

Abstract
The ability to produce isotope-enriched proteins is fundamental to the success of modern protein NMR, and is particularly essential for NMR activities in structural genomics projects. Conventional methods of protein production often prove to be cost prohibitive for obtaining samples, particularly perdeuterated and site-specifically labeled proteins. The condensed single protein production system (cSPP), providing protein expression following condensation of cells 10–40 fold, allows for the production of such samples at a fraction of the cost. The previously described cSPP system is a two plasmid system where both the MazF toxin and ACA-less target gene are coinduced with IPTG. Coinduction results in 10–20% of the target protein produced without isotopic enrichment. Though the unlabeled protein is generally not visible in isotope-filtered NMR experiments, it results in an effective reduction in yield of the observable sample. By altering the cSPP system and separating the induction of the MazF toxin, required to convert cells into a semiquiescent state prior to condensation, from the expression of the target gene, we are now able to eliminate the unlabeled protein fraction and improve the isotope incorporation. Here we describe a series of pCold(tet) vectors with various features that can be used in the dual inducible cSPP(tet) system to obtain high-quality isotopically enriched protein at as little as 2.5% the cost of traditional methods.
Keywords: Anhydrotetracycline, Isotope labeling, MuLV IN, pCold vectors, Structural genomics, Triple resonance NMR
Introduction
A major consideration in protein production for NMR structural analysis is the cost and efficiency of generating isotope-enriched protein samples. The condensed single protein production (cSPP) system utilizes co-expression of the E. coli toxin endoribonuclease MazF, which cuts mRNA at ACA sequences, to allow high-level and selective production of target proteins [1–3]. The target proteins are expressed in the pCold vectors under the control of a cold shock promoter. Previously, expression using pCold vectors was shown to be comparable to that of the pET vectors [4]. The physiological effect of MazF overexpression is to put the E. coli cells into a semi-quiescent state, in which most mRNAs are degraded while the protein synthesis machinery remains intact and capable of translating mRNAs that are engineered to have no ACA sequences. This effectively turns the cell into a “single protein production” factory [1–3], selectively producing a target protein. Importantly, in this semi-quiescent state, cell cultures can be condensed 10–40 fold, or more, without any reduction in protein production levels per cell; i.e. protein production can be done with the same number of cells, producing the same amount of recombinant protein per cell, in much smaller volumes of media. This greatly facilitates the production of recombinant proteins enriched with various isotopes and amino acid analogues at a fraction of the cost of conventional methods [2, 3, W. M. Schneider, personal communication]. Such samples are particularly valuable for NMR studies exploiting isotope-enrichment, as well as for applications in mass spectrometry, neutron diffraction, and other biophysical approaches.
One major limitation of the cSPP system lies in the process of coinducing both the endoribonuclease MazF responsible for degrading cellular mRNAs containing ACA sequences along with the ACA-less target gene, since both proteins are under control of the lac operon [1–3]. The addition of IPTG therefore not only induces MazF expression, but also leads to expression of the ACA-less target gene in the unenriched growth medium during the time required for MazF to establish cell growth arrest prior to culture condensation. Expression of the target gene prior to culture condensation and resuspension in isotope enriched medium leads to heterogeneously labeled protein and results in the accumulation of approximately 10–20% of the total protein completely unlabeled. In order to remedy this problem, the induction of the target protein can be temporally regulated by placing the pCold vectors under control of the tet O1 operator. Here we describe a toolbox of vectors for expression in the cSPP system where temporal regulation of the ACA-less target gene results in greater than 95% isotope enrichment and a reduction in isotopic heterogeneity. This has direct applications to protein NMR for structural genomics and structural biology studies, specifically in applications requiring the production of isotope-enriched proteins.
Materials and methods
Vector cloning
The complete sequence of the pCold(tet) vectors including pColdI(tet), FJ861699; pColdII(tet), FJ861700; pColdIII(tet), FJ861701; pColdIV(tet), FJ861702; pColdX(tet), FJ861703; and pColdTEV(tet), FJ861704 have been deposited in Genbank. Beginning with pColdI(SP-4) as the parental vector pColdI(tet) was generated in the following manner: The lacI gene and upstream region, up to but not including the cspA promoter, was replaced with the tetR gene and a modified version of the promoter-operator region of tetA and tetR from Tn10. The tetA and tetR promoter-operator region was cloned upstream of the existing cspA cold shock promoter in order to avoid disrupting transcription of the target gene by the cold shock promoter. In addition, the DNA sequence of the lac operator region within the 5′ UTR of cspA controlling induction of the target gene was mutated to that of the tet operator tetO1. The Tn10 promoter-operator region consists of two tet repressor binding sites, tetO1 and tetO2 with overlapping promoters driving transcription in opposite directions. Only transcription of the tetR gene was required therefore the −10 and −35 elements of the tetA promoter were mutated to avoid potential interference with the adjacent cspA promoter driving target gene expression. Generation of pCold II, III, and IV(tet) vectors proceeded by cloning the region upstream of the multiple cloning site between NheI and NdeI, containing the various fusion tag sequences from the pCold series into the pColdI(tet) backbone. pColdX(tet) and pColdTEV(tet) inserts were generated by synthesizing the appropriate fusion tag sequences via overlapping PCR and cloning the resulting DNA product into the pColdI(tet) vector between NheI and NdeI.
An ACA-less cspA gene, as previously described [3] was sub-cloned into the pColdI(tet) vector between the NdeI and BamHI restriction sites for protein expression. A truncated C-terminal ACA-less gene construct from Thr287-Gly381 of the Moloney Murine Leukemia Virus (M-MuLV) Integrase (IN) protein was codon optimized for E. coli [1, 5] and synthesized by overlapping PCR [6]. The resulting DNA product was then sub-cloned into the NdeI and BamHI restriction sites of the pCold(tet) series of vectors for protein expression.
Protein expression
pCold(tet) vectors containing the desired insert were transformed into chemically competent BL21(DE3) cells containing the vector pACYCmazF, as previously described [1]. Single colonies were picked and inoculated into 2 ml of M9CAA [1] containing 30 μg/ml chloramphenicol for selection of pACYCmazF and 100 μg/ml carbenicillin for selection of pCold(tet), and grown overnight (20 h). The following day, M9 medium containing 30 μg/ml chloramphenicol and 100 μg/ml carbenicillin was inoculated at 5 μl/ml and grown overnight (20 h). Fresh M9 medium containing 30 μg/ml chloramphenicol and 100 μg/ml carbenicillin was then inoculated at 10% with the overnight culture and grown at 37°C until the OD600 = 0.5 and then the cultures were rapidly cooled while swirling in an ice bath for 10–15 min. The MazF protein was induced with 1 mM IPTG and cultures were incubated at 15°C for an additional 2 h while shaking. Cell cultures are then centrifuged for 10 min at 4°C at 5000×g and resuspended in the desired condensed culture volume of M9 medium or 15N-enriched M9 medium (in which 15NH4Cl is the sole source of nitrogen) containing 30 μg/ml chloramphenicol, 100 μg/ml carbenicillin, 1 mM IPTG, and anhydrotetracycline. The level of anhydrotetracycline varied with the level of condensation; 0.2 μg/ml for 1× cultures, 0.75 μg/ml for 5× cultures, 1.5 μg/ml for 10× cultures, 3.0 μg/ml for 20× cultures, 4.5 μg/ml for 30× cultures, and 6.0 μg/ml for 40× cultures. To directly compare protein expression between different vectors at various degrees of culture condensation, each lane was loaded with the equivalent of 100 μl of un-condensed culture centrifuged and resuspended in lysis buffer (10 mM sodium phosphate pH 7.2; 1% β-mercaptoethanol; 1% SDS; 6 M urea).
Protein purification and mass spectrometry
1 ml of 40-fold condensed, 15N-enriched cultures containing either the expression plasmid pColdI(tet) cspA or pColdI(IPTG) cspA was centrifuged at 10,000×g and resuspended in 1.0 ml of lysis buffer (10 mM sodium phosphate pH 7.2; 1% β-mercaptoethanol; 1% SDS; 6 M urea). Cellular debris was pelleted by centrifugation at 12,000×g for 30 min and supernatant containing the His6 tagged CspA protein was purified by binding the cellular lysate to 40 μl of Ni-NTA agarose resin. Ni-NTA resin was washed twice with 1 ml of wash buffer (50 mM Na2HPO4–NaH2PO4; 300 mM NaCl; 50 mM imidazole; 5 mM β-mercaptoethanol, pH 8.0) and eluted in 100 μl of elution buffer (50 mM Na2HPO4–NaH2PO4; 300 mM NaCl; 250 mM imidazole; 5 mM β-mercaptoethanol, pH 8.0). Purified protein samples were run on a 15% SDS-PAG followed by Coomassie Blue staining and the CspA protein band was excised. In-gel digest with trypsin was performed in 50 mM NH4HCO3, pH 7.9 overnight. Peptides were extracted from the gel with 60% acetonitrile, 5% formic acid, and lyophilized. Samples were then solubilized in 0.1% trifluoroacetic acid (TFA), pH 2.5–3.0 prior to LC-MS/MS mass spectrometry. Chromatography was conducted using an ultimate nano-LC system (Dionex/LC Packings) and a fritless nanoscale column (75 μm × 15 cm) packed in-house with 3 μm, 200 A pore size Magic C18 stationary phase (Michrom Bioresource, Auburn, CA). The column was equilibrated in 0.1% formic acid (Solvent A), and samples were eluted using a linear gradient from 2 to 45% solvent B (0.1% formic acid in acetonitrile) over 30 min at a flow rate of 250 nl/min and analyzed by an LTQ linear ion trap mass spectrometer (ThermoFinnigan, San Jose, CA) equipped with a nanospray source (Proxeon Biosystems).
To select peptides suitable for monitoring, the tryptic peptides were analyzed by MS/Zoom scan/MSMS scan and the peptide GFGFITPDDGSK was selected. For quantitation of isotope incorporation, the analysis was repeated with MS in profile mode. The population of peaks corresponding to the isotope labeled peptides (based on retention time and MSMS information) were integrated for peak area. Mass spectrometry analysis was performed at the Biological Mass Spectrometry Facility of the UMDNJ-Robert Wood Johnson Medical School and Rutgers, The State University of New Jersey.
Results
Tetracycline inducible pCold vectors
A series of pCold vectors were generated, separating the IPTG induction of MazF from that of the ACA-less target gene. Figure 1A graphically describes the various features of pColdI(tet). In the new pCold(tet) vectors, the lacI gene that is responsible for repressing transcription from the lac operator was replaced with the tetR gene. Expression of the tetR gene is controlled by the promoter-operator region of Tn10 that is shared with and overlaps with that of the oppositely oriented tetA gene (Fig. 1B). Modifications were therefore made to the tetR/tetA promoter-operator region in an effort to prevent RNA Polymerase binding and transcription from the tetA promoter. While leaving the −10 and −35 elements from the tetR gene intact, the −10 and −35 elements of the tetA gene, 5′-TTGACA-3′ and 5′-TATTTT-3′ respectively, were mutated away from consensus to 5′-GCTCTA-3′ and 5′-CGCGTT-3′ respectively without disrupting the neighboring tetO1 and tetO2 operator sequences (Fig. 1B). In order to place the cspA promoter-driven target gene under the control of the tet operator, the lac operator sequence within the cspA 5′ UTR was replaced with the tet operator sequence to allow for tet repressor binding to tetO1 (Fig. 1A). The resulting changes successfully allowed for the independent induction of MazF (lacIR) from that of the target protein (tetRR). Figure 1C describes the multiple cloning site. Cloning of an ACA-less synthesized gene is readily obtained through the 5′ NdeI site followed by insertion of an in-frame stop codon. Several enzymes within the multiple cloning site are duplicated. The 3′ terminus of the target gene can utilize the BamHI site or alternative restriction enzyme sites localized in the multiple cloning site.
Fig. 1.
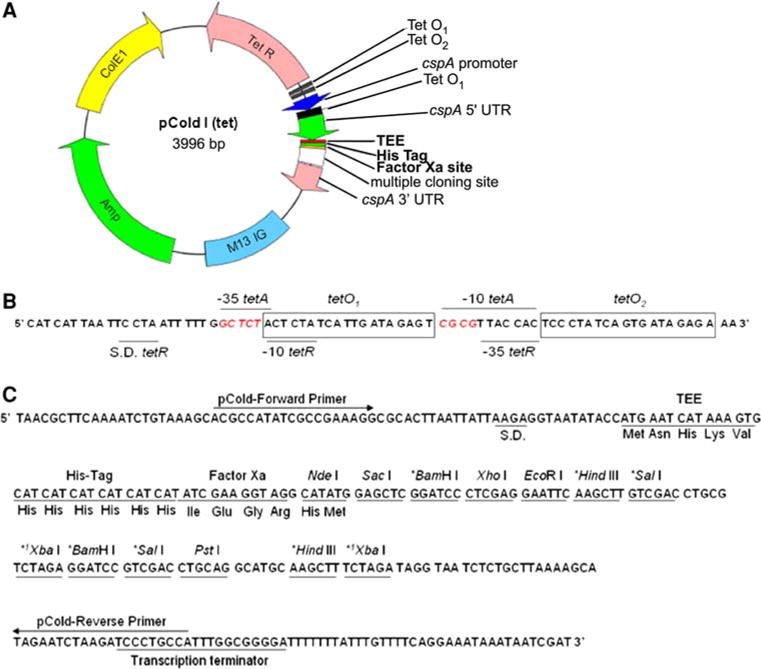
pColdI(tet) vector map. Vector features are highlighted. A pColdI(tet) vector map where the region shown in bold containing TEE (translation enhancing element), His Tag, and Factor Xa vary among the different pCold(tet) vectors. M13 IG intergenic region of M13 bacteriophage, ColE1 colicinogenic factor E1 for plasmid replication. B Expanded tetO1 and tetO2 region depicting the tetA–tetR overlapping promoter-operator. The mutated bases within the −35 and −10 element of the tetA promoter are highlighted in red italicized font. C Expanded multiple cloning site including suggested sequencing primers and fusion tag; S.D. Shine Delgarno sequence. * these restriction sites are duplicated in the multiple cloning region but can be used for 3′ end cloning. 1XbaI is also present in the tetR gene and therefore is not recommended for cloning
pCold(tet) vector series
A series of pCold(tet) vectors have been generated with various features for protein expression. As described in Table 1, each vector provides a unique combination of fusion tags for protein expression and purification purposes. The translational enhancing element (TEE) encoding the amino acids MNHKV, featured in all but two of these vectors [pCold IV(tet) and pCold X(tet)], has previously been shown to increase translational efficiency in cold shock mRNAs [4, 7]. His6 tags are included for purification purposes, where pColdI(tet) encodes a Factor Xa protease cleavage site and pColdTEV(tet) encodes a TEV protease cleavage site. While proteolytic removal of the His6 tag by Factor Xa from protein expressed in pColdI(tet) leaves an N-terminal His-Met as opposed to the three residues (Gly-His-Met) remaining from TEV cleavage of proteins expressed from pColdTEV(tet), the TEV protease efficiently cleaves proteins in a wide range of buffer conditions. In contrast, Factor Xa cleavage requires a specific buffer containing calcium that may decrease the solubility of the target protein. It should be noted that any desired fusion tag can be added in frame to the target gene during gene synthesis and cloned into the pColdIV(tet) vector. Together, the vectors described in Table 1 provide a range of options for protein expression and purification purposes in the cSPP system. As described in Table 1, the various fusion tags encoded in all but the pColdIV(tet) vector will alter the predicted size of the protein product between 0.76 kDa and 2.83 kDa depending on the vector.
Table 1.
Summary of pCold (tet) vectors
| Vector | TEE | His tag | Tag sequence | Protease | kDa (tag) |
|---|---|---|---|---|---|
| pCold I (tet) | + | + | MNHKVHHHHHHIEGR/HM | Factor Xa | 2.04 |
| pCold II (tet) | + | + | MNHKVHHHHHHM | − | 1.45 |
| pCold III (tet) | + | − | MNHKVHM | − | 0.76 |
| pCold IV (tet) | − | − | − | − | − |
| pCold X (tet) | − | + | MGHHHHHHSHM | − | 1.25 |
| pCold TEV (tet) | + | + | MNHKVHHHHHHSSGRENLYFQ/GHM | TEV | 2.83 |
pCold(tet) vector expression
To compare protein expression from the full series of pCold(tet) vectors, an ACA-less gene construct encoding a truncated version of the C-terminal domain of the M-MuLV IN protein was cloned into the multiple cloning site of all six pCold(tet) vectors. Figure 2 confirms that the ACA-less gene product is in fact produced in all six vectors with slight variation in the level of expression. The gel shown in Fig. 2 is representative of several independent inductions where size differences arise from the various fusion tags described in Table 1. Slight variations in culture density can be observed between cultures in Fig. 2 by observing background staining leading to the variability of intensity in the target protein band between samples.
Fig. 2.
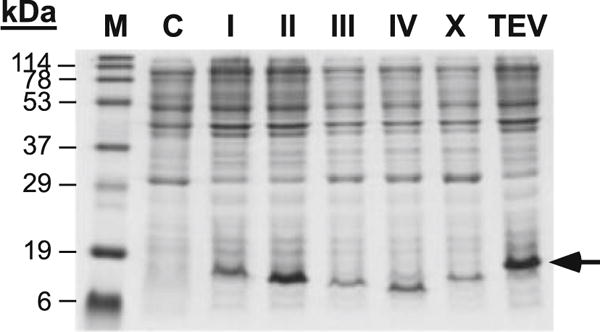
Expression of a truncated C-terminal domain from M-MuLV IN in the full panel of pCold(tet) vectors. M protein marker, C negative control (pColdIV(tet), no aTc induction), I pColdI(tet), II pColdII(tet), III pColdIII(tet), IV pColdIV(tet), X pColdX(tet), TEV pColdTEV(tet)
Comparison of expression and culture condensation in pCold(IPTG) and pCold(tet) vectors
To determine whether proteins expressed from pCold(tet) vectors not only maintained the ability to undergo culture condensation similar to the original IPTG inducible pCold(IPTG) vector, but were improved regarding homogeneity of isotope enrichment (15N), a side by side comparison of expression and culture condensation was carried out. For comparison, the bacterial cold shock cspA gene product was expressed as an ACA-less cassette in the pColdI vector backbone under either IPTG (pColdI(IPTG)) or anhydrotetracycline (pColdI(tet)) induction. Upon reaching the correct cell density, the IPTG inducible MazF toxin was expressed for 2 h prior to culture condensation to allow for MazF mediated degradation of cellular mRNAs and cell growth arrest. After 2 h of MazF induction, cells were centrifuged and resuspended in various volumes of 15N-enriched M9 medium. Figure 3A and B compares the various condensed states of protein expression resulting from the IPTG inducible (a) and tet inducible (b) pCold vectors. In both cases, little difference in expression is observed from uncondensed (1×) to 40-fold (40×) condensation. It can be concluded that expression from the pColdI(tet) vector is comparable to expression from the pColdI(IPTG) vector.
Fig. 3.
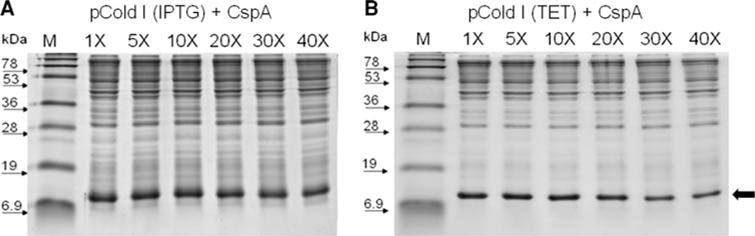
Comparison of protein expression and condensation of pColdI(IPTG) and pColdI(tet). Cultures expressing E. coli protein CspA were condensed 5×, 10×, 20×, 30×, 40×, and compared to uncondensed (1×) cultures in the (A), IPTG inducible pColdI(IPTG), and (B), tetracycline inducible pColdI(tet) vectors
Comparing isotope incorporation in pCold (IPTG) and pCold (tet) vectors
Figure 4A and B shows a graphical representation of the time course of induction and isotope incorporation. For the IPTG induced system, the addition of IPTG required to induce MazF expression prior to culture condensation and introduction of isotope enriched medium leads to a substantial amount of unwanted expression from the pCold(IPTG) vectors. The grey shaded region highlights the period where IPTG induced protein expression occurs in the absence of culture condensation and introduction of isotope enriched medium. The dual IPTG/tet induced system is predicted to eliminate the substantial amount of unlabeled expression from the ACA-less target gene. To confirm the improved yield of isotope enriched protein product expected with the dual induction cSPP(tet) system, mass spectrometry was performed on 40× condensed, 15N-enriched CspA protein. Figure 4C and D, displaying a representative tryptic fragment GFGFITPDDGSK of CspA, highlights the dramatically improved ratio of isotope enriched to unenriched product when using the pColdI(tet) vector as compared to the pColdI(IPTG) vector. The predicted weight of the fully labeled, double charged peptide is 626.70 Da and the unlabeled peak is 621.00 Da. Remarkably, the 621.00 mass unit peak constitutes 20% of the total protein fraction when using the pColdI(IPTG) vector but is nearly absent, representing only 1.3% of the total protein, when using the pColdI(tet) vector. Comparison of the two plots shows that the labeling of the anhydrotetracycline induced protein (Fig. 4D) is dramatically less heterogeneous and more complete than protein expressed from the IPTG inducible pColdI vector (Fig. 4C). The fully labeled peak from the pColdI(tet) product appears at 626.81 mass units, the predicted weight for 100% isotope incorporation, compared to the 625.81 mass unit peak from product produced with the pColdI(IPTG) vector. Furthermore the isotope enrichment is more complete as determined by comparing the width of the half height of the peak. The distribution from product produced with the pColdI(IPTG) vector ranges 2.5 mass units (representing a range of isotopic incorporation of 60–100%), whereas the peak from the pColdI(tet) vector product ranges only 1.5 mass units (representing a range of isotopic incorporation of 90–100%).
Fig. 4.
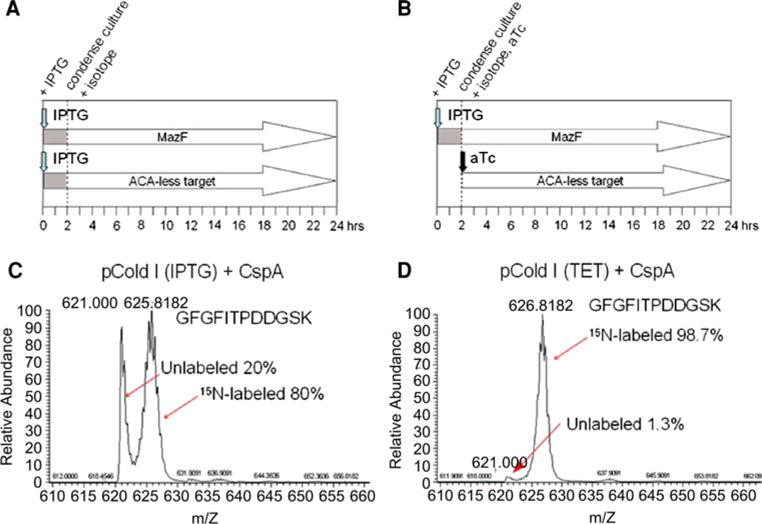
Isotope incorporation in the coinducible versus dual inducible cSPP systems. Panels A and B presents a schematic of the isotope incorporation between (A) the coinducible pCold(IPTG) vectors, and (B) the dual inducible pCold(tet) vectors. Grey shading indicates protein produced in unlabeled medium; blue arrows indicate time of IPTG induction; black arrow indicates time of anhydrotetracycline (aTc) induction. Panels C and D depict representative trypsin fragment of CspA protein expressed at 40× condensation in 15N-labeled minimal medium. Predicted weight of unlabeled and fully labeled, double charged peptide is 621.00 and 626.70 Da, respectively
Discussion
The ability to obtain isotope enriched recombinant protein is critical to the success of structural studies involving protein NMR spectroscopy. While it is possible to achieve protein production and isotope enrichment with conventional protein expression methods, often times the high cost is prohibitive for certain applications, including particularly the production of perdeuterated proteins (W. M. Schneider, personal communication). With the advent of the cSPP system, it is now possible to not only achieve a high level of isotope enriched protein expression but to do so at a fraction of the cost of traditional methods.
Using cSPP, the gene must first be synthesized devoid of ACA sequences, however the cost incurred and time required for the generation of the gene is dwarfed in comparison to the cost incurred in preparing many isotopically enriched samples, such as perdeuterated samples, by conventional methods. Synthesis of genes that encode proteins suitable for NMR analysis (10–25 kDa), as an ACA-less cassette costs between $150 and $300, using commercial sources, whereas growth of 1L of perdeuterated cultures can cost in excess of $2500. Costs for gene synthesis are also expected to drop significantly over the coming years, making the production of ACA-less genes routine.
One weakness of the IPTG inducible set of cSPP vectors, however, was the accumulation of unlabeled protein due to expression prior to culture condensation. Specifically, although the previously described cSPP system [1, 2, W. M. Schneider, personal communication] allowed for protein production and isotope enrichment at a fraction of the cost incurred with traditional methods, a substantial (10–20%) of the target protein remained unlabeled due to the coinduction of the target protein for 2 h with the MazF toxin upon addition of IPTG and prior to culture condensation. While in many NMR applications such as 1H−15N detected triple resonance experiments, this unenriched population of protein is not visible, it contributes effectively to lowering the yield of isotope-enriched species and reducing the effective signal-to-noise of the NMR experiment. These unenriched species in the NMR sample can also create artifacts and complicate interpretation of X-filtered NMR experiments [8, 9].
By temporally separating the induction of the MazF toxin from that of the target protein it is now possible to nearly eliminate contamination from the unlabeled target protein and obtain a protein product that is much more homogeneous with respect to isotopic-enrichment. Furthermore, the wide range of pCold(tet) vectors summarized in Table 1 provide an arsenal of tools to choose from containing a range of features including histidine tags for purification, TEE for improved cold shock expression, and protease cleavable fusion tags. Using this set of vectors, it is now possible to test several different constructs for optimal protein expression in the cSPP(tet) system. The results presented in this manuscript show two proteins, one bacterial (CspA) that can be condensed 40×, and one viral (M-MuLV IN C-terminal domain) that can be condensed 10× (data not shown).
Additional experiments have allowed the N-terminal Zn binding domain of M-MuLV IN to be similarly expressed, condensed 15×, and selectively labeled with amino acid precursors (data not shown). Yields from the cSPP system of the M-MuLV IN protein were between 5–6 mg/l culture. Any loss of yield in using the cSPP system versus a conventional pET vector was compensated by the increase in incorporation of label for NMR analysis (data not shown). Recently, the cSPP system has been applied for production of perdeuterated soluble and membrane proteins (W. M. Schneider, personal communication). Thus the generality of this vectors system is firmly established.
Combining dual induction with the already established cSPP system offers the benefit of increased yield of isotope enriched protein and a significantly improved signal to noise ratios in NMR studies. Previously, the cSPP method was shown to be effective in producing proteins enriched in selenomethionine [2], suitable for X-ray crystallography. The dual inducible cSPP(tet) system extends the established cSPP system to various applications such as 2D NMR on perdeuterated proteins (W. M. Schneider, personal communication) and other systems that would not be possible using the coinduced cSPP system. The condensability of the culture using these systems opens the door to many applications in structural and functional genomics in which high protein yields are required in small sample volumes, including microtiter plate fermentation methods. It is our expectation that the dual inducible expression systems described here, allowing condensation of 10–40 fold in fermentations for isotopic-enrichment, will become the default systems used for producing isotope-enriched proteins in E. coli for NMR, mass spectrometry, neutron diffraction, and a wide range of other applications in molecular biophysics and biotechnology.
Conclusion
This article describes a series of vectors directing the temporal induction of proteins using the condensed single protein production (cSPP) system. The studies overcome a major barrier in protein labeling by suppressing the production of unlabeled target protein species, and results in the production of proteins with greater than 95% isotope enrichment suitable for NMR studies.
Acknowledgments
We thank M. Suzuki, L. Mao, Y. Tang, and P. Rossi for helpful discussions in the course of this work, and for their comments on the manuscript. This work was supported by the National Institutes Health Grants RO1 GM070837 (to M.J.R.), U54 GM074958 (G.T.M. and M.I.), U54 GM75026 (G.T.M. and M.I.), 1R01 GM085449 (M.I.). W.M.S. was supported by NIH training grants T32 GM08360 and T32 A1007403.
Abbreviations
- cSPP
Condensed single protein production
- M-MuLV IN
Moloney Murine Leukemia Virus Integrase
- TEE
Translation enhancing element
Contributor Information
William M. Schneider, Department of Biochemistry, University of Medicine and Dentistry of New Jersey-Robert Wood Johnson Medical School, 675 Hoes Lane Rm 636, Piscataway, NJ 08854, USA
Masayori Inouye, Department of Biochemistry, University of Medicine and Dentistry of New Jersey-Robert Wood Johnson Medical School, 675 Hoes Lane, Piscataway, NJ 08854, USA; New York Consortium for Membrane Protein Structure (NYCOMPS), Piscataway, NJ 08854, USA; Northeast Structural Genomics Consortium, Piscataway, NJ 08854, USA.
Gaetano T. Montelione, Department of Biochemistry, University of Medicine and Dentistry of New Jersey-Robert Wood Johnson Medical School, 675 Hoes Lane, Piscataway, NJ 08854, USA New York Consortium for Membrane Protein Structure (NYCOMPS), Piscataway, NJ 08854, USA; Northeast Structural Genomics Consortium, Piscataway, NJ 08854, USA; Department of Molecular Biology and Biochemistry, Center for Advanced Biotechnology and Medicine, Rutgers University, Piscataway, NJ 08854, USA.
Monica J. Roth, Email: roth@umdnj.edu, Department of Biochemistry, University of Medicine and Dentistry of New Jersey-Robert Wood Johnson Medical School, 675 Hoes Lane Rm 636, Piscataway, NJ 08854, USA.
References
- 1.Suzuki M, Mao L, Inouye M. Single protein production (SPP) system in Escherichia coli. Nat Protoc. 2007;2:1802–1810. doi: 10.1038/nprot.2007.252. [DOI] [PubMed] [Google Scholar]
- 2.Suzuki M, Rohini R, Zheng H, Woychik N, Inouye M. Bacterial bioreactors for high yield production of recombinant protein. J Biol Chem. 2006;281:37559–37565. doi: 10.1074/jbc.M608806200. [DOI] [PubMed] [Google Scholar]
- 3.Suzuki M, Zhang J, Liu M, Woychik NA, Inouye M. Single protein production in living cells facilitated by an mRNA interferase. Mol Cell. 2005;18:253–261. doi: 10.1016/j.molcel.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 4.Qing G, Ma L-C, Khorchid A, Swapna GVT, Mal TK, Takayama MM, Xia B, Phadtare S, Ke H, Acton T, et al. Cold-shock induced high-yield protein production in Escherichia coli. Nat Biotech. 2004;22:877–882. doi: 10.1038/nbt984. [DOI] [PubMed] [Google Scholar]
- 5.Maloy SR, Stewart VJ, Taylor RK. Genetic analysis of pathogenic bacteria: a laboratory manual. Cold Spring Harbor Laboratory Press; NY: 1996. [Google Scholar]
- 6.Holler TP, Foltin SK, Ye QZ, Hupe DJ. HIV1 integrase expressed in Escherichia coli from a synthetic gene. Gene. 1993;136:323–328. doi: 10.1016/0378-1119(93)90488-o. [DOI] [PubMed] [Google Scholar]
- 7.Etchegaray J-P, Inouye M. Translational enhancement by and element downstream of the initiation codon in Escherichia coli. J Biol Chem. 1999;274:10079–10085. doi: 10.1074/jbc.274.15.10079. [DOI] [PubMed] [Google Scholar]
- 8.Clore GM, Omichinski JG, Sakaguchi K, Zambrano N, Sakamoto H, Appella E, Gronenborn AM. High-resolution structure of the oligomerization domain of p53 by multidimensional NMR. Science. 1994;265:386–391. doi: 10.1126/science.8023159. [DOI] [PubMed] [Google Scholar]
- 9.Ikura M, Bax A. Isotope-filtered 2D NMR of a protein peptide complex—study of a skeletal-muscle myosin light chain kinase fragment bound to calmodulin. J Am Chem Soc. 1992;114:2433–2440. [Google Scholar]