

Abstract
Renal coloboma syndrome (RCS) is a rare inherited condition exhibiting a variable clinical phenotype of renal and ocular abnormalities. In 50% of cases, mutations can be found in the transcription factor PAX2. We present three generations of a family with a PAX2 mutation who showed variable eye and renal phenotypes. Renal phenotypes ranged from normal kidneys with the absence of proteinuria to end-stage renal disease (ESRD) at 17 years of age. Eye phenotypes included the typical morning glory anomaly, macular retinal pigment epithelial changes and retinal venous tortuosity. We identified a PAX2 mutation c.228_251dup [p.Ser77_Gly84dup] which segregated with the phenotype in an autosomal dominant fashion. A molecular genetic diagnosis allowed identification and management of at-risk family members. Given the phenotypic variability, clinicians need to consider the possibility of RCS in patients with a family history of chronic kidney disease (CKD) or eye disease.
Keywords: end-stage renal disease, morning glory anomaly, pax2 mutation, renal coloboma syndrome
Introduction
Renal coloboma syndrome (RCS), also known as papillorenal syndrome, is a rare autosomal dominant condition characterized by renal hypodysplasia and optic nerve dysplasia (OMIM 120330). In ∼50% of cases, the disease is caused by a mutation in the PAX2 gene, encoding a transcription factor important for both renal and nervous system development. Clinical presentation and disease progression can be very variable. In proven cases of PAX2 mutations, renal anomalies are found in 92% of cases and eye abnormalities are found in 77% of cases [1]. High-frequency hearing loss can be associated with RCS, and indeed was found in 7% of cases [1]. Genotype–phenotype correlations have not been identified [1]. Highly variable clinical features are found in patients with identical mutations [2–4], which suggests that epigenetic factors, environmental factors and modifier genes may play a role. Here, we present three generations of a family with RCS with variable clinical presentations and features. This family emphasizes the importance of recognition of this rare condition, and the need for involvement of nephrological and ophthalmological services to help diagnose and manage these patients. Genetic testing of at-risk individuals can allow appropriate management and follow-up of patients with milder sub-clinical phenotypes.
Case report
A 62-year-old female presented with chronic kidney disease (CKD) stage 4 (serum creatinine 178 µmol/L) and proteinuria (200 mg/mmol creatinine), hypertension and tophaceous gout. Fractional excretion of urate was normal at 7.7% (reference range 7–10%). Her past medical history included breast cancer which was successfully treated with a right mastectomy and tamoxifen. Of note, she had a significant family history of renal disease, with both her son and grandson affected by end-stage renal disease (ESRD) (Figure 1). Her son had also been diagnosed with congenital unilaternal optic nerve hypoplasia. The association between eye and renal disease in her son prompted a formal ophthalmological evaluation, which revealed a pseudo-exfoliation syndrome and a morning glory anomaly of her optic discs with tortuous retinal venules that emerged from the edge of the discs (Figure 2A and B). Her vision was relatively well preserved with 6/9 visual acuity in her right eye and 6/5 acuity in the left. ISCEV (International Society for the Clinical Electrophysiology of Vision) standard full field and pattern electroretinograms demonstrated normal generalized retinal and macular function. The pattern reversal visual evoked potential suggested that a mild bilateral optic neuropathy was present. She was also noted to have a high-frequency hearing loss. Abdominal ultrasound revealed bilateral small kidneys (left 8.5 cm, right 7 cm) but no other structural anomaly of the urinary tract.
Fig. 1.

Family pedigree and mutational analysis. (A) The family pedigree is shown. Males are represented by squares, females by circles. Half-shaded symbols indicated heterozygous mutation status. The proband is marked with an arrow. The genetic diagnosis in II:1 is presumed. (B) Sequence chromatograms of the wild-type and affected patient showing the heterozygous insertion mutation c.228_251dup, p.Ser77_Gly84dup.
Fig. 2.
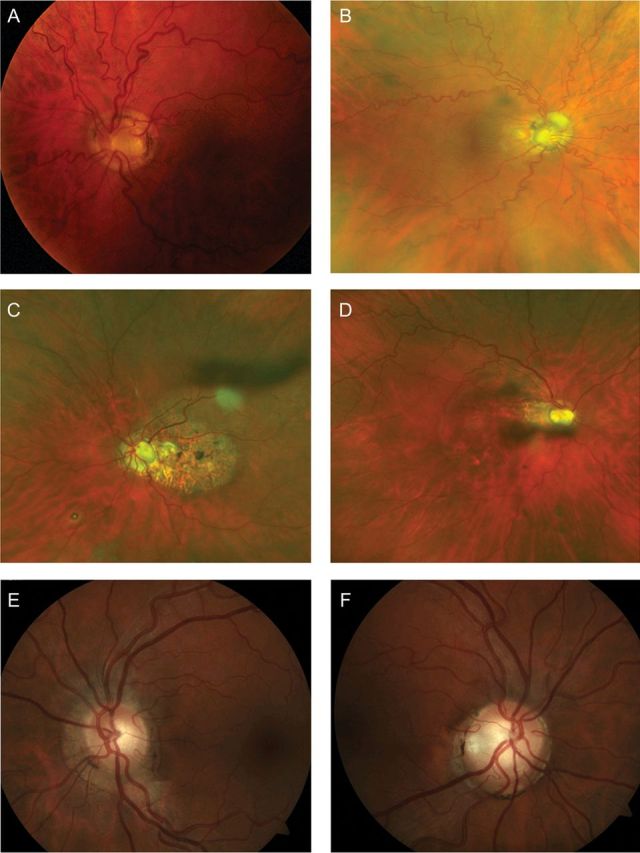
Retinal abnormalities observed in the proband and her two daughters. (A and B) Retinal photographs of the proband showing (A) retinal vessel tortuosity and dysplastic optic discs. The discs have a morning glory appearance with deeply excavated cups, central gliotic tissue and multiple cilioretinal vessels that emerge from the edge of the disc margin. (B) Wide angle fundus photograph demonstrating marked retinal vascular tortuosity. (C and D) Colour fundus photographs of proband's eldest daughter demonstrating pale, cupped optic discs and retinal pigment epithelium changes in the macular area, due to previous central serous retinopathy (left eye, C) and demonstrating pale cupped optic discs (right eye, D). (E and F) Colour retinal photographs of proband's youngest daughter, demonstrating pale, cupped optic discs in both eyes and mildly tortuous blood vessels.
These features prompted genetic investigation of the proband for PAX2 mutations and further investigation of other family members.
The proband's son (II:1, Figure 1) had presented at 35 years of age with monocular visual loss, accelerated hypertension and abnormal renal function with creatinine 339 µmol/L and severe proteinuria (24 h urinary protein 4.4 g). He had small kidneys on renal ultrasound scanning (left kidney 5 cm, right kidney 7 cm). A renal biopsy revealed marked tubular atrophy, evidence of tubular hypertrophy and enlargement of the remaining tubules together with interstitial fibrosis. Foam cells were noted in the interstitium. His CKD progressed to ERSD and he commenced haemodialysis at the age of 39 years. He had known congenital optic disc anomalies and papilloedema, as well as a left homonymous haemianopia. Six months after commencing haemodialysis he died from an intracerebral bleed following an emergency appendicectomy.
The proband's grandson (III:1, Figure 1) presented at birth with hypospadias which was corrected surgically at 2 years of age. He also had documented reflux nephropathy, demonstrated on a micturating cystogram. He developed CKD with proteinuria at 2 years of age. A renal biopsy at this stage was suggestive of mesangiocapillary glomerulonephritis. Despite the use of antiproteinuric agents, including angiotensin-converting enzyme (ACE) inhibition, he progressed to ESRD by 17 years of age. He received a deceased donor renal transplant at 18 years of age, with good function. He awaits a formal ophthalmological review.
Genetic investigations in the proband revealed a heterozygous c.228_251dup [p.(Ser77_Gly84dup)] mutation in exon 3 of the PAX2 gene (Figure 1B). This in frame duplication mutation is predicted to affect the highly conserved paired domain of the PAX2 transcription factor [1]. The heterozygous mutation was confirmed in the patient's grandson. DNA from the proband's son was not available for testing.
Given this genetic diagnosis, the proband's daughters (II:2, II:3, Figure 1) were also invited for review. The proband's eldest daughter had entirely normal renal function with no proteinuria, normal blood pressure and a normal renal ultrasound scan appearance of her kidneys. However, she was noted to have bilateral optic disc pits with associated bilateral macular atrophy secondary to central serous chorioretinopathy (Figure 2C and D). She also had suffered a left retinal detachment previously. Her vision was significantly impaired with 6/18 vision in her right eye and finger counting on the left. The PAX2 mutation was confirmed in this patient.
The youngest daughter had normal renal function with mild proteinuria (protein/creatinine ratio 55 mg/mmol creatinine). Her blood pressure was normal, and her renal ultrasound scan showed no structural anomaly of the renal tract. On ophthalmoscopy, she has cupped optic disc and mildly tortuous blood vessels with only slightly reduced visual acuity in her left eye (Figure 2E and F). She also suffered from recurrent erosion syndrome of her left eye which was presumed to be unrelated. The PAX2 mutation was also confirmed in this patient.
Discussion
Within this family, all affected members whose DNA samples were available had the same heterozygous PAX2 mutation, consistent with an autosomal dominant inheritance pattern. A brief outline of the genotypic and phenotypic data from the proband, son and grandson (I:1, II:1, III:1) has been previously reported [1]. We deduce that the proband's son had the identical genetic change in PAX2, consistent with transmission of this change to her grandson.
There is a remarkable clinical variability of phenotypes within this family, as is often recognized in RCS [2–4]. Interestingly, previous reports suggest that those with subtle or absent renal involvement often have marked ophthalmic dysplasia.
The intriguing clinical variability of RCS is exemplified by a unique case of monozygotic twins with a PAX2 mutation, one twin had predominant renal disease, while the other had predominant eye disease [5]. This allowed the authors to make some insightful observations. First, given the identical genotype of the twins, the effect of modifier genes can be excluded. The discordant phenotypes were present prenatally, suggesting that the intrauterine environment of the twins may have played a role, perhaps with different placental blood flow. A hypothesis of somatic cell mosaicism was excluded by quantifying wild-type and mutant DNA in cell samples from different tissues in both twins. Finally, epigenetic phenomena, such as DNA methylation, were discussed [5]. This process, which can randomly silence one of the two alleles in a cell, may have accounted for the differences in phenotype, given that different tissue patterns of methylation have been noted in identical twins [6, 7]. In the family we present, the variability suggests that a careful family history and ophthalmological examination is required in all cases of renal dysplasia.
In a recent publication of 68 individuals in 33 families with RCS [1], non-renal genitourinary anomalies (hypospadias and unicornate uterus) were reported in just two cases. Thus, hypospadias, as seen in the grandson of the proband, could be incidental to the PAX2 mutation.
The detailed family history of the proband presented in this case has led to the discovery of an inherited condition, which has implications for the wider family. Indeed, other family members had been under the care of nephrologists for many years. This approach has allowed a precise molecular diagnosis in the proband and the detection of the PAX2 mutation in other at-risk family members who are now under regular surveillance of their renal function and blood pressure. Earlier detection of hypertension and CKD in these family members should afford them a better outlook. RCS remains an uncommon disease, and is potentially underdiagnosed due to its variable phenotype.
Funding
We acknowledge funding from the Northern Counties Kidney Research Fund.
Conflict of interest statement
None declared.
Acknowledgements
We thank the family for their permission to publish this report.
References
- 1.Bower M, Salomon R, Allanson J, et al. Update of PAX2 mutations in renal coloboma syndrome and establishment of a locus-specific database. Hum Mutat. 2012;33:457–466. doi: 10.1002/humu.22020. doi:10.1002/humu.22020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schimmenti LA, Cunliffe HE, McNoe LA, et al. Further delineation of renal-coloboma syndrome in patients with extreme variability of phenotype and identical PAX2 mutations. Am J Hum Genet. 1997;60:869–878. [PMC free article] [PubMed] [Google Scholar]
- 3.Fletcher J, Hu M, Berman Y, et al. Multicystic dysplastic kidney and variable phenotype in a family with a novel deletion mutation of PAX2. J Am Soc Nephrol. 2005;16:2754–2761. doi: 10.1681/ASN.2005030239. doi:10.1681/ASN.2005030239. [DOI] [PubMed] [Google Scholar]
- 4.Thomas R, Sanna-Cherchi S, Warady BA, et al. HNF1B and PAX2 mutations are a common cause of renal hypodysplasia in the CKiD cohort. Pediatr Nephrol. 2011;26:897–903. doi: 10.1007/s00467-011-1826-9. doi:10.1007/s00467-011-1826-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iatropoulos P, Daina E, Mele C, et al. Discordant phenotype in monozygotic twins with renal coloboma syndrome and a PAX2 mutation. Pediatr Nephrol. 2012;27:1989–1993. doi: 10.1007/s00467-012-2205-x. doi:10.1007/s00467-012-2205-x. [DOI] [PubMed] [Google Scholar]
- 6.Machin G. Non-identical monozygotic twins, intermediate twin types, zygosity testing, and the non-random nature of monozygotic twinning: a review. Am J Med Genet C Semin Med Genet. 2009;151C:110–127. doi: 10.1002/ajmg.c.30212. doi:10.1002/ajmg.c.30212. [DOI] [PubMed] [Google Scholar]
- 7.Fraga MF, Ballestar E, Paz MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. doi:10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]