system. This can be attributed to the forces contributing to DNA double helix stability. More specifically, the DNA double helix is stabilized by π–π stacking between each two adjacent bases and the hydrogen bond between complementary base pairs such as A-T and C-G. These two forces, which are highly interrelated and mutually reinforcing, finally lead to the thermostability of the DNA double helix. Reviewing the beacon complex, as shown in Figure 1, we can see that the reporter strand and the byproduct strand each hybridizes with the quencher strand through 22 base pairs, but together they form a long double helix of 44 base pairs. According to the two forces that stabilize the DNA double helix, the three-stranded double helix should have similar thermostability as the two-stranded double helix of 44 base pairs. Using the open source software Mfold and NUPACK, we could calculate the melting temperature of a double helix with 44 base pairs to be ∼80 °C and the ΔG value to be −64.6 kcal/mol. Both experimental and theoretical results suggested that (i) the three-stranded beacon complex had high thermostability and (ii) Ebeacon could function well over a wide range of temperatures.
Figure 5.
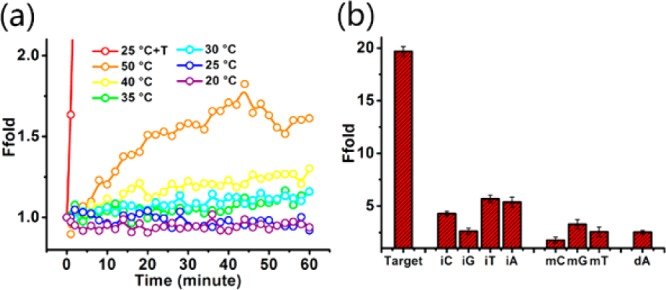
(a) Thermostability of the Ebeacon system at different temperatures from 20 °C to 50 °C. As the reaction temperature increased from 20 °C to 50 °C, the background remained relatively stable for 1 h. Even at 50 °C, the background signal enhancement was less than 2-fold, compared with the initial signal. (b) Selectivity toward single-base substitution (mC, mG, mT), insertion (iC, iG, iT, iA) and deletion (dA).
Selectivity toward Single Base Mutation
The sensitive and selective detection of nucleic acid fragments is important in biological studies, clinical diagnostics, and biodefense applications.2,4,5 To test the selectivity of the Ebeacon probes, we used various oligonucleotides, including matched, mismatched, deleted, and inserted targets (mutant sites are shown in Table S2, red bases). Only the matched DNA triggered the reaction, while the mutant targets led to only very weak signals. (Fluorescence intensities with error bars are shown in Figure 5b, and fluorescence kinetics plots are shown in Figure S5 in the Supporting Information.) In contrast, we designed and synthesized a conventional molecular beacon (MB) that could hybridize with the target strand in the loop and therefore be opened with an increased fluorescence signal. As reported previously, MBs have been widely used for SNP assays. However, according to our results, the ability of the MB to distinguish SNPs is negligible when compared with that of the Ebeacon system (see Figure S6 in the Supporting Information).
According to our thermodynamics calculations, Ebeacon has a huge potential to convert to waste complex and release the reporter strand. The function of target DNA in this system can be regarded as a catalyst and all mutant targets here (matched, mismatched, deleted, and inserted targets) possess very high similarity. The ability of Ebeacon to distinguish a matched target from a mutant target might be explained at the base level. As previously demonstrated, the conversion of such a system is based on strand migration and strand breathing.19,22 For a full complementary target strand, only one base-pair breath is needed in each base migration, which accounts for a high migration rate. For a mutant target strand, two adjacent base pairs should breathe simultaneously in base migration at the mutant site, leading to a low migration rate. It is supposed that the difference in breathing rate between a single base pair and two adjacent base pairs explains the high selectivity of the system. To illustrate our hypothesis, a test was carried out to demonstrate this conclusion.
The Relationship between Strand Breathing Rate and System Selectivity
We constructed a simple two-stranded probe with a blunt end composed of a fluorophore-modified signal strand (S) and quencher modified complementary strand (C) (Figure 6a). The probe has a 6 nt toehold, enabling strand-displacement reaction after the addition of a full-length invading strand (I). As a result, the complementary strand is released, and the fluorescence signal recovers. This is a conventional toehold-mediated strand displacement reaction, and the result is shown in Figure 6d (red line). When a mutant site—in this case, a mismatch—is introduced on an invading strand (mI) (Figure 6b, open circle), the displacement reaction was largely restrained (Figure 6d, green line). These results partially prove that the difference of breathing rate between a single base pair and two adjacent base pairs accounts for selectivity. However, to further demonstrate that the difference in breathing rate between a single base pair and two adjacent base pairs accounts for the different migration rate, we introduced a mismatch site on the complementary strand (mC) next to the previously mentioned mutant site (shown in Figure 6c). Although the invading strand was still mismatched, we predicted that the reaction rate would be comparable to the completely hybridized system, because the different breathing rate between a single base pair and two adjacent base pairs was eliminated. The result shown in Figure 6d (orange line) finally demonstrates this conclusion, proving the relationship between strand breathing rate and system selectivity.
Figure 6.
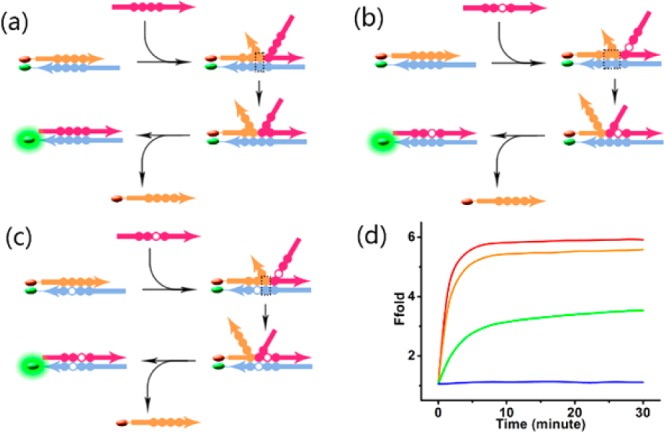
Scheme of the two-mismatch system for mechanism study. (a) Traditional strand displacement reaction with a fully complementary I strand. (b) Strand displacement reaction with mismatched I strand (mI). (c) Strand displacement reaction with mismatched I strand (mI) and mismatched C strand (mC). Solid circles indicate adjacent bases while open circles indicate mismatched bases. Dashed boxes in panels a, b, and c contain base pairs that breathe in matched or mismatched sites when efficient strand migrations occur. (d) Fluorescence results of the two-mismatch system.
Compared with traditional nuclease-based signal amplification strategies, it is an obvious advantage that enzyme-free systems can be used in some complex biological environments. To test the performance of Ebeacon under such conditions, we further carried out assays in cellular homogenate. The fluorescence kinetics is shown in Figure 7a, and a calibration curve with different target concentrations is shown in Figure 7b. According to these results, despite a visible background signal caused by nuclease digestion, Ebeacon still showed great reliability when functioning in cellular homogenate, and the detection limit was calculated to be 50 pM. We also studied the stability of Ebeacon in a complex environment consisting of a 10-fold excess of 30 nt DNA library. The result showed that Ebeacon was undisturbed and functioned normally (see Figure S7 in the Supporting Information).
Figure 7.
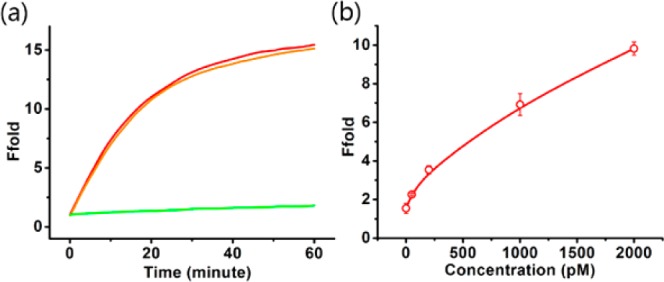
Detection of target DNA in cellular homogenate. (a) Time-dependent fluorescence signal for 10 nM target DNA in cellular homogenate. Red line and orange line correspond to two independent assays which showed the difference between batches. (b) Calibration curve of the Ebeacon system for target DNA with different concentrations in cellular homogenate. From bottom to top: 0 pM, 50 pM, 200 pM, 1 nM, and 2 nM. Error bars show the standard deviations of measurements taken from three independent experiments.
In order to illustrate the potential of Ebeacon as a universal biosensing platform, we designed two types of triggering mechanisms to generate the initial sequence so that Ebeacon could be applied for the detection of a wider range of targets (Figure S8 in the Supporting Information). Both electrophoresis and fluorescence kinetics results showed that Ebeacon was successfully triggered under both regimes and gave a low background and highly amplified signal.
Conclusion
In summary, we have designed an entropy-driven enzyme-free DNA amplification system called the entropy beacon, or Ebeacon. The system is driven forward by the entropy increase of all species, instead of the free energy released by the formation of new base pairs. Benefiting from this unique and exclusive driving force, the base pairs of Ebeacon remain unchanged during the amplification process. This DNA amplification system has several advantages over enzyme-free systems previously reported, including (1) rapid and effective amplification performance with a low detection limit of 5 pM DNA, (2) the capacity to resist disturbance from a complex environment, (3) high thermostability, (4) the ability to distinguish SNPs, (5) the potential for constructing a cascading amplification platform (see Figure S9 in the Supporting Information), and (6) flexible sequence design and decreased reversible conversion. Taken together, this efficient and reliable enzyme- and hairpin-free DNA amplification platform should find broad applications as an efficient signal amplification element in the construction of various biosensing systems.
Acknowledgments
This work is supported by NIH grants (Nos. GM079359, CA133086), the National Key Scientific Program of China (No. 2011CB911000), the Foundation for Innovative Research Groups of the NSFC (No. 21221003), the China National Instrumentation Program (No. 2011YQ03012412).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.5b02654.
Additional information about the Ebeacon system, including fluorescence kinetics and electrophoresis characterization data and a table containing sequences used in this experiment (PDF)
Author Contributions
The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Yan L.; Zhou J.; Zheng Y.; Gamson A. S.; Roembke B. T.; Nakayama S.; Sintim H. O. Mol. BioSyst. 2014, 10, 970–1003. 10.1039/c3mb70304e. [DOI] [PubMed] [Google Scholar]
- Irizarry K.; Kustanovich V.; Li C.; Brown N.; Nelson S.; Wong W.; Lee C. J. Nat. Genet. 2000, 26, 233–236. 10.1038/79981. [DOI] [PubMed] [Google Scholar]
- Choi H. M.; Chang J. Y.; Trinh L. A.; Padilla J. E.; Fraser S. E.; Pierce N. A. Nat. Biotechnol. 2010, 28, 1208–1212. 10.1038/nbt.1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das J.; Ivanov I.; Montermini L.; Rak J.; Sargent E. H.; Kelley S. O. Nat. Chem. 2015, 7, 569–575. 10.1038/nchem.2270. [DOI] [PubMed] [Google Scholar]
- Newman A. M.; Bratman S. V.; To J.; Wynne J. F.; Eclov N. C.; Modlin L. A.; Liu C. L.; Neal J. W.; Wakelee H. A.; Merritt R. E.; Shrager J. B.; Loo B. W. Jr.; Alizadeh A. A.; Diehn M. Nat. Med. 2014, 20, 548–554. 10.1038/nm.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong R. M.; Zhang X. B.; Zhang L. L.; Huang Y.; Lu D. Q.; Tan W.; Shen G. L.; Yu R. Q. Anal. Chem. 2011, 83, 14–17. 10.1021/ac1025072. [DOI] [PubMed] [Google Scholar]
- Saiki R.; Gelfand D.; Stoffel S.; Scharf S.; Higuchi R.; Horn G.; Mullis K.; Erlich H. Science 1988, 239, 487–491. 10.1126/science.2448875. [DOI] [PubMed] [Google Scholar]
- Kiesling T.; Cox K.; Davidson E. A.; Dretchen K.; Grater G.; Hibbard S.; Lasken R. S.; Leshin J.; Skowronski E.; Danielsen M. Nucleic Acids Res. 2007, 35, e117. 10.1093/nar/gkm654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly A. R.; Trau M. Angew. Chem., Int. Ed. 2010, 49, 2720–2723. 10.1002/anie.200906992. [DOI] [PubMed] [Google Scholar]
- Zuo X.; Xia F.; Xiao Y.; Plaxco K. W. J. Am. Chem. Soc. 2010, 132, 1816–1818. 10.1021/ja909551b. [DOI] [PubMed] [Google Scholar]
- Zhou W.; Chen Q.; Huang P. J.; Ding J.; Liu J. Anal. Chem. 2015, 87, 4001–4007. 10.1021/acs.analchem.5b00220. [DOI] [PubMed] [Google Scholar]
- Wu C.; Cansiz S.; Zhang L.; Teng I. T.; Qiu L.; Li J.; Liu Y.; Zhou C.; Hu R.; Zhang T.; Cui C.; Cui L.; Tan W. J. Am. Chem. Soc. 2015, 137, 4900–4903. 10.1021/jacs.5b00542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. B.; Kong R. M.; Lu Y. Annu. Rev. Anal. Chem. 2011, 4, 105–128. 10.1146/annurev.anchem.111808.073617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Cao Z.; Lu Y. Chem. Rev. 2009, 109, 1948–1998. 10.1021/cr030183i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirks R. M.; Pierce N. A. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 15275–15278. 10.1073/pnas.0407024101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin P.; Choi H. M.; Calvert C. R.; Pierce N. A. Nature 2008, 451, 318–322. 10.1038/nature06451. [DOI] [PubMed] [Google Scholar]
- Furukawa K.; Abe H.; Hibino K.; Sako Y.; Tsuneda S.; Ito Y. Bioconjugate Chem. 2009, 20, 1026–1036. 10.1021/bc900040t. [DOI] [PubMed] [Google Scholar]
- Eckhoff G.; Codrea V.; Ellington A. D.; Chen X. J. Syst. Chem. 2010, 1, 13. 10.1186/1759-2208-1-13. [DOI] [Google Scholar]
- Zhang D. Y.; Seelig G. Nat. Chem. 2011, 3, 103–113. 10.1038/nchem.957. [DOI] [PubMed] [Google Scholar]
- Seelig G.; Soloveichik D.; Zhang D. Y.; Winfree E. Science 2006, 314, 1585–1588. 10.1126/science.1132493. [DOI] [PubMed] [Google Scholar]
- Seelig G.; Yurke B.; Winfree E. J. Am. Chem. Soc. 2006, 128, 12211–12220. 10.1021/ja0635635. [DOI] [PubMed] [Google Scholar]
- Zhang D. Y.; Winfree E. J. Am. Chem. Soc. 2009, 131, 17303–17314. 10.1021/ja906987s. [DOI] [PubMed] [Google Scholar]
- Rudchenko M.; Taylor S.; Pallavi P.; Dechkovskaia A.; Khan S.; Butler V. P. Jr.; Rudchenko S.; Stojanovic M. N. Nat. Nanotechnol. 2013, 8, 580–586. 10.1038/nnano.2013.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bois J. S.; Venkataraman S.; Choi H. M.; Spakowitz A. J.; Wang Z. G.; Pierce N. A. Nucleic Acids Res. 2005, 33, 4090–4095. 10.1093/nar/gki721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Briggs N.; McLain J. R.; Ellington A. D. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 5386–5391. 10.1073/pnas.1222807110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Su X.; Li Z. Anal. Chem. 2012, 84, 5939–5943. 10.1021/ac3004727. [DOI] [PubMed] [Google Scholar]
- Huang J.; Wu Y.; Chen Y.; Zhu Z.; Yang X.; Yang C. J.; Wang K.; Tan W. Angew. Chem., Int. Ed. 2011, 50, 401–404. 10.1002/anie.201005375. [DOI] [PubMed] [Google Scholar]
- Wang F.; Elbaz J.; Orbach R.; Magen N.; Willner I. J. Am. Chem. Soc. 2011, 133, 17149–17151. 10.1021/ja2076789. [DOI] [PubMed] [Google Scholar]
- Qing Z.; He X.; Huang J.; Wang K.; Zou Z.; Qing T.; Mao Z.; Shi H.; He D. Anal. Chem. 2014, 86, 4934–4939. 10.1021/ac500834g. [DOI] [PubMed] [Google Scholar]
- Niu S.; Jiang Y.; Zhang S. Chem. Commun. (Cambridge, U.K.) 2010, 46, 3089–3091. 10.1039/c000166j. [DOI] [PubMed] [Google Scholar]
- Shimron S.; Wang F.; Orbach R.; Willner I. Anal. Chem. 2012, 84, 1042–1048. 10.1021/ac202643y. [DOI] [PubMed] [Google Scholar]
- Lu L. M.; Zhang X. B.; Kong R. M.; Yang B.; Tan W. J. Am. Chem. Soc. 2011, 133, 11686–11691. 10.1021/ja203693b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B.; Jiang Y.; Chen X.; Ellington A. D. J. Am. Chem. Soc. 2012, 134, 13918–13921. 10.1021/ja300984b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y. S.; Bhadra S.; Li B.; Ellington A. D. Angew. Chem., Int. Ed. 2014, 53, 1845–1848. 10.1002/anie.201307418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y. S.; Li B.; Milligan J. N.; Bhadra S.; Ellington A. D. J. Am. Chem. Soc. 2013, 135, 7430–7433. 10.1021/ja4023978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green S. J.; Lubrich D.; Turberfield A. J. Biophys. J. 2006, 91, 2966–2975. 10.1529/biophysj.106.084681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan F.; Hsing I. M. J. Am. Chem. Soc. 2014, 136, 9810–9813. 10.1021/ja502904s. [DOI] [PubMed] [Google Scholar]
- Zhang D. Y.; Turberfield A. J.; Yurke B.; Winfree E. Science 2007, 318, 1121–1125. 10.1126/science.1148532. [DOI] [PubMed] [Google Scholar]
- Lu C. H.; Wang F.; Willner I. J. Am. Chem. Soc. 2012, 134, 10651–10658. 10.1021/ja3037838. [DOI] [PubMed] [Google Scholar]
- Wang F.; Elbaz J.; Willner I. J. Am. Chem. Soc. 2012, 134, 5504–5507. 10.1021/ja300616w. [DOI] [PubMed] [Google Scholar]
- Zhao X. H.; Gong L.; Zhang X. B.; Yang B.; Fu T.; Hu R.; Tan W.; Yu R. Anal. Chem. 2013, 85, 3614–3620. 10.1021/ac303457u. [DOI] [PubMed] [Google Scholar]
- Han D.; Zhu Z.; Wu C.; Peng L.; Zhou L.; Gulbakan B.; Zhu G.; Williams K. R.; Tan W. J. Am. Chem. Soc. 2012, 134, 20797–20804. 10.1021/ja310428s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L.; Winfree E. Science 2011, 332, 1196–1201. 10.1126/science.1200520. [DOI] [PubMed] [Google Scholar]
- Qian L.; Winfree E.; Bruck J. Nature 2011, 475, 368–372. 10.1038/nature10262. [DOI] [PubMed] [Google Scholar]
- Zadeh J. N.; Steenberg C. D.; Bois J. S.; Wolfe B. R.; Pierce M. B.; Khan A. R.; Dirks R. M.; Pierce N. A. J. Comput. Chem. 2011, 32, 170–173. 10.1002/jcc.21596. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.